

Abstract
Cancer stem cells (CSC) are resistant to radiation and chemotherapy and play a significant role in cancer recurrence and metastatic disease. It is therefore important to identify alternative strategies, such as immunotherapies that can be used to control this refractory population. A CD44+CD24−/low subpopulation of cells within the B6 PyMT-MMTV transgenic mouse-derived AT-3 mammary carcinoma cell line was identified, which had CSC-like characteristics, including pluripotency and a resistance to chemo- and radiotherapy. Therefore, unlike xenograph models that require immunocompromised settings, this novel system may provide a means to study immune-mediated responses against CSC-like cells. The immunobiology of the AT-3 CSC-like cell population was studied by their surface molecule expression profile and their sensitivity to specified cell death pathways. Comparable levels of Rae-1, CD155, CD54 and higher levels of Fas and DR5 were expressed on the AT-3 CSC-like cells compared to non-CSC-like tumor cells. Expression correlated with an in vitro sensitivity to cell death by NK cells or through the ligation of the death receptors (Fas or DR5), by their ligands or anti-Fas and anti-DR5 mAbs. Indeed, compared to the rest of the AT-3 tumor cells, the CD44+CD24−/low subpopulation of cells were more sensitive to both Fas- and TRAIL-mediated cell death pathways. Therefore, despite the refractory nature of CSC to other conventional therapies, these CSC-like cells were not inherently resistant to specified forms of immune-mediated cell death. These results encourage the continued investigation into immunotherapeutic strategies as a means of controlling breast CSC, particularly through their cell death pathways.
Keywords: Cancer stem cells, Immunotherapy, TNF-related cell death, Breast cancer, Fas, TRAIL
Introduction
In cancer, a lack of a curative treatment for metastatic disease and the high level of disease recurrence following standard therapies support increasing evidence of the importance of cancer stem cells (CSC) in disease progression [1, 2]. Since CSC are refractory to conventional radiation and chemotherapies [3–6], it is important for the design of better treatment strategies that the CSC population of tumors be eliminated or otherwise controlled. Along with many other cancer types, a population of putative CSC have been identified in both primary and metastatic breast tumors from patient samples as well as in breast cancer cell lines [7]. These CSC undergo both self-renewal and differentiation, and have a high tumorigenic potential. The pluripotent nature of CSC may be responsible for driving tumor initiation, progression and the development of solid mammary tumors that comprises of the cell diversity seen in breast cancer [8]. Disease recurrence is thought to be due to the quiescent character of CSC and their resistance to chemo- and radiation therapies [4, 8]. For example, a study of breast tumor biopsy samples found that, following standard chemotherapy, the proportion of cells expressing CSC markers increased sharply compared to other tumor cell types within the sample [9]. Moreover, breast cancer progression to metastatic disease may be initiated by invasive CSC, which is supported by studies showing that tumors rich in CSC have a significantly worse clinical outcome [10]. Indeed, it is thought by a growing number of oncologists that it is only by controlling the CSC component, that cancer can be truly cured [11]. Therefore, although CSC comprise only a small proportion of cells within a tumor, they should be considered an important target for therapeutic intervention.
Given the resistance of CSC to conventional radio- and chemotherapies; the growing number of immunotherapeutic strategies that have been approved or are being trialed, for the treatment of cancer; and the fact that some standard of care radio- and chemotherapies may actually prime immune responses [12, 13]; the susceptibility of CSC to immune-based therapies needs to be investigated as an alternative strategy to combat CSC-mediated cancer progression. To date, the majority of CSC studies have utilized cells from patient samples or from human cancer cell lines and have concentrated on investigation into their biology, such as the dysregulation of self-renewal pathways. However, the use of xenograft models limits our ability to investigate the complexity of in vivo immunological interactions. We have therefore identified and validated a murine CD44+CD24−/low cell subpopulation that exhibits many of the reported characteristics of human breast CSC. A similar CD44+CD24−/low subpopulation of cells was also identified in the primary tumors of B6 PyMT-MMTV transgenic mice, from which the AT-3 tumor cell line was derived. Therefore, in contrast to currently used xenograft models, these cells provide a novel model system to study interactions between breast CSC and a competent immune system and to investigate their susceptibility to immunotherapeutic strategies.
The immunological characterization of CSC is essential to identify immune-associated molecules that are expressed by CSC and that can be exploited for immune-mediated responses against this refractory population. It was found that these CSC-like cells expressed a number of immunologically relevant cell surface molecules at similar, if not higher levels, to those of other non-CSC-like cell populations within the tumor line. Correlating with surface expression was an increased sensitivity of the AT-3 CSC-like cells to Fas-mediated and TRAIL-mediated cell death induced by either ligand-expressing effector cells or mAbs, when compared to other AT-3 tumor cells. Therefore, despite these CSC-like cells being inherently resistant to chemotherapy and radiation therapy, they remained susceptible, and in some cases more sensitive, to immune-mediated cell death pathways.
Materials and methods
Mice
Inbred C57BL/6 mice at 8–10 weeks of age were obtained from The Walter and Eliza Hall Institute of Medical Research. C57BL/6 RAG-1-deficient (RAG-1 −/−) mice and C57BL/6 RAG-2.common-gamma-chain receptor-deficient (Rag-2.cγR −/−) mice were bred and maintained at the Peter MacCallum Cancer Centre (PMCC). Only female mice were used in all experiments. PyMT-MMTV transgenic (MTAG) mice, which were used to source primary tumors, were bred onsite [14]. Mice were maintained under specific pathogen-free conditions and used in accordance with the institutional guidelines of the PMCC and in compliance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes.
Cell lines and culture conditions
The AT-3 mammary carcinoma tumor line was derived as previously described from a primary tumor of a PyMT-MMTV transgenic mouse [15]. The respective 2PK-3 and L5178Y, vector and mock mouse TRAIL (mTRAIL) and mFasL-transfected B cell lymphoma lines have been previously described [16] and were a kind gift of H. Yagita (Juntendo University, Tokyo). P815 is a FcR-expressing mastocytoma cell line [17]. All lines were cultured in DMEM media supplemented with 10% heat-inactivated FBS (Moregate Biotech, QLD, Australia), 1 mM sodium pyruvate, 0.1 mM non-essential amino acids and 1 mM l-glutamine and incubated at 37°C, 10% CO2. Culture of mammosphere/tumorspheres was conducted in non-adherent 24-well plates (FALCON® #351147) using either complete DMEM media or serum-free MammoCult® media (StemCell Technologies) after seeding at 1 × 105 cells/ml/well and 3 days of culture at 37°C, 10% CO2.
Antibodies and flow cytometry
The profile of mouse cell surface molecule expression was conducted on unsorted AT-3 cells and gated using CD44/CD24 staining. The following antibodies were used: CD24 (M1/69), CD44 (IM7), CD326 (G8.8), CD133 (13A4), CD34 (RAM34), CD80 (B7-1), CD86 (GL1), H-2Kb (AF6-88.5), I-Ae (KH74), CD54 (3E2), CD262 (MD5.1), CD95 (15A7), CD274 (M1H5), CD178 (MFL3), TRAIL (N2B2), Rae-1 (186107) and CD155 (4.24.1). Appropriate isotype control Abs were used and are represented as gray histograms. Agonistic mAbs to mouse DR5 (MD5.1) and anti-mouse CD95 (Jo2) were used to induce cell death pathways. Flow cytometric analysis was performed on a LSRII cytometer (BD Biosciences) and data analyzed using Flowjo software (Tree Star Incorporated). Gating of single cells using FSC/W and SSC/W and exclusion of dead cells were routinely performed. A FACSAriaII (BD Biosciences) was used to sort specific cell populations.
Tumor challenge
AT-3 tumor cells were cultured and sorted, based on CD44/CD24 staining, for AT-3 subpopulations using a FACSAriaII. The CD44+CD24− and “other” cells (AT-3 minus CD44+CD24− cells) were resuspended in PBS at stated doses in 100 μl volumes. Groups of 5–6 female mice were challenged in the 4th mammary fatpad. Mice were monitored for tumor growth for 10 weeks or until tumors reached a size of 1 cm2.
CSC within primary MTAG tumors
Primary tumors of at least 1 cm2 were excised from MTAG mice and manually disrupted using fine dissection scissors. This preparation was resuspended in a PBS digestion solution containing 1 mg/ml Collagenase, 0.1 mg/ml Hyaluronidase and 30 U/ml DNase. After approximately 1-h incubation at 37°C with shaking, the cell suspension was passed through a sieve to remove any remaining clumped material, washed twice before the suspension in FACs buffer (PBS + 2% FBS) and stained using anti-CD24 and anti-CD44 mAbs.
CFSE-based proliferation assay
Differences in AT-3 subpopulation proliferation rates were investigated using CFSE dilution assays. AT-3 tumor cells were suspended in PBS + 0.1% BSA and labeled with CFSE (5-6-carboxyfluorescein diacetate succinimidyl ester; Molecular Probes) at a final concentration of 5 μM and incubated at 37°C for 10 min. Cells were then washed 3 times with DMEM + 2.5% FBS to remove excess CFSE and resuspended in complete DMEM culture media. After 72-h incubation, cells were collected and stained with anti-CD24 and anti-CD44 to identify the AT-3 subpopulations, which were analyzed for remaining CFSE levels using flow cytometry.
Cytotoxicity assays
Doxorubicin
Sensitivity to the chemotherapeutic drug Doxorubicin hydrochloride [InterPharma Pty Ltd, Australia manufactured by EBEWE Pharma Ges.m.b.H Nfg.KG (Unterach, Austria)] was tested using the CellTiter-Blue® viability assay (Promega) as per manufacturer instructions. Briefly, sorted cell populations were placed in a 96-well flat-bottomed plate at 1 × 104 cells per well in 100 μl of complete medium containing a dose titration of doxorubicin. After 20-h incubation, the CellTiter-Blue® reagent was added to the wells and incubated for a further 2 h. The fluorescence produced is proportional to the number of viable cells.
TRAIL mediated
Tumor cell sensitivity to TRAIL-mediated cytotoxicity was assessed using the murine TRAIL-transfected or mock-transfected 2PK-3 lines as “effector” cells at the indicated E/T ratios. Cytotoxic activity was tested in both standard 51Cr release and Annexin/7AAD staining assays. Briefly, AT-3 CSC or other AT-3 target cells were placed in either a 96-well flat-bottomed plate at 1 × 104 51Cr-labeled target cells per well (Cr release assay) or a 48-well flat-bottomed plate at 5 × 104 cells per well (Annexin assay). 2PK-3 control or 2PK-3mTRAIL cells were added at stated E/T ratios and the plates incubated for 20 h. Cells were harvested and percentage-specific lysis was calculated as previously described [18]. In the Annexin cytotoxicity assay, measurement of cell death was taken as per manufacturers’ instructions using PE-Annexin V Apoptosis Detection Kit I (BD Pharmingen) following cell harvest using trypsin. To preclude 2PK-3 effector cells from the analysis for cell death, harvested cells were stained for CD19 prior to Annexin staining. Ab-mediated: MD5-1-mediated cytotoxicity was tested in the presence of FcR-expressing P815 cells at a 10:1 E/T ratio in the presence of 10 μg/ml MD5.1 mAb. Control wells consisted of tumor cells incubated with isotype-matched Ab. Cells were incubated in a 48-well plate at 5 × 104 target cells per well for 20 h and the induction of cell death measured using the aforementioned Annexin apoptosis kit. Harvested cells were stained with anti-CD16 prior to Annexin staining, to preclude the CD16+ effector cells from the analysis for Annexin/7AAD-positive cell death staining.
Fas/FasL mediated
Fas-mediated killing assays were conducted similar to the TRAIL-mediated assays, except that L5178Y-mFasL-transfected or mock-transfected cells were used as effectors. Ab-mediated: Cells were placed in a 48-well plate with 10 μg/ml anti-mouse Fas (Jo2) at 5 × 104 cells per well in a final volume of 400 μl complete media. After 48-h incubation, Fas-mediated killing was analyzed by Annexin/7AAD staining as described above. Control wells consisted of tumor cells incubated with isotype-matched Ab.
NK cell killing
NK cell enrichment from wild-type B6 mouse spleens was performed as previously described [19]. Briefly, NK cells were isolated using a NK cell isolation kit (Miltenyi Biotec) and stimulated for 4 days with 1000 U/ml IL-2. These effector cells were then used in a standard 4 h 51Cr release assay against sorted AT-3 subpopulations at a range of E/T ratios.
Statistical analysis
Unless otherwise stated, graphed values represent the mean ± SEM and a Mann–Whitney test was used to calculate the P values at specified E/T. A P value of less than 0.05 (*) was considered significant, with other P values represented as 0.001–0.01 (**) and 0.0001–0.001 (***).
Results
Identification of a CSC-like population based on CD44/CD24 expression
Since many CSC-like cell populations within human tumors have been identified as being CD44+CD24−, these markers were first used to screen a number of mouse mammary carcinoma cell lines for a corresponding murine CSC-like subpopulation. This screen revealed that the AT-3 cell line possessed a small subpopulation with a CD44+CD24− phenotype that comprised <1% (0.2–0.8%) of the bulk population (Fig. 1a). As AT-3 was derived from a primary tumor of a MTAG (PyMT-MMTV transgenic) mouse, we looked for a similar population within more than 25 ex vivo primary tumors from MTAG mice to establish that this CD44+CD24− subpopulation was not an artifact of cell line establishment or culturing. A CD44+CD24− population was identified within all ex vivo tumors analyzed, the proportion of which ranged from 0.32% to 1.12% of cells (Mean 0.71% ± 0.27 SD) within the tumors. Other murine mammary carcinoma cell lines that were screened either did not possess a CD44+CD24− subpopulation (e.g., 4T1) or else the identification of a CD44+CD24− cell subpopulation was not necessarily associated with CSC-like properties as outlined in this manuscript (e.g., H2N113). Of the 12 mouse mammary carcinoma lines screened, only AT-3 contained a CD44+CD24− subpopulation with CSC-like characteristics similar to what has been described in human cancers.
Fig. 1.

a A putative population of CSC-like cells was identified within the AT-3 mammary carcinoma cell line or within single cell preparations of primary MTAG tumors (n > 25) following staining for CD44 and CD24, and flow cytometric analysis. b An analysis of other commonly used cancer stem cell markers, namely CD326, CD133 and CD34 (black line), was conducted on the putative AT-3 CSC-like (CD44+CD24−) subpopulation compared to the major CD44−CD24+ subpopulation of AT-3 cells. Results are illustrative of at least 4 separate experiments with the gray histogram representing isotype control staining. c The three subpopulations of cells within the AT-3 cell line, based on CD44/CD24 staining, were purified by FACS and cultured under normal AT-3 cell line culturing conditions. The purity of these initial cell preparations is stated in brackets at line 0 week. A sample of each cell culture was taken at weekly intervals over 3 weeks (1–3 weeks) and reassessed for CD44/CD24 expression. Results are representative of 2 separate experiments
An examination of other commonly used CSC markers, namely CD326 (EpCAM), CD133 or CD34, showed that this CD44+CD24− putative AT-3 CSC population did not express these molecules (Fig. 1b). The lack of CD326 expression on the CD44+CD24− subpopulation may explain the observation that these cells detached earlier, from the culture flask following treatment with trypsin, compared to the other AT-3 cells, which are CD326 positive. The AT-3 CD44+CD24− subpopulation was further investigated to verify the CSC-like behavior of these cells.
The CD44+CD24− subpopulation of AT-3 cells demonstrate a pluripotent capacity
A property of CSC-like cells is their capacity to differentiate and repopulate a tumor. To investigate the differentiation capacity of the AT-3 cell line subpopulations, AT-3 cells were sorted into three distinct subpopulations based on CD44 and CD24 expression. These CD44+CD24−/low, CD44+CD24+ and CD44lowCD24+ subpopulations were then placed in culture and further analyzed at weekly intervals to determine their path of differentiation (Fig. 1c). It was observed that the major AT-3 subpopulation of CD44lowCD24+ cells appeared to be a relatively stable/terminally differentiated subset and demonstrated very little phenotypic change after 3 weeks in culture. The CD44+CD24+ subset, however, gradually lost their CD44 expression to eventually differentiate into a cell line consisting of mostly CD44lowCD24+ cells. The percentage of CD44+CD24− cells rapidly decreased with continued culture and correlated with an increase in the proportion of CD44lowCD24+ cells and a repopulation of the CD44+CD24+ subset. Eventually, after 5–6 weeks of culture (data not shown), the cultures originally composed of just CD44+CD24− cells differentiated and repopulated all the CD44/CD24 cell subsets to resemble the subset proportions present in the parent AT-3 cell line. These data demonstrate the differentiation capacity of the CD44+CD24− cell subset and support their putative CSC-like status.
CD44+CD24− cells have a reduced rate of proliferation
A further common characteristic of many reported CSC populations is their quiescent nature and slow rate of proliferation. The CD44+CD24− AT-3 subpopulation of cells was shown to have the least amount of proliferation of the three AT-3 CD44/CD24 subpopulations based on the highest retention of CFSE (Fig. 2a). The major AT-3 subpopulation consisting of CD44lowCD24+ cells were highly proliferative, whereas the CD44+CD24+ cells demonstrated an intermediate proliferative rate. This correlates with the differentiation data showing that CD44+CD24− cells differentiate to CD44+CD24+ cells and then to the CD44lowCD24+ population of cells that make up the majority of the AT-3 cell line. The demonstrated low proliferation rate of the CD44+CD24− subpopulation, when compared to the major CD44lowCD24+ cell population, supports the quiescent nature and CSC-like character of the CD44+CD24− subpopulation.
Fig. 2.

a An AT-3 cell line that had been enriched for CD44+CD24− cells (approx. 3%) was labeled with CFSE (See histogram inset). After 72-h incubation, cells were harvested and analyzed by flow cytometry to determine CFSE dilution within the three CD44/CD24 cell subpopulations. Data represent one of three similar experiments. b The AT-3 cell line, enriched for CD44+CD24− cells (approx. 5%), was plated in non-adherent plates either in normal DMEM complete media or in serum-free MammoCult® media. After 3-day incubation, the resulting cells were harvested and analyzed for CD44/CD24 expression. Experiment was repeated with similar results. c CD44+CD24− cells were purified by FACS and their sensitivity to Doxorubicin compared to that of the “other” AT-3 cells (i.e., AT-3 minus CD44+CD24− cells). Cells were incubated in triplicate wells, with a dose titration of Doxorubicin for 20 h and the amount of viable cells remaining determined by fluorescence intensity of the CellTiter-Blue® viability assay reagent. Results represent the mean ± SEM of fluorescence readings of two similar experiments. Significance determined by Student’s t test, where ** represents a P value between 0.01 and 0.001. d Similar to (c), cells were purified and irradiated for various exposure times from 5 to 50 Gy. The CD44+CD24− and other AT-3 cell population were then plated and incubated for 20 h before harvesting and Annexin/7-AAD staining. Cells were analyzed by flow cytometry and the percentage of Annexin and 7-AAD double-positive cells determined. Results are representative of two separate experiments, and the P value determined using linear regression
CD44+CD24− cells survive better than other AT-3 cells under nutrient-starved culture conditions
Characterization of cancer stem cells in human systems has been facilitated by the development of in vitro culture systems that allow for the propagation of stem cells in an undifferentiated state. These systems have been employed in the study of breast CSC, which can be enriched and purified from “mammospheres” that form from the unattached culture of breast cancer cells [20]. Another property of CSC related to their quiescent nature is their ability to survive in low-nutrient conditions. It was found that culturing of the AT-3 cell line using low nutrient/serum-free MammoCult® culture media resulted in the enrichment of the CD44+CD24− cell subpopulation through the elimination of the major AT-3 CD44−/lowCD24+ cell subpopulation (Fig. 2b). These surviving cells also formed tumorspheres/mammospheres when grown in non-adherent plates. The CD44+CD24− cells, although they do not proliferate/expand very well, could continue to be passaged in low-nutrient media and remain at a higher proportion of CD44+CD24− cells. When these cells were transferred into complete media, they were shown to differentiate and repopulate the whole cell line (data not shown), similarly to what was described above (refer to Fig. 1c). Therefore, the CD44+CD24− subpopulation can be expanded and maintained in an undifferentiated CSC-like phenotype that form mammospheres, when grown under low-nutrient culture conditions. These cells can then be induced to pluripotent differentiation, based on CD44/CD24 markers, once exposed to complete media.
CD44+CD24− cells are more resistant to radiation and chemotherapy
An important and highly reported characteristic of CSC is their resistance to chemotherapy. This characteristic resistance to chemotherapy in the CD44+CD24− subpopulation of AT-3 cells compared to “other” AT-3 cells was verified following treatment with the chemotherapeutic drug, Doxorubicin. Doxorubicin treatment of CD44+CD24− cells compared to other AT-3 cells (i.e., AT-3 minus CD44+CD24− cells) showed a significantly greater number of metabolically active/viable cells in the CD44+CD24− cell population compared to other AT-3 cells at a range of Doxorubicin concentrations (Fig. 2c), despite their slow proliferation rate. A similar trend was observed following 6- or 46-h treatment (data not shown). Therefore, CD44+CD24− AT-3 cells were shown to be more resistant to a standard chemotherapy than other AT-3 cells, which is further evidence of the CSC-like properties of this population.
CSC are also resistant to radiation-induced cell death. To test whether this is a characteristic of our putative CSC-like population, the CD44+CD24− and CD44−CD24+ AT-3 subpopulations were purified and exposed to a range of radiation doses from 5 to 50 Gy. After an overnight incubation, the percentage of radiation-induced cell death, measured by Annexin/7-AAD double-positive cells, was found to be significantly lower in the CD44+CD24− subpopulation than within the major CD44−CD24+ AT-3 population (Fig. 2d). Similar trends were observed at 44- or 70-h time points following irradiation (data not shown). Comparable results were also obtained if the whole AT-3 line was irradiated and the percentage of Annexin/7-AAD cells within the CD44+CD24− and CD44−CD24+ AT-3 subpopulations determined. These data are further evidence that the CD44+CD24− subpopulation of AT-3 is, indeed, CSC-like and will be from now on referred to as AT-3 CSC-like cells.
CD44+CD24− cells are sensitive to immune control and are more tumorigenic
An investigation into whether a competent immune response can affect the establishment of tumors from the CD44+CD24− AT-3 subpopulation compared to the remaining AT-3 cells was conducted by comparing the establishment of these two tumor cell populations in B6 wild-type and immune-deficient mice (Fig. 3a). RAG-1−/− mice lack T and B cells, whereas RAG-2.cγR−/− are deficient in T, B and NK cells. Tumors were found to establish at much lower doses in both these immune-deficient strains of mice compared to wt, demonstrating a role for adaptive immune responses in controlling the ability of AT-3 cell subpopulations to establish a tumor. Based on the lower cell number required for establishing tumors in RAG-2.cγR−/− compared to RAG-1−/− mice, there was an indication that an innate response by NK cells can also effect tumor growth. It was also evident, particularly in wt mice, that the number of CD44+CD24− AT-3 cells required to form tumors was much less than the population of other AT-3 cells. Therefore, the lower initiating dose of CD44+CD24− AT-3 cells demonstrates an increased tumorigenic potential and supports the CSC likeness of this subpopulation. The role for an immune component in influencing the tumorigenic potential of both CSC-like and other AT-3 cells is evidence that utilizing components of the immune system may be a viable option in controlling tumor growth.
Fig. 3.
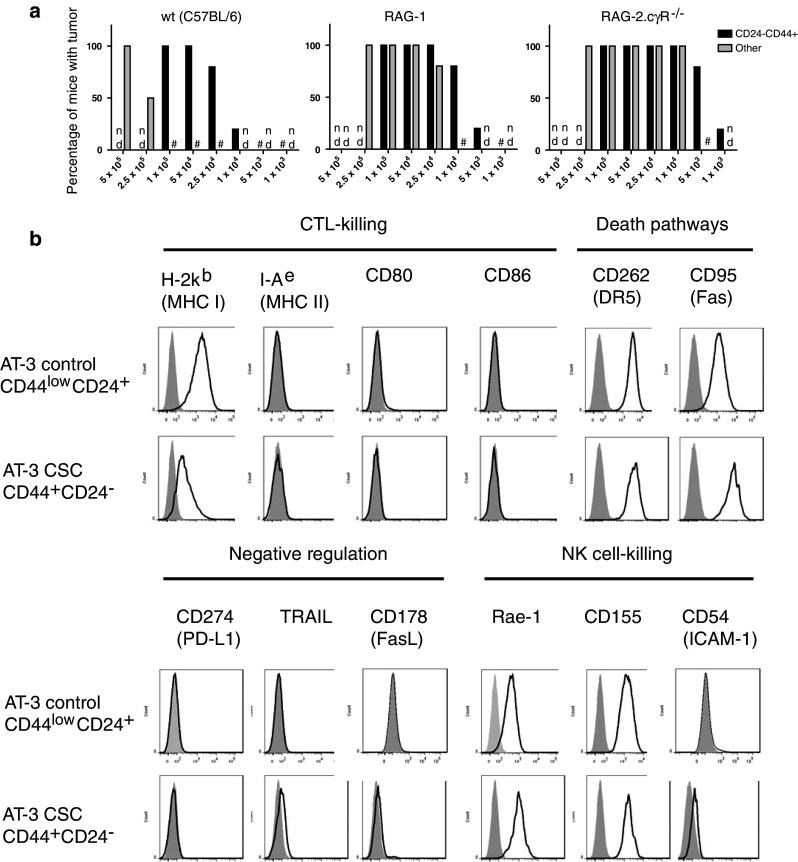
a AT-3 cells were sorted into two cell populations based on CD44/CD24 staining and used to challenge wt, RAG-1 and RAG-2.cγR−/− (common-gamma-chain receptor null) mice in the mammary fatpad at a range of cell doses (n = 5–6 mice per group). The “other” population of cells consists of the remaining AT-3 cells minus the CD44+CD24− subpopulation. Mice were monitored for progressive tumor growth. # represents groups that did not develop tumors; nd = not determined. b Sorted AT-3 CD44/CD24 subpopulations were stained for a range of immunologically relevant surface markers and their expression levels determined. Specific marker expression is represented by the black line and isotype staining by the shaded gray area. Results are representative of at least 3 individual experiments
Expression profile of immunologically relevant surface molecules
Immune-mediated killing of cancer cells can occur through the recognition of certain surface molecules by components of the immune system that initiate intracellular death pathways. Therefore, to explore whether CSC are sensitive to immune-mediated death pathways, an expression profile of immunologically relevant surface markers was analyzed using flow cytometry. These surface markers included molecules associated with T-cell recognition, co-stimulation and killing [MHC I and II, CD80 and CD86]; negative immune regulation [CD178 (FasL), TRAIL, CD274 (PD-L1/B7-H1)]; NK cell-mediated killing [Rae-1 (NKG2D ligand), CD155 (DNAM ligand), CD54 (ICAM-1)]; and other death pathway–associated molecules [DR5 (TRAIL-mediated killing), CD95 (Fas/FasL-mediated killing)]. None of the AT-3 subpopulations were found to express the CD80 and CD86 costimulatory molecules or MHC class II molecules (Fig. 3b). AT-3 cells were positive for MHC class I expression (H-2Kb), although the AT-3 CSC-like cells did express lower levels of H-2Kb. Both the CSC-like population and other AT-3 cells expressed DR5, CD95 (Fas), Rae-1 and CD155. Indeed, the CSC-like population appeared to express slightly higher levels of all these molecules particularly Fas. In support of this statement was the higher mean fluorescent intensity values (MFI) provided in Table 1. Based on the immunological expression profile of the AT-3 CSC-like cells, it appeared that these cells may be potentially sensitive to various pathways of immune-mediated cell death, including by NK cells and in particular TRAIL- and Fas-mediated pathways.
Table 1.
Expression levels (MFI) of immunologically relevant surface markers on AT-3 CSC with or without irradiation
| Characterization profile | AT-3 CSC | |||
|---|---|---|---|---|
| CD44lowCD24+ control cells | CD44+CD24− cells (CSC) | Non-irrad. | Irradiated* | |
| H-2Kb | 1,999 | 356 | 2,123 | 330 |
| DR5 | 3,285 | 4,087 | 2,422 | 3,314 |
| Fas | 1,378 | 7,415 | 36 | 654 |
| Rae-1 | 382 | 1,078 | 1,252 | 1,769 |
| CD155 | 1,600 | 1,802 | 2,646 | 4,690 |
Mean fluorescent intensity (MFI) was determined for specific molecule and background isotype staining. Values in table have been adjusted by subtracting isotype staining MFI from the specific staining level
* AT-3 cells were irradiated at 50 Gy and cultured a further 96 h before analysis by flow cytometry
Changes in surface marker expression profile following irradiation
Irradiation is a common treatment strategy for breast cancer patients and it has been reported that irradiation of tumor cells may result in the upregulation of some immunological surface molecules. This upregulation may therefore generate tumor cells that are more attractive targets for immunological or immunotherapeutic recognition or control. To investigate whether irradiation induced similar immunogenic changes in the CSC-like population of cells, the AT-3 cell line was irradiated and the surface marker expression profile of AT-3 CSC-like cells analyzed by flow cytometry.
Irradiation was found to have no effect on the expression levels of I-Ae, CD86, PD-L1, TRAIL, FasL or CD54 on the AT-3 CSC-like subpopulation (data not shown). However, as a consequence of irradiation, the levels of DR5, Fas, Rae-1 and CD155 were modestly increased on the AT-3 CSC-like cell population (Fig. 4; Table 1). There also appears to be a loss in MHC H-2Kb expression on the AT-3 CSC-like cell population following irradiation, which may enhance their potential as NK cell targets. Therefore, although resistant to radiation-induced cell death, the AT-3 CSC-like cells could still be induced to upregulate various immunological surface molecules that may potentiate their sensitivity to immunotherapy.
Fig. 4.
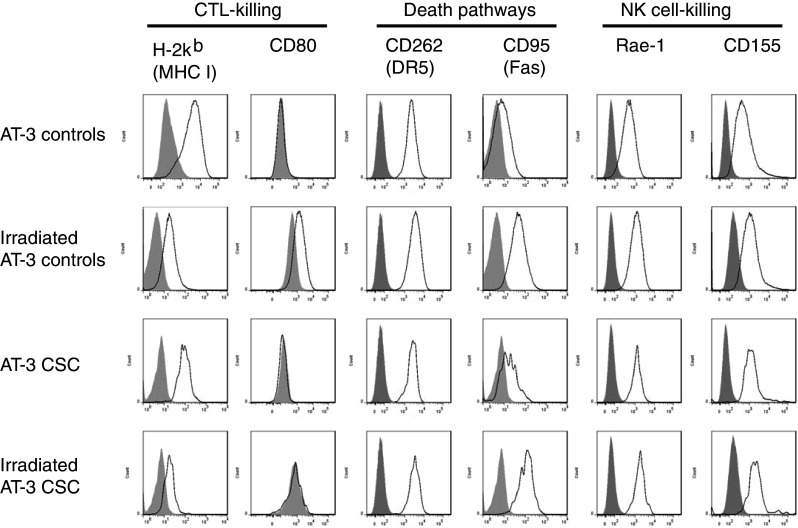
The AT-3 cell line was irradiated with 50 Gy, washed and cultured a further 96 h. Cells were harvested and stained for CD44/CD24 and a range of other immunologically relevant surface markers. Cells were then gated on either CD44+CD24− (AT-3 CSC) or CD44−/lowCD24+ (control) populations and their surface marker expression levels determined. Specific marker expression is represented by the black line and isotype staining by the shaded gray area. Results are representative of at least 3 separate experiments
AT-3 CSC-like cells are more sensitive to Fas-/FasL-mediated killing
Since we have demonstrated that AT-3 CSC-like cells express Fas molecules on their surface, an investigation into their sensitivity to Fas-/FasL-mediated cell death was undertaken. To test their sensitivity to this form of cell death, we used either the L5178Y cell line that has been transfected to express mouse FasL as “effector” cells that can bind and induce cell death in Fas-expressing target cells or the Jo2 anti-Fas mAb. Following exposure to FasL-expressing effector cells, the percentage of Annexin/7-AAD-positive cells was significantly higher (P = 0.0046 at 10:1) in the AT-3 CSC-like subpopulation compared to exposure to control-transfected L5178Y cells or to other AT-3 cells (i.e., AT-3 minus CD44+CD24− cells) exposed to L5178Y-mFasL effector cells (Fig. 5a). Their susceptibility to Fas-mediated cell death was verified with the anti-Fas mAb. Addition of the anti-Fas mAb (10 μg/ml), was found to induce significantly greater cell death in AT-3 CSC-like cells compared to other AT-3 cells (P = 0.0052) based on Annexin+ staining (Fig. 5b). Therefore, the AT-3 CSC-like cells were found to be more sensitive than the remainder of the AT-3 population of tumor cells to both cell- and Ab-mediated induction of the Fas-mediated cell death pathway.
Fig. 5.

a L5178Y-mFasL or control “effectors” were added at stated ratios to AT-3 CSC or other (AT-3 minus CSC) target cells and incubated 20 h. Cells were harvested from wells and stained with Annexin and 7-AAD before analysis by flow cytometry. Graph represents the mean ± SEM of Annexin/7-AAD double-positive cells from duplicate wells and where ** equals a P value of 0.005. Data are representative of three separate killing assays. b Anti-Fas mAb or its isotype control (10 μg/ml) was added to triplicate wells of sorted AT-3 CSC or other AT-3 cells at 5 × 104 cells/well and incubated for 20 h. Cells were harvested and stained for Annexin before flow cytometric analysis. Graph represents the mean ± SEM of Annexin positive cells and where ** equals a P value of 0.0052 calculated using a Student’s t test. Data are representative of three separate experiments. c Standard 4-h chromium release assays were conducted using 2PK3-mTRAIL and control “effector” cells against the AT-3 CSC or other AT-3 target cell populations. Graph represents the mean ± SEM of % lysis from triplicate wells. Data are representative of three separate killing assays and * equals a P value of 0.019 when comparing mTRAIL-induced killing of CSC and other AT-3 cells. d The MD5.1 (anti-DR5) mAb, or its Ig control, was added to P815 cells (FcR+CD16+) at 10 μg/ml and combined with AT-3 CSC or other AT-3 target cell populations at an E:T of 10:1. Following an overnight incubation, cells were harvested and stained for CD16, Annexin and 7AAD. The percentage of Annexin/7AAD double-positive cells within the CD16− target cell populations was determined. The graphed values are the mean ± SEM of duplicate wells that have had the background Ig percentage subtracted. The results are representative of 3 separate MD5.1-induced killing assays. *** equals a P value of <0.0001. e NK effector cells were purified from syngeneic spleens and stimulated in vitro with IL-2 prior to mixing with 51Cr-labeled AT-3 CSC and other AT-3 target cells in a standard 4-h chromium release assay. Graph represents the mean ± SEM of % lysis from triplicate wells at a range of E/T ratios. Supernatants were harvested, the amount of 51Cr release counted and the percentage lysis determined. Results represent at least 3 similar NK killing assays
CSC-like cells are susceptible to TRAIL-mediated killing
AT-3 CSC-like cells also expressed the TRAIL death receptor DR5, and therefore have the potential to be attacked through the TRAIL-mediated cell death pathway. To demonstrate that the AT-3 CSC-like cells could be killed through the TRAIL pathway, TRAIL-expressing effector cells (2PK3-mTRAIL) were used to trigger DR5 on the target cells and stimulate this cell death pathway. 51Cr release, mediated by 2PK3-mTRAIL cells, was significantly higher (P = 0.019) for AT-3 CSC-like cells than other AT-3 cells (Fig. 5c) at the 10:1 E/T ratio but not at lower ratios. Therefore, the CSC-like population of AT-3 cells appeared to be modestly more sensitive to TRAIL-mediated cell death than the remaining population of AT-3 tumor cells. An anti-DR5 mAb, MD5-1, has been developed as a means of immunotherapeutic control of TRAIL-sensitive tumors [21], both in vitro and in vivo. Given the apparent sensitivity of the AT-3 CSC-like cells to TRAIL-mediated killing, we therefore went on to investigate whether this form of therapy could induce apoptosis in the AT-3 CSC-like population. Because this mAb has been shown to require cross-linking for efficacy [21], FcR-expressing P815 cells were added to the wells to facilitate mAb cross-linking. It was demonstrated that the level of cell death was greater in AT-3 CSC-like cells compared to other AT-3 cells following exposure to the MD5-1 mAb (anti-DR5). Therefore, the AT-3 CSC-like cells were sensitive, if not more so, than other AT-3 tumor cells to both cell- and Ab-mediated induction of the TRAIL-mediated cell death pathway.
Sensitivity of CSC-like cells to NK cell-mediated cell death
The sensitivity of AT-3 CSC-like cells to NK cell-mediated cell death was tested in a standard in vitro 51Cr release cytotoxicity assay using activated splenic NK cells as effectors and FACS-sorted AT-3 CSC-like cells and other AT-3 tumor cells as targets. At different E/T ratios, the AT-3 CSC-like cells underwent NK cell-mediated apoptosis just as effectively as the other AT-3 cells (Fig. 5e). Therefore, the AT-3 CSC-like cells were equivalently sensitive to NK cell-mediated cell death as other AT-3 tumor cells, suggesting that there is no inherent-resistant capacity of these CSC-like cells to NK cell-mediated cytotoxicity.
Discussion
Given the resistance of CSC to conventional chemotherapeutics and radiotherapy and their pluripotent capacity, it is becoming increasingly evident that improvement in cancer treatment responses and cures may not be possible without control of the CSC component of tumors. It is therefore important to identify alternative strategies whereby CSC can be therapeutically mitigated. It has been suggested that several factors contribute to the resistance of CSC to conventional therapies including a relatively slow cell cycling, active efflux of drugs by ABC transporters and the upregulation of pro-survival genes [4, 22].
One approach that may bypass the refractory nature of CSC to current therapies is to attack these cells using immunotherapeutic strategies. Most CSC investigation models have utilized cells from patient samples or from human cancer cell lines and have focused on self-renewal pathways and therapy resistance. These xenograft model systems necessitate the use of immunocompromised animal hosts and severely limit our ability to study complex in vivo immune responses to CSC. In addition to being in an immunocompetent setting, it may also be important that these CSC models be conducted in an autologous model system where CSC biology, or the formation of CSC niches, is not aberrantly influenced by non-species-specific environment or factors [23, 24].
We have therefore identified and validated a murine cell population that exhibits many of the reported characteristics of human CSC and can be used as a model system to study interactions between breast CSC-like cells and a competent immune system. This autologous model can be used in immunocompetent settings and will permit the study of the CSC-like populations’ susceptibility to immunotherapeutic strategies. The CSC-like population within the AT-3 mammary carcinoma cell line was initially identified based on the commonly used CD44 and CD24 breast cancer stem cell markers. This CD44+CD24− CSC-like population was also found in the primary tumors of MTAG mice, from which the AT-3 cell line was derived, indicating that the AT-3 CSC-like cells were not just an artifact of in vitro culturing. In the first instance, given the reported variety of markers used to define CSC populations not only between different tumor types but also different tumor subtypes [25, 26], a CSC-like population cannot be defined by cell markers alone. Considerable effort was therefore made to verify the CSC-like characteristics of this putative AT-3 CSC-like population. These characteristics included a quiescent nature, the ability to undergo pluripotent differentiation and a resistance to irradiation and chemotherapy. Variety in defining a CSC-like population based on surface marker expression was evident from our screening of other mouse mammary carcinoma cell lines. The CD44/CD24 markers, although commonly used in human breast cancers, did not always either identify a CD44+CD24− population or if a population could be identified, it may not have possessed the further characteristics associated with CSC-like cells. Based on the number of lines screened (i.e. 12), a well-defined population with a CD44+CD24− phenotype and with CSC-like properties may not be a common occurrence in mouse mammary carcinoma lines. The difficulty in identifying a CSC-like population may explain the paucity of reported CSC-like experimental models. It is therefore important that a CSC-like population has been identified that mirrors reported human CSC and can be utilized as an experimental model to test therapeutic responses in autologous and immunocompetent settings.
Results represented in Fig. 1c suggest that purified CD44+CD24− cells have a pluripotent capacity that permits them to differentiate and reconstitute the whole cell line. Another possibility is that differences in proliferation rates (Fig. 2a) may be responsible for the changes in the CD44/CD24 subpopulation ratios following culture of the purified subpopulations. However, it was evident that the CD44+CD24−-purified subpopulation went through an intermediate phenotype (CD44+CD24+), which was shown to have a slower proliferation rate than that of the CD44lowCD24+ subpopulation (Fig. 2a). This intermediate population, after 1 week of culture, had a much higher percentage of cells than the faster proliferating CD44lowCD24+ subpopulation, suggesting that a higher proliferation of “contaminating cells” could not entirely account for the observed changes in subpopulation ratios. The percentage of this intermediate population then decreased as the CD44lowCD24+ subpopulation increased until the ratio of the three CD44/CD24 cell subpopulations returned to that of the original parental cell line. The CD44+CD24− purified cells were also shown to be the only subpopulation that was eventually able to reconstitute, in similar ratios, the whole AT-3 line, even after long-term culturing. This supports our contention that the accumulation of CD44lowCD24+ cells from a culture of CD44+CD24−-purified cells was not due, in the main, to the faster proliferation of other contaminating cells but rather was a result of differentiation and proliferation.
The immunological expression profile of specific cell surface molecules on the CSC-like cells compared to other AT-3 tumor cells suggested that there was no inherent barrier to utilizing specified immunological recognition and cell death pathways, based on the expression levels. Indeed, some molecules, particularly those associated with NK cell, Fas/FasL- or TRAIL-mediated cell death pathways, appeared to be more highly expressed on the CSC-like subpopulation than on the remainder of the AT-3 tumor cells. The CSC-like cells also did not express the negative regulatory molecules TRAIL, FasL and PD-L1, which could inhibit immune responses. The zero-to-low expression levels of MHC I and II, CD80 and CD86 suggest that they would not be good targets for CTL killing, although low MHC levels would permit NK cell-mediated killing.
Induction of the Fas/FasL- or TRAIL-mediated cellular death pathways was achieved through receptor/ligand interactions using either mFasL- or mTRAIL-expressing cells or anti-Fas and anti-DR5 mAbs, respectively. The level of sensitivity appeared to correlate with differences in receptor expression, such that Fas expression on AT-3 CSC-like cells was much higher than on other AT-3 cells and resulted in significantly greater sensitivity to Fas-mediated killing. The difference in DR5 expression between the CSC-like population and the rest of the AT-3 cells was not as great and correlated with only a modestly higher sensitivity to TRAIL-mediated killing. In the case of NK cell-mediated killing, however, increased NK cell ligand expression did not result in an increased sensitivity to killing by activated NK cells. This may be due to the balance between expressions of inhibitory versus activating receptors on the tumor target cells. However, there was no inherent resistance of the CSC-like cells to NK cell-mediated killing.
It was also demonstrated that although the CSC-like cells were resistant to radiation-induced cell death, irradiation could still be used to upregulate various immunological surface molecules such as Fas and DR5, on the CSC-like cell population. Irradiation also led to a downregulation of MHC class I, which may enhance their susceptibility to NK cell killing. Therefore, tumor irradiation has the potential to render the CSC population an even better target for immunotherapy.
It was demonstrated that the induction of apoptosis in the CSC fraction of tumor cells by an agonistic mAb to either Fas or DR5, or death receptor ligand–expressing cells, is a potential strategy to bypass the refractory nature of CSC to conventional therapy. The use of Ab-based immunotherapies is steadily growing; however, directly targeting CSC through their defining antigens, such as CD44, is difficult because they are often expressed on normal stem cells or other host tissues, and the absence of certain markers (e.g., CD24), which is also used to classify many CSC, makes their use as therapeutic targets impractical. Ab-based therapies that induce tumor cell apoptosis are being actively pursued [27], and the previously unreported higher activity against CSC-like cells demonstrated here may be an important consideration in the development of new combination strategies. The importance of Fas-mediated killing as a means of targeting CSC is supported by a recent report showing a role for Fas/FasL signaling in CSC apoptosis as a bystander effect of using IL-8/CXCR1 interference to target breast CSC [28]. The Jo2 anti-Fas is a strong agonistic mAb that mimics the action of natural FasL by binding Fas and inducing cytoplasmic signaling that results in target cell apoptosis. There was considerable clinical potential for humanized anti-Fas Abs, although realizing this potential has been hindered by the reported induction of severe hepatotoxic side effects [29].
Preclinical studies have demonstrated the potential of TRAIL to selectively induce apoptosis of tumor cells and have driven the development of TRAIL-based cancer therapies [30]. Their specific effect on the CSC population is yet unclear; however, our data suggest that TRAIL-based therapies may prove effective at not only selectively killing “normal” tumor cells but also attacking the CSC component. Translation of TRAIL-based therapies into clinical trials has been promising and without significant toxicity [31, 32], suggesting that this form of therapy may indeed prove an effective strategy to kill CSC-like cells. The use of TRAIL-based therapies may be of particular value given the reported synergistic effect observed in preclinical settings of modulators of the death receptor pathways and chemotherapy [32, 33]. Other ways to target CSC by Ab-based therapies are being investigated [34]; however, the use of Abs in inducing cell death pathways in the CSC, which express higher levels of specific cell death-associated molecules, may be a novel strategy to mitigate these cells. In addition to Ab-mediated induction of cell death, cell death pathways can be activated by FasL- or TRAIL-expressing cells. The adoptive transfer or induction of immune effector cells that express death receptor ligands may therefore be an alternative strategy for the induction of apoptosis in CSC.
Therefore, although CSC are resistant to radiation and chemotherapies, it is encouraging to demonstrate that CSC-like cells may not be inherently resistant to at least some forms of immune-mediated killing. This indicates that CSC-like cells in tumors can be eliminated or controlled through the use of immunotherapeutic approaches and supports the investigation of such approaches to enhance current treatment strategies. The observation that CSC-like cells may actually be more sensitive to some forms of immunotherapy is interesting, although this may not be sufficient to specifically target this population, the use of a therapy that controls both CSC and other tumor cells would be advantageous. Investigations into the sensitivity of CSC-like cells to various forms of immunotherapy in vivo are currently underway, although their rarity within tumors presents an analysis challenge. It could be envisaged that debulking the tumor through radio- and/or chemotherapy, and potentially enhancing the immunogenicity and proportion of CSC within the tumor, be used in combination with immunotherapies, such as Fas- or TRAIL-based therapies that could then be used to reduce the refractory CSC population. These investigations will have implications for current treatment strategies and for the design of next-generation therapies that include immunotherapeutic modalities that can target otherwise resistant CSC-like cells and lead to better outcomes for patients.
Acknowledgments
We would like to thank members of the Flow Cytometry core facility for their patience in obtaining the high number of CSC required for these investigations. This work is supported by the National Breast Cancer Foundation (Australia) and the Cancer Council of Victoria.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Cobaleda C, Cruz JJ, Gonzalez-Sarmiento R, Sanchez-Garcia I, Perez-Losada J. The emerging picture of human breast cancer as a stem cell-based disease. Stem Cell Rev. 2008;4:67–79. doi: 10.1007/s12015-008-9012-6. [DOI] [PubMed] [Google Scholar]
- 2.Dontu G, Liu S, Wicha MS. Stem cells in mammary development and carcinogenesis: implications for prevention and treatment. Stem Cell Rev. 2005;1:207–213. doi: 10.1385/SCR:1:3:207. [DOI] [PubMed] [Google Scholar]
- 3.Baumann M, Krause M, Hill R. Exploring the role of cancer stem cells in radioresistance. Nature Rev Cancer. 2008;8:545–554. doi: 10.1038/nrc2419. [DOI] [PubMed] [Google Scholar]
- 4.Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol Off J Am Soc Clin Oncol. 2008;26:2839–2845. doi: 10.1200/JCO.2007.15.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li X, Lewis MT, Huang J, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 6.Woodward WA, Bristow RG. Radiosensitivity of cancer-initiating cells and normal stem cells (or what the Heisenberg uncertainly principle has to do with biology) Semin Radiat Oncol. 2009;19:87–95. doi: 10.1016/j.semradonc.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oliveira LR, Jeffrey SS, Ribeiro-Silva A. Stem cells in human breast cancer. Histol Histopathol. 2010;25:371–385. doi: 10.14670/HH-25.371. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt C. Lapatinib study supports cancer stem cell hypothesis, encourages industry research. J Natl Cancer Inst. 2008;100:694–695. doi: 10.1093/jnci/djn168. [DOI] [PubMed] [Google Scholar]
- 10.Shipitsin M, Polyak K. The cancer stem cell hypothesis: in search of definitions, markers, and relevance. Lab Invest. 2008;88:459–463. doi: 10.1038/labinvest.2008.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zitvogel L, Kepp O, Kroemer G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol. 2011;8:151–160. doi: 10.1038/nrclinonc.2010.223. [DOI] [PubMed] [Google Scholar]
- 13.Locher C, Conforti R, Aymeric L, et al. Desirable cell death during anticancer chemotherapy. Ann N Y Acad Sci. 2010;1209:99–108. doi: 10.1111/j.1749-6632.2010.05763.x. [DOI] [PubMed] [Google Scholar]
- 14.Stewart TJ, Greeneltch KM, Reid JE, Liewehr DJ, Steinberg SM, Liu K, Abrams SI. Interferon regulatory factor-8 modulates the development of tumour-induced CD11b+Gr-1+ myeloid cells. J Cell Mol Med. 2009;13:3939–3950. doi: 10.1111/j.1582-4934.2009.00685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stewart TJ, Abrams SI. Altered immune function during long-term host-tumor interactions can be modulated to retard autochthonous neoplastic growth. J Immunol. 2007;179:2851–2859. doi: 10.4049/jimmunol.179.5.2851. [DOI] [PubMed] [Google Scholar]
- 16.Nakayama M, Harada N, Okumura K, Yagita H. Characterization of murine TWEAK and its receptor (Fn14) by monoclonal antibodies. Biochem Biophys Res Commun. 2003;306:819–825. doi: 10.1016/S0006-291X(03)01051-9. [DOI] [PubMed] [Google Scholar]
- 17.Latour S, Fridman WH, Daeron M. Identification, molecular cloning, biologic properties, and tissue distribution of a novel isoform of murine low-affinity IgG receptor homologous to human Fc gamma RIIB1. J Immunol. 1996;157:189–197. [PubMed] [Google Scholar]
- 18.Stewart TJ, Drane D, Malliaros J, Elmer H, Malcolm KM, Cox JC, Edwards SJ, Frazer IH, Fernando GJ. ISCOMATRIX adjuvant: an adjuvant suitable for use in anticancer vaccines. Vaccine. 2004;22:3738–3743. doi: 10.1016/j.vaccine.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 19.Chan CJ, Andrews DM, McLaughlin NM, Yagita H, Gilfillan S, Colonna M, Smyth MJ. DNAM-1/CD155 interactions promote cytokine and NK cell-mediated suppression of poorly immunogenic melanoma metastases. J Immunol. 2010;184:902–911. doi: 10.4049/jimmunol.0903225. [DOI] [PubMed] [Google Scholar]
- 20.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takeda K, Yamaguchi N, Akiba H, et al. Induction of tumor-specific T cell immunity by anti-DR5 antibody therapy. J Exp Med. 2004;199:437–448. doi: 10.1084/jem.20031457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keysar SB, Jimeno A. More than markers: biological significance of cancer stem cell-defining molecules. Mol Cancer Ther. 2010;9:2450–2457. doi: 10.1158/1535-7163.MCT-10-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sneddon JB, Werb Z. Location, location, location: the cancer stem cell niche. Cell Stem Cell. 2007;1:607–611. doi: 10.1016/j.stem.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borovski T, De Sousa EMF, Vermeulen L, Medema JP. Cancer stem cell niche: the place to be. Cancer Res. 2011;71:634–639. doi: 10.1158/0008-5472.CAN-10-3220. [DOI] [PubMed] [Google Scholar]
- 25.Hurt EM, Farrar WL. Purification and characterization of cancer stem cells. In: Farrar WL, editor. Cancer stem cells. 1. NY: Cambridge University Press, New York; 2010. pp. 1–14. [Google Scholar]
- 26.Nakshatri H, Srour EF, Badve S. Breast cancer stem cells and intrinsic subtypes: controversies rage on. Curr Stem Cell Res Ther. 2009;4:50–60. doi: 10.2174/157488809787169110. [DOI] [PubMed] [Google Scholar]
- 27.Mahalingam D, Szegezdi E, Keane M, de Jong S, Samali A. TRAIL receptor signalling and modulation: are we on the right TRAIL? Cancer Treat Rev. 2009;35:280–288. doi: 10.1016/j.ctrv.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 28.Ginestier C, Liu S, Diebel ME, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Investig. 2010;120:485–497. doi: 10.1172/JCI39397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ogasawara J, Watanabe-Fukunaga R, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y, Itoh N, Suda T, Nagata S. Lethal effect of the anti-Fas antibody in mice. Nature. 1993;364:806–809. doi: 10.1038/364806a0. [DOI] [PubMed] [Google Scholar]
- 30.Cretney E, Takeda K, Smyth MJ. Cancer: novel therapeutic strategies that exploit the TNF-related apoptosis-inducing ligand (TRAIL)/TRAIL receptor pathway. Int J Biochem Cell Biol. 2007;39:280–286. doi: 10.1016/j.biocel.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 31.Wu GS. TRAIL as a target in anti-cancer therapy. Cancer Lett. 2009;285:1–5. doi: 10.1016/j.canlet.2009.02.029. [DOI] [PubMed] [Google Scholar]
- 32.Abdulghani J, El-Deiry WS. TRAIL receptor signaling and therapeutics. Expert Opin Ther Targets. 2010;14:1091–1108. doi: 10.1517/14728222.2010.519701. [DOI] [PubMed] [Google Scholar]
- 33.Rajeshkumar NV, Rasheed ZA, Garcia-Garcia E, Lopez-Rios F, Fujiwara K, Matsui WH, Hidalgo M. A combination of DR5 agonistic monoclonal antibody with gemcitabine targets pancreatic cancer stem cells and results in long-term disease control in human pancreatic cancer model. Mol Cancer Ther. 2010;9:2582–2592. doi: 10.1158/1535-7163.MCT-10-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deonarain MP, Kousparou CA, Epenetos AA. Antibodies targeting cancer stem cells: a new paradigm in immunotherapy? MAbs. 2009;1:12–25. doi: 10.4161/mabs.1.1.7347. [DOI] [PMC free article] [PubMed] [Google Scholar]