

Abstract
The natural history of a patient’s cancer is often characterised by genetic diversity and sequential sweeps of clonal dominance. It is therefore not surprising that identifying the most appropriate tumour-associated antigen for targeted intervention is challenging. The 5T4 oncofoetal antigen was identified by searching for surface molecules shared between human trophoblast and cancer cells with the rationale that they may function to allow survival of the foetus as a semi-allograft in the mother or a tumour in its host. The 5T4 protein is expressed by many different cancers but rarely in normal adult tissues. 5T4 molecules are 72 kD, heavily N-glycosylated proteins with several leucine-rich repeats which are often associated with protein–protein interactions. 5T4 expression is associated with the directional movement of cells through epithelial mesenchymal transition, potentiation of CXCL12/CXCR4 chemotaxis and inhibition of canonical Wnt/beta-catenin while favouring non-canonical pathway signalling; all processes which help drive the spread of cancer cells. The selective pattern of 5T4 tumour expression, association with a tumour-initiating phenotype plus a mechanistic involvement with cancer spread have underwritten the clinical development of different immunotherapeutic strategies including a vaccine, a tumour-targeted superantigen and an antibody drug conjugate. In addition, a chimeric antigen receptor T cell approach targeting 5T4 expressing tumour cells is in pre-clinical development. A key challenge will include how best to combine each 5T4 targeted immunotherapy with the most appropriate standard of care treatment (or adjunct therapy) to maximise the recovery of immune control and ultimately eliminate the tumour.
Keywords: 5T4 oncofetal antigen, Trophoblast glycoprotein (TBPG), Cancer vaccine, Chimeric antigen receptors, Antibody drug targeting, Superantigen therapy
Identifying tumour-associated antigens with therapeutic relevance
There has been considerable effort directed at the development of immunotherapeutic approaches for the treatment of cancer; many of which depend on targeting TAAs. While numerous TAAs have been identified, not all have the appropriate properties to enable safe and effective immune targeting. Such properties include a highly restricted expression profile on normal tissues but broad expression across many different cancer types. Furthermore, cell surface expression is an important property for antibody-targeted therapies while the lack of a tolerised repertoire providing for suitable antibody or cell-mediated adaptive immune responses is critical to vaccine approaches.
Cancers arise through genetic changes and in some cases TAAs are generated through the specific driver mutations, translocations or viral oncogenes associated with particular cancers [1, 2]. These genetic changes provide for the initial development of the clonal progeny but over time additional mutations, frequently reflecting increased genomic instability, produce a plethora of diverse sub-clones. Thus, individual tumours show a variable mutational burden resulting in extensive inter- and intra-tumour heterogeneity [3]. Some clones may become more dominant, but overall this process generates a reservoir of independent lineages of branched evolution associated with a range of fitness for survival in the face of natural immune mechanisms or any exogenous therapy. Independently evolving sub-clones also provide the opportunity for differential seeding and metastatic spread. In spite of this individual patient tumour heterogeneity, there is increasing evidence of a significant association between survival and CD8 T cell infiltration in tumours [4]. It seems most likely that effective immune control must include recognition of many different idiotypic TAAs expressed by the heterogeneous and independent tumour cell lineages in a patient. It is equally clear that multiple immune-escape mechanisms are important in allowing for tumour progression [5]. Controversially, it is possible that relapse may not always represent drug resistance alone, but instead the growth of pre-existing distinct tumour clones released from existing natural immune control as a result of the treatments which inadvertently damage the immune system.
Given that the mutational landscape of an individual tumour and indeed different cancers is variable, it is perhaps not surprising that immune checkpoint inhibitors, such as anti-CTLA-4 and PD-1, appear to show increased efficacy in patients with a higher mutational load and a larger array of neo-antigens [6]. The intratumoural genetic diversity is difficult to accurately document, as it will reflect sequential sweeps of clonal dominance in the natural history of the patient’s cancer. Given the complex network of interacting factors, it is perhaps not surprising that identifying the most appropriate TAA for immunotherapy is challenging.
A further complication is the role of stem cell/developmental processes relevant to normal tissue renewal that are also functional in tumours. Thus, teratocarcinomas, small cell lung and mammary carcinomas all show evidence of normal tissue differentiation consistent with a hierarchical tissue organisation derivative from a “cancer stem cell” function. There is now evidence for such stem cells defined by tumour-initiating cell (TIC) properties recapitulating the primary tissue/tumour architecture in xenograft models in many different cancer types [3]. In normal tissues, stem cells remain quiescent until requisitioned for the replacement of the tissue structure through differentiation and stem cell renewal. Thus, while not all tumour cells have TIC function, there is evidence of dormant TIC sub-populations that drive tumour growth in different cancers including breast, melanoma and leukaemia [3]. It is these subpopulations of cancer stem cells which can evade or survive the toxicity of most conventional chemotherapies which are mostly active on dividing cells. Surviving clonogenic cells provide the opportunity for acquisition of new mutations or further selection of different pre-existing clones and critically seed tumour progression. As such, the ability to eliminate TICs would likely have a profound impact on tumour progression.
This review is focused on the utility of the 5T4 oncofoetal antigen as a target for cancer therapy in the context of the likely properties required of a tumour-associated antigen to deliver safe and effective treatment. In 2009, the National Cancer Institute (NCI) published results from a project which aimed “to develop a well-vetted, priority-ranked list of cancer vaccine target antigens based on pre-defined and pre-weighted objective criteria” [2]. The NCI project ranked 75 cancer antigens (but not including 5T4) using a published series of weighted criteria for comparison of potential vaccine targets. Our conservative evaluation of 5T4 antigen in this process is summarised in Table 1; the overall score of 0.671 ranks at position 9 (of 75 TAAs) which falls above NY-ESO-1, CEA (another oncofoetal antigen), gp100, PSA and p53. (N.B. Other therapeutic modalities that use antibodies would have different weighted properties).
Table 1.
5T4 properties in relation to the characteristics of an ideal cancer antigen
| Criteria (weight) | 5T4 weighting sub-criteria (allocated score) | Overall score |
|---|---|---|
|
Therapeutic function (0.32) Controlled vaccine trial suggestive |
Fair data controlled showing vaccine-induced clinical responses (75 %) | 75 % × 0.32 |
|
Immunogenicity (0.17) Immunogenic in clinical trials |
T cell and/or antibody responses in clinical trials (100 %) | 100 % × 0.17 |
|
Oncogenicity (0.15) Oncogenic “self” protein |
Uncertain function but increased expression correlated with decreased survival (25 %) | 25 % × 0.15 |
|
Specificity (0.15) Absolutely specificity |
Oncofoetal antigen with little expression in adult tissue (54 %) | 54 % × 0.15 |
| Expression level and % of positive cells (0.07) | Highly expressed on most cancer cells in patients designated for treatment (37 %) | 37 % × 0.07 |
| Stem cell expression (0.05) | Evidence for expression on putative cancer stem cells (100 %) | 100 % × 0.05 |
| Number of patients with antigen-positive cancers (0.04) | High/Low-level expression in many patients of particular tumour types (16 %) | 16 % × 0.04 |
| Number of epitopes (0.04) | Ability to bind to most MHC molecules (100 %) | 100 % × 0.04 |
| Cellular expression location (0.02) | Normally expressed on cell surface with no circulating antigen (100 %) | 100 % × 0.02 |
| Total score | 0.6708 | |
Based upon the criteria outlined in Table 1, 5T4 is an attractive target for immune intervention as it has many of the favourable properties of a TAA including a good tumour/normal tissue expression profile, an association with tumour-initiating subpopulations and several functional attributes that promote cancer spread. As such, targeting 5T4 expressing tumour cells could potentially have a significant impact on cancer progression.
5T4 trophoblast glycoprotein (TBPG)
Antigen identification, structure and expression
5T4 Trophoblast glycoprotein was discovered in the context of trying to identify shared cell surface molecules that may function to allow survival of the foetus as a semi-allograft in the mother, or a tumour in its host. The rationale was that such shared expression would reflect common functions relevant to growth, invasion or altered immune surveillance in the host. Murine monoclonal antibodies were raised against purified glycoproteins from trophoblast membrane preparations from term human placenta and initially screened against different cancer cell lines and human peripheral blood mononuclear cells. Further screening using the 5T4 monoclonal antibody (mAb) by immunohistochemistry indicated the antigen was expressed by many different cancers but with a restricted normal tissue distribution [7]. A series of biochemical and genomic studies identified 5T4 molecules as N-glycosylated proteins with an apparent molecular size of 72 kD and are encoded on chromosome 6q14-15 [8–10]. The human gene encodes a 42 kD transmembrane protein core which contains several leucine-rich repeats (LRRs) [11] that are associated with protein–protein interactions of a functionally diverse set of molecules [12]. The extracellular part of the molecule has several LRRs in two domains separated by a short hydrophilic sequence; there is a transmembrane domain and a short cytoplasmic sequence. Importantly, the 5T4-specific mAb recognises a conformationally dependent epitope that relies on the integrity of the intramolecular S–S bonds and the indirect presence of the complex N-linked glycosylation [9]. When human 5T4 was overexpressed in murine fibroblasts, the cells became more spindle shaped and had reduced adherence [13] while in normal epithelial cells there was E-Cadherin down-regulation, increased motility and cytoskeletal disruption [14]. The cytoskeletal disruption through 5T4 overexpression is dependent on the 5T4 cytoplasmic domain, which interacts with TIP2/GIPC, known to mediate links to the actin cytoskeleton [15]. These studies were the first to indicate a possible association of 5T4 expression with cancer spread.
Immunohistochemistry (IHC) of frozen sections established that the 5T4-specific monoclonal antibody detected antigen expression by many different types of carcinoma but demonstrated only low-level expression in some normal adult tissue epithelia [16]. Importantly, 5T4 was expressed in many different primary and metastatic cancers, frequently at high levels; in some cases, there was an additional stromal expression. Linking a role for 5T4 in tumour development and spread, it has been shown that 5T4 expression in colorectal, gastric and ovarian cancers correlates with poorer clinical outcome [21–23, 27]. A summary of the 5T4 expression by different cancers evaluated using IHC of cryostat sections with the original 5T4 mAb is shown in Table 2.
Table 2.
5T4 expression in cancer
| Cancer | Incidence | % Positive 5T4 expression (N) | References |
|---|---|---|---|
| Bladder |
EU5: 80,431a US: 76,960b |
100 (4/4) | [16] |
| Breast |
EU5: 248,658a US: 249,260b |
96 (52/54) | OXB unpublished data |
| Cervical |
EU5: 15,945a US: 12,990b |
100 (5/5) 97 (64/66) |
[16] [19] |
| Colorectal |
EU5: 225,502a US: 134,490b |
100 (17/17) 54 (39/72) |
[20] [21] |
| Gastric |
EU5: 50,017a US: 26,370b |
56 (15/27) 40 (35/86) 52 (32/62) |
[22] [23] [24] |
| Non-small cell lung cancer |
EU5: 165,912a US: 190,731b |
100 (30/30) >95 (N/A) |
OXB unpublished data [25] |
| Mesothelioma |
UK: 2,215c US: 3,000d |
100 (31/31) | [26] |
| Ovarian |
EU5: 27,104a US: 22,280b |
71 (51/72) 100 (19/19) 79 (26/33) |
[27] OXB unpublished data OXB unpublished data |
| Pancreatic |
EU5: 51,402a US: 53,070b |
100 (3/3) >95 (N/A) |
[16] [25] |
| Prostate |
EU5: 242,887a US: 180,890b |
100 (23/23) 84 (32/38) |
OXB unpublished data OXB unpublished data |
| Renal |
EU5: 57,126a US: 62,700b |
95 (20/21) | [28] |
Unpublished data are provided by Oxford BioMedica (OXB)
The table summarises the frequency of 5T4 expression in different cancers. All immunohistochemistry was performed using the same monoclonal antibody which was used to identify 5T4 initially. The number of tumours stained for 5T4 expression is shown in parentheses. Incidence figures are from
aEUCAN website (http://eco.iarc.fr/EUCAN/Default.aspx)
b [17]
c http://www.hse.gov.uk/statistics/causdis/mesothelioma/
d[18]
Several “5T4-specific” antibodies made against specific peptides or sequences of the 5T4 molecule have become available commercially. However, the specificity may not be identical to the original mAb as the precise epitopes have often not been identified and could include parts of the 5T4 molecule which contains leucine-rich repeats which are shared by a large number of proteins of diverse function and expression. However, for antibody-targeted therapies, the individual property of the reagent in the context of live tissues is the only significant relevant property.
5T4 and tumour-initiating cells
There is increasing evidence for key sub-populations of tumour-initiating cells reflecting normal tissue renewal properties retained and exploited for advantage by developing cancers. Poorly differentiated tumours in NSCLC have been associated with shorter patient survival and shorter time to recurrence following treatment. Using multiple experimental models with clinico-pathologic analysis of patient tumours to delineate a cellular hierarchy in NSCLC, it has been shown that 5T4 is expressed on tumour-initiating cells and associated with worse clinical outcome. Despite heterogeneous expression of 5T4 in NSCLC patient–derived xenografts, treatment with an anti-5T4 antibody–drug conjugate resulted in complete and sustained tumour regression. Thus, the aggressive growth of heterogeneous solid tumours can be blocked by therapeutic agents that target a 5T4 expressing subpopulation of cells near the top of the cellular hierarchy [29].
Using gene expression profiling of diagnostic paediatric B-cell precursor (BCP)-ALL bone marrow samples stratified by cytogenetics for risk of relapse, it was shown that 5T4 protein expression correlates with risk of relapse [30]. A recent study using leukaemias from patients stratified by treatment response (minimal residual disease MRD), and established as mouse primagrafts, explored the role of 5T4 in tumour initiation and concluded that 5T4 is a marker of either leukaemia-initiating cells or at least more immature clonogenic cells [30]. The relative resistance to chemotherapy and an increased incidence of relapse in BCP-leukaemia could be attributable to 5T4 expression leading to increased clonogenicity, such that a few cells can reconstitute disease burden.
5T4-associated functions
5T4 and epithelial mesenchyme transition (EMT)
Overexpression of 5T4 in normal murine epithelial cells is associated with E-cadherin down-regulation [14] which is a key component of EMT; this occurs during embryonic development and is important for the metastatic spread of epithelial tumours [31]. 5T4 was shown to be a marker of the early differentiation of mouse and human embryonic stem (ES) cell, and this process involves an E- to N-cadherin switch, upregulation of E-cadherin repressor molecules (Snail and Slug proteins), increased matrix metalloproteinase (MMP-2 and MMP-9) activity and motility, all classic EMT features [32–35]. Undifferentiated knock-out (KO) E-Cadherin ES cells constitutively express cell surface 5T4 molecules while antibody induced down-regulation of E-Cadherin in ES cells induced 5T4 membrane expression, increased motility, altered actin cytoskeleton arrangement and a mesenchymal phenotype. These observations are consistent with E-cadherin somehow preventing 5T4 cell surface expression; the possible mechanism being through stabilisation of cortical actin cytoskeletal organisation. Co-expression of 5T4 and factors involved in the epithelial-to-mesenchymal transition was also observed in undifferentiated but not in differentiated lung tumour cells [29].
5T4 modulation of chemokine signalling
5T4 molecules have been shown to be involved in the functional expression of CXCR4 at the cell surface in some embryonic and tumour cells [36, 37]. Both CXCL12 and CXCR4 expression have been associated with tumourigenesis in many cancers, and it is believed that CXCR4 expression facilitates the spread to tissues that highly express CXCL12 including lung, liver, lymph nodes and bone marrow [38, 39]. 5T4 is expressed by putative leukaemia initiating cells in BCP-ALL, and these cells show the associated property of CXCL12/CXCR4 chemotaxis [30]. 5T4-positive leukaemia-initiating cells are likely attracted by CXCL12 produced by extramedullary sites where there is decreased therapeutic bioavailability leading to disease relapse following treatment.
Interestingly, in the absence of 5T4 expression, CXCR7 (the other receptor for CXCL12 with tenfold higher affinity) is upregulated and activates a distinct signal transduction pathway with slower kinetics involving transactivation of the EGFR, with downstream consequences for proliferation/anti-apoptosis rather than chemotaxis [37]. Such differential responses can potentially impact the behaviour of a primary tumour. Thus, while 5T4 surface expression by cells at the periphery would result in chemotactic responses to CXCL12, from, for example, secreting endothelial cells and facilitate metastatic spread where there is preferential CXCR7 expression with higher affinity for the ligand, detection of lower chemokine levels could lead to stimulation of cell growth/survival. It appears that cell surface 5T4 associates with the use of the CXCR4 rather than the CXCR7 receptor, and the balance of the receptor expression impacts through CXCL12 exposure to support both the spread and growth of a tumour [40].
5T4 modulation of Wnt signalling
Wnt protein intracellular signalling is a central component of many aspects of cellular regulation critical to normal development, homoeostasis and regeneration, while misregulation can lead to disease, including cancer [41]. There are two pathways, the most characterised being the canonical Wnt/β-catenin pathway while non-canonical Wnt signalling through a cell autonomous planar cell polarity (PCP) type pathway can drive the modulation of actin and microtubular skeletons facilitating cell movement in development of cancer. 5T4 has been shown to interfere with Wnt/β-catenin signalling and concomitantly activate non-canonical Wnt pathways. 5T4 binds to the Wnt co-receptor LRP6 and inhibits Wnt-induced LRP6 internalisation into endocytic vesicles, a process that is required for pathway activation thereby modulating Wnt/β-catenin signalling by regulating LRP6 subcellular localisation. In addition, 5T4 enhances β-catenin-independent Wnt signalling in promoting a non-canonical function of Dickkopf1 [42]. These results suggest that 5T4 facilitates pathway selection in Wnt-receiving cells, inhibiting Wnt/β-catenin canonical while concomitantly activating the non-canonical Wnt signalling pathway associated with increased motility [40]. 1.8 Å resolution crystal of the extracellular domain of 5T4 and associated cell biological studies have provided a structural basis for inhibition of Wnt/β-catenin signalling [43].
Exploiting 5T4 expression for cancer therapy
The selective pattern of 5T4 tumour expression, association with a tumour-initiating phenotype plus a mechanistic involvement with cancer spread have underwritten the development of several different immunotherapeutic strategies (see Fig. 1). The approaches which have reached clinical development include a vaccine, a tumour-targeted superantigen and an antibody drug conjugate; each is discussed below.
Fig. 1.
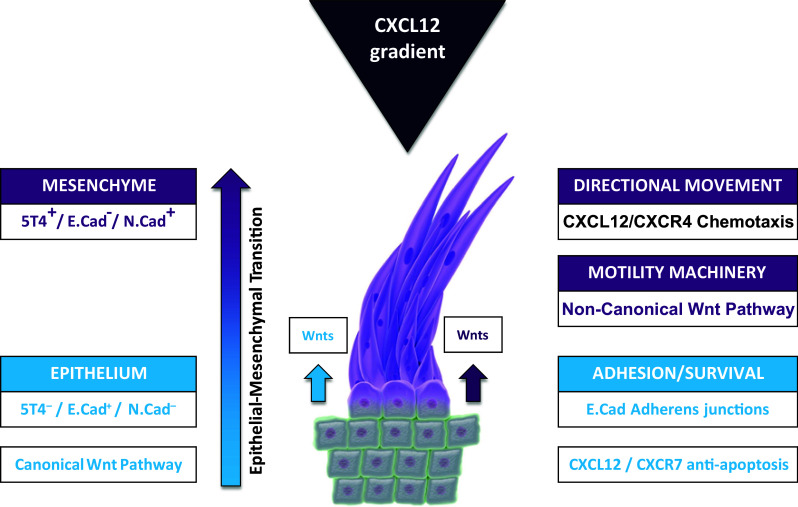
Integrated 5T4 regulation of both chemokine and Wnt pathways acts to promote tumour cell migration and spread. Cell surface 5T4 expression is associated with a mesenchymal phenotype, a Wnt non-canonical influenced cytoskeletal organisation for motility and facilitation of CXCL12/CXCR4-directed chemotaxis (purple pathways and phenotypes). 5T4 negativity is associated with epithelial morphology with E-Cadherin-mediated adhesions, canonical Wnt-responsive pathways and a CXC12/CXCR7 favoured proliferation/anti-apoptopic outcome (blue pathways and phenotypes). Both non-canonical (purple) and canonical (blue) Wnt ligands are made by tumour-associated tissues; CXCL12 is often made by vascular epithelium
5T4 vaccine development
Preclinical studies
Prior to the clinical development of a vaccine approach targeting 5T4, it is important to show therapeutic activity in animal tumour models and the presence of a T cell repertoire in humans; results from such studies are described below. The availability of a 5T4 KO mouse provided an opportunity to analyse the mechanisms by which endogenous expression of 5T4 influences autologous T cell immunity and tolerance. 5T4 is expressed in the murine thymus and could potentially influence the repertoire and/or induction of specific regulatory T cells (Tregs) impacting either natural or vaccine-induced immunity. Vaccination of 5T4KO mice with a recombinant adenovirus vector expressing murine 5T4 produced strong responses to CD8 and CD4 m5T4 T cell epitopes, while in wild-type (WT) mice the responses were either significantly reduced (only low avidity CD8) or absent (CD4). It appears that in WT mice naturally occurring 5T4-specific Tregs are preferentially generated whereas in 5T4KO mice 5T4-specific CD4 T helper cells are produced. [44]. Therefore, clonal deletion and/or active suppression by Tregs is involved in the development of 5T4 tolerance in WT animals and this might potentially limit the immunogenicity of 5T4 vaccines in humans.
The use of a viral vector such as modified vaccinia Ankara (MVA) to deliver 5T4 may overcome lack of 5T4-specific T helper cells and was chosen to evaluate immunogenicity and anti-tumour activity in pre-clinical studies [45]. Importantly, immunisation of mice with MVA expressing human or mouse 5T4 constructs (MVA-5T4 or MVA-m5T4, respectively) induced 5T4-specific antibody responses. Vaccination of mice with MVA-5T4 protected them against challenge with syngeneic tumour cells expressing human 5T4, and this effect was mediated by 5T4-specific antibodies and was dependent on the presence of CD4 T cells. Immunisation of mice with MVA-m5T4 was able to protect against challenge with B16 cells expressing m5T4 and, importantly, there was no sign of autoimmune toxicity. Vaccination with MVA-5T4 was also able to treat established CT26-h5T4 lung tumours and to some extent B16.h5T4 subcutaneous tumours.
In terms of an immune repertoire in humans, CD8 and CD4 T cells recognising HLA-restricted 5T4 peptides were identified by methods using monocyte-derived dendritic cells (DC) to stimulate peripheral blood lymphocytes from healthy individuals with the absence of CD4 T cells or depletion of T regulatory cells, respectively, a requirement [46, 47]. Collectively, these data supported the clinical development of MVA-5T4 (TroVax®).
Clinical studies of TroVax®
TroVax has been tested in 10 phase I and II studies in renal, colorectal and prostate cancer; all of which demonstrated that TroVax was well tolerated and induced 5T4-specific antibody and/or cellular immune responses in the majority of patients. Surprisingly, while only low-level 5T4-specific T cell responses were detectable in mice after multiple rounds of in vitro restimulation, 5T4-specific responses could be detected ex vivo in many patients vaccinated with TroVax. Indeed, ex vivo IFN-gamma ELISPOT responses in excess of 1 5T4-specific T cell per 10000 PBMCs were seen in patients with colorectal cancer [49, 50], prostate cancer [52] and renal cancer [55–57]. Importantly, such immune responses were associated with signs of clinical benefit in 7 of the 10 phase I and II clinical studies (Table 3).
Table 3.
Correlation of 5T4-specific immune responses with clinical benefit in phase I, II and III trials of TroVax in patients with colorectal, prostate and renal cancer
| Indication | Trial phase | Treatment regimen | Nos. | Correlation between 5T4 immune response and clinical benefit | References | ||
|---|---|---|---|---|---|---|---|
| Overall survival | Progression-free survival | Tumour response | |||||
| Colorectal cancer | I | None | 22 | P < 0.01 | – | – | [48] |
| II | FOLFOX | 17 | – | – | P < 0.05 | [49] | |
| II | FOLFIRI | 19 | – | – | – | [50] | |
| II | None | 20 | P < 0.05 | – | – | [51] | |
| Prostate cancer | II | ±GM-CSF | 27 | – | P < 0.05 | – | [52] |
| II | Docetaxel | 25 | – | P < 0.1 | – | [53] | |
| Renal cancer | II | High-dose IL-2 | 25 | – | – | – | [54] |
| II | Low-dose IL-2 | 25 | P < 0.05 | – | – | [55] | |
| II | ±IFN-α | 28 | – | P < 0.05 | – | [56] | |
| II | IFN-α | 11 | – | – | – | [57] | |
| III | IL-2, IFN-α or Sunitinib | 733 | P < 0.01 | [58] | |||
Following these encouraging results, TroVax was tested in a phase III trial in 733 patients with metastatic renal cancer. While TroVax was shown to be safe and well tolerated in these patients, it failed to meet its primary endpoint as there was no significant difference in the survival of TroVax and placebo-treated groups. However, in the subset of patients with a good prognosis (MSKCC grade 0) and receiving IL-2, a significant increase in overall survival was seen in the TroVax group compared to the placebo group (P < 0.05) [58]. Furthermore, as demonstrated previously in numerous phase I and II studies [59], a strong association between 5T4 antibody response and enhanced survival was detected (P < 0.01) [60]. In addition, exploratory analyses identified a number of pre-treatment haematological factors which predicted those patients who were more likely to (a) mount the strongest 5T4-specific antibody responses and (b) show improved survival when treated with TroVax. The key factors identified were all associated with an inflammatory “signature” and included C-reactive protein (CRP), platelets, IL-6, vascular endothelial growth factor (VEGF), monocytes and haemoglobin levels [61].
From the outset, the clinical development of cancer vaccines has largely followed the paradigm used for cytotoxic agents; namely studies have been performed in patients with late-stage metastatic disease who have large tumour burdens and may have already failed one or more lines of treatment. Furthermore, tumour responses have often been used as indicators of efficacy. Such development paradigms may well be inappropriate for immunotherapy agents (including the new wave of promising immune checkpoint inhibitors).
A better understanding of the complex, multi-faceted immune interactions which occur in oncogenesis, and in particular the negative influence of a highly inflammatory environment on induction of a 5T4 responses or treatment benefit for TroVax-treated patients has led to changes to the clinical development of TroVax. The approach uses an enrichment strategy through a trial design utilising biomarker data to provide inclusion/exclusion criteria so as to target patients most likely to benefit from a cancer vaccine. This approach has been applied to the phase I and II investigator-led studies of TroVax in early-stage prostate cancer patients (VANCE; NCT02390063), malignant pleural mesothelioma (SKOPOS; NCT01569919), colorectal cancer (TaCTiCC; ISRCTN54669986) and ovarian cancer (TRIOC; NCT01556841).
5T4 antibody superantigen therapy
The principle underwriting this approach is to harness the ability of bacterial superantigens to activate large numbers of human T cells by binding to the T cells through the TCR vbeta chain family and replacing the interaction with MHC class II with the specificity of a TAA-targeting antibody. Therapeutic efficacy comes from direct lysis of tumour cells by the engaged T cells along with secretion of cytokines providing for bystander antitumour activity [62].
Preclinical studies
Early pre-clinical studies utilised a 5T4-specific antibody derived Fab–SEA fusion (ABR-214936) which contained a point mutation in the staphylococcal enterotoxin A (SEA) sequence. This mutation reduced binding to MHC class II while showing 5T4 specific superantigen antibody-dependent cellular cytotoxicity, reduced toxicity via class MHC II and demonstrated efficacy in murine xenograft tumour models [63].
Clinical studies
Initially, an open-label phase I study of ABR-214936 was undertaken in patients with non-small cell lung, renal cell or pancreatic cancer. In this study, a maximum tolerated dose (MTD) was determined and was a function of pre-existing anti-SEA antibody levels [64]. A subsequent phase II trial in RCC patients showed encouraging survival data relative to historical comparators [65]. Importantly, patients receiving higher drug exposure exhibited better disease control and survival than expected and this correlated with sustained IL-2 production at day 2. These data supported the development of a next-generation therapeutic molecule (ABR-217620, also called ANYARA) that aimed to further reduce any residual toxicity through MHC class II binding and reduction in antigenicity of the superantigen. This version is a hybrid SEA/E-120 superantigen sequence showed reduced binding to pre-formed anti-superantigen antibodies, lower toxicity, higher affinity for 5T4, and improved tumour cell killing [66, 67]. Further studies established a MTD and dose–dependent induction of biomarkers for T cell activation (IL-2 and interferon-gamma), selective expansion of ANYARA reactive T cells, infiltration of T cells and selective retention of ANYARA in tumour tissue [68]. These data collectively supported the initiation of a phase II/III trial of ANYARA in combination with interferon-alpha versus interferon-alpha alone in 513 patients with advanced RCC. The safety profile was similar to previous studies but the primary endpoint of overall survival was not reached [69]. A major confounding factor was believed to be the fact that the majority of the patients showed higher levels of pre-formed antibodies against the superantigen component of ANYARA than had been previously documented in patient populations in the UK and USA. A subgroup analysis, excluding patients with high levels of pre-formed antibodies, resulted in a trend for survival benefit with ANYARA treatment. Of note was the observation that high baseline levels of IL-6 correlated with a poorer outcome; this was also seen in trials of RCC patients treated with TroVax. When the outcome of approximately 25 % of patients with low/normal levels of baseline IL-6 and low anti-superantigen antibody levels were assessed, a statistically significant treatment advantage for overall survival was seen (P = 0.02, HR = 0.59). This is of importance since in North America and Western Europe, this represents 40–50 % of all the advanced renal cell cancer patients. The potential of combining ANYARA with a tyrosine kinase inhibitor in the favourable RCC subgroup is being explored.
5T4 antibody drug conjugate: development and clinical testing
Antibody drug conjugates (ADCs) chemically combine a cytotoxic drug with the targeting specificity of an antibody. Preliminary research by Boghaert et al. [70] aimed to combine the tumour specificity of a 5T4 monoclonal antibody with double-strand DNA break inducing toxin calicheamicin. The toxic effector moiety is only released after engagement of the antibody with its target antigen and subsequent internalisation of the ADC. This drug conjugate showed efficacy in several tumour models including an orthotopic model for 5T4-positive lung cancer. As documented earlier, 5T4 is expressed on tumour-initiating cells (TICs) in NSCLC and its expression on primary tumours correlates with worse clinical outcome [29]. Anti-5T4-ADC caused tumour regression, and no regrowth in two patient-derived xenografts, independently of heterogeneity or different levels of 5T4 expression predominantly at the lung tumour–stroma interface. Further studies showed the superior long-term efficacy of an ADC which can target TICs when compared with a conventional cisplatin treatment [29].
A subsequent development utilised a different internalising 5T4 humanised monoclonal antibody (A1) conjugated to the tubulin inhibitor, monomethyllauristan F (MMAF) via a maleimidocaproyl linker. A1mcMMAF has proven very effective in vivo with induction of long-term regression in several tumour models [71]. Drug release specifically in the tumour tissue with associated mitotic arrest has been demonstrated. Once again, a complete pathogenic response was independent of the degree of heterogeneity in 5T4 expression consistent with the targeting of TICs. Recent studies have provided a biological rationale for combination treatments of 5T4-ADC with PI3 K/mTOR pathway inhibitors or taxanes where the synergies observed may be derivative from the cellular impact of the auristatin [72]. Toxicity and pharmacokinetics of the conjugate have been analysed in the cynomolgus monkey where 5T4 is recognised by the A1 antibody. The A1mcMMAF was not toxic up to 10 mg/kg/cycle × 2 and displayed a half-life of 5 days with the free drug remaining very low in the plasma of monkeys. These observations suggest that the A1mcMMAF provides sufficient targeted payload to the tumour tissue with limited non-specific exposure of the cytotoxic agent. A full clinical evaluation of this drug is awaited.
Utilising chimeric antigen receptor (CAR)-T cells to target 5T4
Genetic modification of T cells to express chimeric antigen receptors (CARs) can produce effector populations with defined antigen specificities that function independently of the natural T cell receptor (TCR). Typically, an immunoglobulin-derived single-chain variable fragment (scFv) is fused to the TCR CD3ζ signalling domain for T cell activation and sometimes co-stimulatory elements such as CD28 or 4-1BB or inducible IL-12 production are incorporated to promote survival and expansion of CAR-T cells in the patient. Recent clinical testing of such modified T cells has been encouraging; indeed, the CAR-T cell field was catalysed by the stunning responses seen in patients with haematological malignancies (particularly B-ALL) [73, 74]. While these early results in haematological malignancies await formal confirmation in larger registration track studies, trials are ongoing to determine whether these results can also be replicated in solid tumours. Early results in solid tumours have been less compelling than the response rates reported in B-ALL [75]. However, it is clear that treating solid tumours, compared to haematological cancers, presents a different proposition both physically and technically which may require changes to the technology, clinical trial design and efficacy assessments. Like cancer vaccines before them, it is perhaps more realistic to expect that CAR-T cells used to treat solid tumours will deliver relatively slow tumour regression resulting in long-term stable disease and therefore increased survival rather than induction of rapid complete responses. Improvements to the CAR-T cell technologies may also be required to optimise cell growth, persistence and efficacy in the presence of large, hypoxic solid tumours which have multiple mechanisms to subvert host immune responses [76].
Human T cells expressing a first-generation 5T4-specific CAR construct showed specific cytokine release and cytotoxicity in vitro against 5T4-positive targets [77]. The construct required an extracellular spacer region in contrast to CARs using scFv against CEA and CD19 which might relate to the relative accessibility of the target antigen epitopes. This 5T4 CAR was used to modify T cells from patients with renal cell carcinoma (RCC) and kill RCC cells in vitro [28]. Furthermore, treatment of mice bearing 5T4-expressing B16 tumours with syngeneic T cells transduced with the first-generation CAR resulted in enhanced survival; the survival benefit was enhanced by combining CAR-T cell treatment with a vaccine targeting 5T4, thus providing evidence of synergy when used in combination [78].
Targeting 5T4 using a CAR-T cell approach is attractive for a multitude of reasons (many of which are listed in Table 1); these include the fact that 5T4 is expressed on the cell surface (and is therefore accessible to a CAR), is present on a wide range of solid tumours (in which expression often correlates with poor prognosis) and has been shown to be a marker of tumour-initiating cells in some cases.
Future perspectives
The key future challenge will be how best to combine available 5T4 immunotherapy(s) with standard of care treatments to maximise the recovery of the immune control and ultimately eliminate the tumour [79–81]. Development of treatment protocols will need to address the different mechanisms that contribute to immune evasion and which impact antigenicity (e.g. MHC downregulation), immunogenicity (e.g. ineffective APC function) and generate an immunosuppressive microenvironment in a cancer (e.g. Tregs and immunosuppressive macrophages). Radiation treatment can promote some useful potential synergies with immunotherapies such as increased MHC expression, activation of DCs, useful inflammatory cytokines (TNFα, IL-1, IL-2) and increased tumour infiltrating lymphocyte densities but can also induce immunosuppressive cytokines, (TGFβ), Tregs and induction of checkpoint inhibitor expression by tumour cells. Subsequent induction of T cell anti-tumour immunity could be achieved by a TAA vaccine possibly combined in a temporally favourable way with chemo- or radiotherapy to induce immunogenic tumour cell death, or through the use of immune checkpoint inhibitors. The use of an immune checkpoint inhibitor in combination with a 5T4-targeted vaccine is especially interesting given the clear correlation between the magnitude of 5T4-specific immune responses and clinical benefit seen in multiple clinical trials. Adoptive transfer of CAR-T cells is an attractive means for changing the balance of immune factors in favour of at least short-term tumour control. However, longer term control may also require the reversal of mechanisms of immune tolerance by inhibiting suppressive factors like TGFβ, IL10, etc. and/or by using checkpoint inhibitors. This can provide the means for recovery of the immune repertoire of the patient to deal with the heterogeneity of any residual tumour through activation of new and/or recovery of existing anti-tumour CTL targeting multiple TAA. Through the maturation of existing technologies and the selection of combinatorial therapies which lead to synergistic effects, it is hoped that immune-based therapies will become the central therapeutic modality in the fight against cancer.
Abbreviations
- ADC
Antibody drug conjugate
- ALL
Acute lymphoblastic leukaemia
- BCP
B-cell precursor
- CAR
Chimeric antigen receptor
- CRP
C-reactive protein
- EMT
Epithelial mesenchyme transition
- ES
Embryonic stem
- IHC
Immunohistochemistry
- kD
Kilodalton
- KO
Knock-out
- LRR
Leucine-rich repeat
- MSKCC
Memorial Sloan Kettering Cancer Center
- MTD
Maximum tolerated dose
- MVA
Modified vaccinia Ankara
- NCI
National Cancer Institute
- NSCLC
Non-small cell lung cancer
- RCC
Renal cell carcinoma
- SEA
Staphylococcal enterotoxin A
- TBPG
Trophoblast glycoprotein
- TIC
Tumour-initiating cell
- Treg
Regulatory T cell
- VEGF
Vascular endothelial growth factor
Compliance with ethical standards
Conflict of interest
Author Richard Harrop is employed by Oxford BioMedica who is developing 5T4-targeted therapies. Peter L. Stern is a consultant for Oxford BioMedica and has received speaker honoraria from Pfizer.
References
- 1.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 2.Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15(17):5323–5337. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Mlecnik B, Bindea G, Kirilovsky A, Angell HK, Obenauf AC, Tosolini M, et al. The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci Transl Med. 2016;8(327):327ra26. doi: 10.1126/scitranslmed.aad6352. [DOI] [PubMed] [Google Scholar]
- 5.Beatty GL, Gladney W. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015;21:687–692. doi: 10.1158/1078-0432.CCR-14-1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hole N, Stern PL. A 72 kD trophoblast glycoprotein defined by a monoclonal antibody. Br J Cancer. 1988;57(3):239–246. doi: 10.1038/bjc.1988.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hole N, Stern PL. Isolation and characterization of 5T4, a tumour-associated antigen. Int J Cancer. 1990;45(1):179–184. doi: 10.1002/ijc.2910450132. [DOI] [PubMed] [Google Scholar]
- 9.Shaw DM, Woods AM, Myers KA, Westwater C, Rahi-Saund V, Davies MJ, et al. Glycosylation and epitope mapping of the 5T4 glycoprotein oncofoetal antigen. Biochem J. 2002;363(Pt 1):137–145. doi: 10.1042/bj3630137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyle JM, Grzeschik KH, Heath PR, Morten JE, Stern PL. Trophoblast glycoprotein recognised by monoclonal antibody 5T4 maps to human chromosome 6q14-q15. Hum Genet. 1990;84(5):455–458. doi: 10.1007/BF00195819. [DOI] [PubMed] [Google Scholar]
- 11.Myers KA, Rahi-Saund V, Davison MD, Young JA, Cheater AJ, Stern PL. Isolation of a cDNA encoding 5T4 oncofetal trophoblast glycoprotein. An antigen associated with metastasis contains leucine-rich repeats. J Biol Chem. 1994;269(12):9319–9324. [PubMed] [Google Scholar]
- 12.Bella J, Hindle KL, McEwan PA, Lovell SC. The leucine-rich repeat structure. Cell Mol Life Sci. 2008;65(15):2307–2333. doi: 10.1007/s00018-008-8019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carsberg CJ, Myers KA, Evans GS, Allen TD, Stern PL. Metastasis-associated 5T4 oncofoetal antigen is concentrated at microvillus projections of the plasma membrane. J Cell Sci. 1995;108(Pt 8):2905–2916. doi: 10.1242/jcs.108.8.2905. [DOI] [PubMed] [Google Scholar]
- 14.Carsberg CJ, Myers KA, Stern PL. Metastasis-associated 5T4 antigen disrupts cell-cell contacts and induces cellular motility in epithelial cells. Int J Cancer. 1996;68(1):84–92. doi: 10.1002/(SICI)1097-0215(19960927)68:1<84::AID-IJC15>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 15.Awan A, Lucic MR, Shaw DM, Sheppard F, Westwater C, Lyons SA, et al. 5T4 interacts with TIP-2/GIPC, a PDZ protein, with implications for metastasis. Biochem Biophys Res Commun. 2002;290(3):1030–1036. doi: 10.1006/bbrc.2001.6288. [DOI] [PubMed] [Google Scholar]
- 16.Southall PJ, Boxer GM, Bagshawe KD, Hole N, Bromley M, Stern PL. Immunohistological distribution of 5T4 antigen in normal and malignant tissues. Br J Cancer. 1990;61(1):89–95. doi: 10.1038/bjc.1990.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 18.Ai J, Stevenson JP. Current issues in malignant pleural mesothelioma evaluation and management. Oncologist. 2014;19(9):975–984. doi: 10.1634/theoncologist.2014-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Connor ME, Stern PL. Loss of MHC class-I expression in cervical carcinomas. Int J Cancer. 1990;46(6):1029–1034. doi: 10.1002/ijc.2910460614. [DOI] [PubMed] [Google Scholar]
- 20.Elkord E, Burt DJ, Drijfhout JW, Hawkins RE, Stern PL. CD4+ T-cell recognition of human 5T4 oncofoetal antigen: implications for initial depletion of CD25+ T cells. Cancer Immunol Immunother. 2008;57(6):833–847. doi: 10.1007/s00262-007-0419-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Starzynska T, Marsh PJ, Schofield PF, Roberts SA, Myers KA, Stern PL. Prognostic significance of 5T4 oncofetal antigen expression in colorectal carcinoma. Br J Cancer. 1994;69(5):899–902. doi: 10.1038/bjc.1994.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Starzynska T, Rahi V, Stern PL. The expression of 5T4 antigen in colorectal and gastric carcinoma. Br J Cancer. 1992;66(5):867–869. doi: 10.1038/bjc.1992.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Starzynska T, Wiechowska-Kozlowska A, Marlicz K, Bromley M, Roberts SA, Lawniczak M, et al. 5T4 oncofetal antigen in gastric carcinoma and its clinical significance. Eur J Gastroenterol Hepatol. 1998;10(6):479–484. doi: 10.1097/00042737-199806000-00008. [DOI] [PubMed] [Google Scholar]
- 24.Naganuma H, Kono K, Mori Y, Takayoshi S, Stern PL, Tasaka K, et al. Oncofetal antigen 5T4 expression as a prognostic factor in patients with gastric cancer. Anticancer Res. 2002;22(2):1033–1038. [PubMed] [Google Scholar]
- 25.Hedlund G, Forsbery G, Sundstedt A, Axellson B, Celander M (2008) Poster presentation at IBC 6th annual antibody therapeutics, San Diego, USA, December 9–11
- 26.Al-Taei S, Salimu J, Lester JF, Linnane S, Goonewardena M, Harrop R, et al. Overexpression and potential targeting of the oncofoetal antigen 5T4 in malignant pleural mesothelioma. Lung Cancer. 2012;77(2):312–318. doi: 10.1016/j.lungcan.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 27.Wrigley E, McGown AT, Rennison J, Swindell R, Crowther D, Starzynska T, et al. 5T4 oncofetal antigen expression in ovarian carcinoma. Int J Gynecol Cancer. 1995;5(4):269–274. doi: 10.1046/j.1525-1438.1995.05040269.x. [DOI] [PubMed] [Google Scholar]
- 28.Griffiths RW, Gilham DE, Dangoor A, Ramani V, Clarke NW, Stern PL, et al. Expression of the 5T4 oncofoetal antigen in renal cell carcinoma: a potential target for T-cell-based immunotherapy. Br J Cancer. 2005;93(6):670–677. doi: 10.1038/sj.bjc.6602776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Damelin M, Geles KG, Follettie MT, Yuan P, Baxter M, Golas J, et al. Delineation of a cellular hierarchy in lung cancer reveals an oncofetal antigen expressed on tumour-initiating cells. Cancer Res. 2011;71(12):4236–4246. doi: 10.1158/0008-5472.CAN-10-3919. [DOI] [PubMed] [Google Scholar]
- 30.Castro FV, McGinn OJ, Krishnan S, Marinov G, Li J, Rutkowski AJ, et al. 5T4 oncofetal antigen is expressed in high risk of relapse childhood pre-B acute lymphoblastic leukemia and is associated with a more invasive and chemotactic phenotype. Leukemia. 2012;26(7):1487–1498. doi: 10.1038/leu.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nieto MA, Cano A. The epithelial-mesenchymal transition under control: global programs to regulate epithelial plasticity. Semin Cancer Biol. 2012;22(5–6):361–368. doi: 10.1016/j.semcancer.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 32.Ward CM, Barrow K, Woods AM, Stern PL. The 5T4 oncofoetal antigen is an early differentiation marker of mouse ES cells and its absence is a useful means to assess pluripotency. J Cell Sci. 2003;116(Pt 22):4533–4542. doi: 10.1242/jcs.00767. [DOI] [PubMed] [Google Scholar]
- 33.Ward CM, Eastham AM, Stern PL. Cell surface 5T4 antigen is transiently upregulated during early human embryonic stem cell differentiation: effect of 5T4 phenotype on neural lineage formation. Exp Cell Res. 2006;312(10):1713–1726. doi: 10.1016/j.yexcr.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 34.Eastham AM, Spencer H, Soncin F, Ritson S, Merry CL, Stern PL, et al. Epithelial-mesenchymal transition events during human embryonic stem cell differentiation. Cancer Res. 2007;67(23):11254–11262. doi: 10.1158/0008-5472.CAN-07-2253. [DOI] [PubMed] [Google Scholar]
- 35.Spencer HL, Eastham AM, Merry CL, Southgate TD, Perez-Campo F, Soncin F, et al. E-cadherin inhibits cell surface localization of the pro-migratory 5T4 oncofetal antigen in mouse embryonic stem cells. Mol Biol Cell. 2007;18(8):2838–2851. doi: 10.1091/mbc.E06-09-0875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Southgate TD, McGinn OJ, Castro FV, Rutkowski AJ, Al-Muftah M, Marinov G, et al. CXCR4 mediated chemotaxis is regulated by 5T4 oncofetal glycoprotein in mouse embryonic cells. PLoS ONE. 2010;5(4):e9982. doi: 10.1371/journal.pone.0009982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGinn OJ, Marinov G, Sawan S, Stern PL. CXCL12 receptor preference, signal transduction, biological response and the expression of 5T4 oncofoetal glycoprotein. J Cell Sci. 2012;125(Pt 22):5467–5478. doi: 10.1242/jcs.109488. [DOI] [PubMed] [Google Scholar]
- 38.Balkwill F. The significance of cancer cell expression of the chemokine receptor CXCR4. Semin Cancer Biol. 2004;14(3):171–179. doi: 10.1016/j.semcancer.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 39.Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumour cells and their microenvironment. Blood. 2006;107(5):1761–1767. doi: 10.1182/blood-2005-08-3182. [DOI] [PubMed] [Google Scholar]
- 40.Stern PL, Brazzatti J, Sawan S, Wan Y-L, McGinn O. Understanding & exploiting 5T4 oncofoetal glycoprotein expression. Semin Cancer Biol. 2014;29:13–20. doi: 10.1016/j.semcancer.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 41.Nusse R. Wnt signaling in disease and in development. Cell Res. 2005;15:28–32. doi: 10.1038/sj.cr.7290260. [DOI] [PubMed] [Google Scholar]
- 42.Kagermeier-Schenk B, Wehner D, Ozhan-Kizil G, Yamamoto H, Li J, Kirchner K, et al. Waif1/5T4 inhibits Wnt/β-catenin signaling and activates noncanonical Wnt pathways by modifying LRP6 subcellular localization. Dev Cell. 2011;21(6):1129–1143. doi: 10.1016/j.devcel.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 43.Zhao Y, Malinauskas T, Harlos K, Jones EY. Structural insights into the inhibition of Wnt signaling by cancer antigen 5T4/Wnt-activated inhibitory factor 1. Structure. 2014;22(4):612–620. doi: 10.1016/j.str.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Castro FV, Al-Muftah M, Mulryan K, Jiang HR, Drijfhout JW, Ali S, et al. Regulation of autologous immunity to the mouse 5T4 oncofoetal antigen: implications for immunotherapy. Cancer Immunol Immunother. 2012;61(7):1005–1018. doi: 10.1007/s00262-011-1167-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mulryan K, Ryan MG, Myers KA, Shaw D, Wang W, Kingsman SM, et al. Attenuated recombinant vaccinia virus expressing oncofetal antigen (tumour-associated antigen) 5T4 induces active therapy of established tumours. Mol Cancer Ther. 2002;1(12):1129–1137. [PubMed] [Google Scholar]
- 46.Smyth LJ, Elkord E, Taher TE, Jiang HR, Burt DJ, Clayton A, et al. CD8 T-cell recognition of human 5T4 oncofetal antigen. Int J Cancer. 2006;119(7):1638–1647. doi: 10.1002/ijc.22018. [DOI] [PubMed] [Google Scholar]
- 47.Redchenko I, Harrop R, Ryan MG, Hawkins RE, Carroll MW. Identification of a major histocompatibility complex class I-restricted T-cell epitope in the tumour-associated antigen, 5T4. Immunology. 2006;118(1):50–57. doi: 10.1111/j.1365-2567.2006.02338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harrop R, Connolly N, Redchenko I, Valle J, Saunders M, Ryan MG, et al. Vaccination of colorectal cancer patients with modified vaccinia Ankara delivering the tumor antigen 5T4 (TroVax) induces immune responses which correlate with disease control: a phase I/II trial. Clin Cancer Res. 2006;12(11 Pt 1):3416–3424. doi: 10.1158/1078-0432.CCR-05-2732. [DOI] [PubMed] [Google Scholar]
- 49.Harrop R, Drury N, Shingler W, Chikoti P, Redchenko I, Carroll MW, et al. Vaccination of colorectal cancer patients with modified vaccinia ankara encoding the tumor antigen 5T4 (TroVax) given alongside chemotherapy induces potent immune responses. Clin Cancer Res. 2007;13(15 Pt 1):4487–4494. doi: 10.1158/1078-0432.CCR-07-0704. [DOI] [PubMed] [Google Scholar]
- 50.Harrop R, Drury N, Shingler W, Chikoti P, Redchenko I, Carroll MW, et al. Vaccination of colorectal cancer patients with TroVax given alongside chemotherapy (5-fluorouracil, leukovorin and irinotecan) is safe and induces potent immune responses. Cancer Immunol Immunother. 2008;57(7):977–986. doi: 10.1007/s00262-007-0428-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elkord E, Dangoor A, Drury NL, Harrop R, Burt DJ, Drijfhout JW, et al. An MVA-based vaccine targeting the oncofetal antigen 5T4 in patients undergoing surgical resection of colorectal cancer liver metastases. J Immunother. 2008;31(9):820–829. doi: 10.1097/CJI.0b013e3181876ab3. [DOI] [PubMed] [Google Scholar]
- 52.Amato RJ, Drury N, Naylor S, Jac J, Saxena S, Cao A, et al. Vaccination of prostate cancer patients with modified vaccinia ankara delivering the tumor antigen 5T4 (TroVax): a phase 2 trial. J Immunother. 2008;6:577–585. doi: 10.1097/CJI.0b013e31817deafd. [DOI] [PubMed] [Google Scholar]
- 53.Harrop R, Chu F, Gabrail N, Srinivas S, Blount D, Ferrari A. Vaccination of castration-resistant prostate cancer patients with TroVax (MVA-5T4) in combination with docetaxel: a randomized phase II trial. Cancer Immunol Immunother. 2013;62(9):1511–1520. doi: 10.1007/s00262-013-1457-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaufman HL, Taback B, Sherman W, Kim DW, Shingler WH, Moroziewicz D, et al. Phase II trial of Modified Vaccinia Ankara (MVA) virus expressing 5T4 and high dose Interleukin-2 (IL-2) in patients with metastatic renal cell carcinoma. J Transl Med. 2009;7:2. doi: 10.1186/1479-5876-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Amato RJ, Shingler W, Goonewardena M, de Belin J, Naylor S, Jac J, et al. Vaccination of renal cell cancer patients with modified vaccinia Ankara delivering the tumor antigen 5T4 (TroVax) alone or administered in combination with interferon-alpha (IFN-alpha): a phase 2 trial. J Immunother. 2009;32(7):765–772. doi: 10.1097/CJI.0b013e3181ace876. [DOI] [PubMed] [Google Scholar]
- 56.Amato RJ, Shingler W, Naylor S, Jac J, Willis J, Saxena S, et al. Vaccination of renal cell cancer patients with modified vaccinia ankara delivering tumor antigen 5T4 (TroVax) administered with interleukin 2: a phase II trial. Clin Cancer Res. 2008;14(22):7504–7510. doi: 10.1158/1078-0432.CCR-08-0668. [DOI] [PubMed] [Google Scholar]
- 57.Hawkins RE, Macdermott C, Shablak A, Hamer C, Thistlethwaite F, Drury NL, et al. Vaccination of patients with metastatic renal cancer with modified vaccinia Ankara encoding the tumor antigen 5T4 (TroVax) given alongside interferon-alpha. J Immunother. 2009;32(4):424–429. doi: 10.1097/CJI.0b013e31819d297e. [DOI] [PubMed] [Google Scholar]
- 58.Amato RJ, Hawkins RE, Kaufman HL, Thompson JA, Tomczak P, Szczylik C, et al. Vaccination of metastatic renal cancer patients with MVA-5T4: a randomized, double-blind, placebo-controlled phase III study. Clin Cancer Res. 2010;16(22):5539–5547. doi: 10.1158/1078-0432.CCR-10-2082. [DOI] [PubMed] [Google Scholar]
- 59.Harrop R, Shingler W, Kelleher M, de Belin J, Treasure P. Cross-trial analysis of immunologic and clinical data resulting from phase I and II trials of MVA-5T4 (TroVax) in colorectal, renal, and prostate cancer patients. J Immunother. 2010;33(9):999–1005. doi: 10.1097/CJI.0b013e3181f5dac7. [DOI] [PubMed] [Google Scholar]
- 60.Harrop R, Shingler WH, McDonald M, Treasure P, Amato RJ, Hawkins RE, et al. MVA-5T4-induced immune responses are an early marker of efficacy in renal cancer patients. Cancer Immunol Immunother. 2011;60(6):829–837. doi: 10.1007/s00262-011-0993-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harrop R, Treasure P, de Belin J, Kelleher M, Bolton G, Naylor S, et al. Analysis of pre-treatment markers predictive of treatment benefit for the therapeutic cancer vaccine MVA-5T4 (TroVax) Cancer Immunol Immunother. 2012;61(12):2283–2294. doi: 10.1007/s00262-012-1302-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dohlsten M, Hansson J, Ohlsson L, Litton M, Kalland T. Antibody-targeted superantigens are potent inducers of tumour-infiltrating T lymphocytes in vivo. Proc Natl Acad Sci USA. 1995;92(21):9791–9795. doi: 10.1073/pnas.92.21.9791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Forsberg G, Ohlsson L, Brodin T, Björk P, Lando PA, Shaw D, et al. Therapy of human non-small-cell lung carcinoma using antibody targeting of a modified superantigen. Br J Cancer. 2001;85(1):129–136. doi: 10.1054/bjoc.2001.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cheng JD, Babb JS, Langer C, Aamdal S, Robert F, Engelhardt LR, et al. Individualized patient dosing in phase I clinical trials: the role of escalation with overdose control in PNU-214936. J Clin Oncol. 2004;22(4):602–609. doi: 10.1200/JCO.2004.12.034. [DOI] [PubMed] [Google Scholar]
- 65.Shaw DM, Connolly NB, Patel PM, Kilany S, Hedlund G, Nordle O, et al. A phase II study of a 5T4 oncofoetal antigen tumour-targeted superantigen (ABR-214936) therapy in patients with advanced renal cell carcinoma. Br J Cancer. 2007;96(4):567–574. doi: 10.1038/sj.bjc.6603567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Erlandsson E, Andersson K, Cavallin A, Nilsson A, Larsson-Lorek U, Niss U, et al. Identification of the antigenic epitopes in staphylococcal enterotoxins A and E and design of a superantigen for human cancer therapy. J Mol Biol. 2003;333(5):893–905. doi: 10.1016/j.jmb.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 67.Forsberg G, Skartved NJ, Wallén-Ohman M, Nyhlén HC, Behm K, Hedlund G, et al. Naptumomab estafenatox, an engineered antibody-superantigen fusion protein with low toxicity and reduced antigenicity. J Immunother. 2010;33(5):492–499. doi: 10.1097/CJI.0b013e3181d75820. [DOI] [PubMed] [Google Scholar]
- 68.Borghaei H, Alpaugh K, Hedlund G, Forsberg G, Langer C, Rogatko A, et al. Phase I dose escalation, pharmacokinetic and pharmacodynamic study of naptumomab estafenatox alone in patients with advanced cancer and with docetaxel in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2009;27(25):4116–4123. doi: 10.1200/JCO.2008.20.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hawkins RE, Gore M, Shparyk Y, Bondar V, Gladkov O, Ganev T, et al. A randomized phase II/III study of naptumomab estafenatox + IFNα versus IFNα in renal cell carcinoma: final analysis with baseline biomarker subgroup and trend analysis. Clin Cancer Res. 2016;22(13):3172–3181. doi: 10.1158/1078-0432.CCR-15-0580. [DOI] [PubMed] [Google Scholar]
- 70.Boghaert ER, Sridharan L, Khandke KM, Armellino D, Ryan MG, Myers K, et al. The oncofetal protein, 5T4, is a suitable target for antibody-guided anti-cancer chemotherapy with calicheamicin. Int J Oncol. 2008;32(1):221–234. doi: 10.3892/ijo.32.1.221. [DOI] [PubMed] [Google Scholar]
- 71.Sapra P, Damelin M, Dijoseph J, Marquette K, Geles KG, Golas J, et al. Long-term tumour regression induced by an antibody-drug conjugate that targets 5T4, an oncofetal antigen expressed on tumour-initiating cells. Mol Cancer Ther. 2013;12:38–47. doi: 10.1158/1535-7163.MCT-12-0603. [DOI] [PubMed] [Google Scholar]
- 72.Shor B, Kahler J, Dougher M, Xu J, Mack M, Rosfjord E, et al. Enhanced antitumour activity of an anti-5T4 antibody-drug conjugate in combination with PI3 K/mTOR inhibitors or Taxanes. Clin Cancer Res. 2016;22:383–394. doi: 10.1158/1078-0432.CCR-15-1166. [DOI] [PubMed] [Google Scholar]
- 73.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol. 2016;13:273–290. doi: 10.1038/nrclinonc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Whilding LM, Maher J. CAR T-cell immunotherapy: the path from the by-road to the freeway. Mol Oncol. 2015;9:1994–2018. doi: 10.1016/j.molonc.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med. 2016;22(1):26–36. doi: 10.1038/nm.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Spear TT, Nagato K, Nishimura MI. Strategies to genetically engineer T cells for cancer immunotherapy. Cancer Immunol Immunother. 2016;65:631–649. doi: 10.1007/s00262-016-1842-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guest RD, Hawkins RE, Kirillova N, Cheadle EJ, Arnold J, O’Neill A, et al. Relative position of scFv binding to target proteins influences the optimal design of chimeric immune receptors for four different scFvs and antigens. J Immunother. 2005;28:203–211. doi: 10.1097/01.cji.0000161397.96582.59. [DOI] [PubMed] [Google Scholar]
- 78.Jiang H-R, Mulryan K, Kirillova N, Hawkins RE, Gilham D, Stern PL. Combination of vaccination and chimeric receptor expressing T cells provides improved active therapy of tumours. J Immunol. 2006;177:4288–4298. doi: 10.4049/jimmunol.177.7.4288. [DOI] [PubMed] [Google Scholar]
- 79.Beatty GL, Glagney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015;21:687–692. doi: 10.1158/1078-0432.CCR-14-1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sharabi AB, Lim M, DeWesse TL, Drake CG. Radiation and checkpoint blockade immunotherapy: radiosensitisation and potential mechanisms of synergy. Lancet Oncol. 2015;16(13):e498–e509. doi: 10.1016/S1470-2045(15)00007-8. [DOI] [PubMed] [Google Scholar]
- 81.Spranger S. Mechanisms of tumor escape in the context of the T-cell-inflamed and the non-T-cell-inflamed tumor microenvironment. Int Immunol. 2016;28(8):383–391. doi: 10.1093/intimm/dxw014. [DOI] [PMC free article] [PubMed] [Google Scholar]