

Abstract
We have previously reported two trials in non-small cell lung cancer (NSCLC) evaluating vaccine therapy with the telomerase peptide GV1001. The studies demonstrated considerable differences in survival among immune responders, highlighting that an immune response is not necessarily beneficial. In the present study, we conducted long-term clinical follow-up and investigated immunological factors hypothesized to influence clinical efficacy. Peripheral blood mononuclear cells from 33 NSCLC trial patients and 15 healthy donors were analyzed by flow cytometry for T regulatory cells (Tregs, CD4+CD25+CD127low/−FOXP3+) and two types of myeloid-derived suppressor cells (MDSCs, HLA-DR low CD14 + or Lin −/lo HLA-DR − CD33 + CD11b +). T cell cultures were analyzed for 17 cytokines. The results demonstrated that immune responders had increased overall survival (OS, p < 0.001) and progression-free survival (p = 0.003), compared to subjects without immunological response. The mean OS advantage was 54 versus 13 months. Six patients were still alive at the last clinical update, all belonging to the immune responders. No serious toxicity had developed (maximum observation 13 years). Most patients developed a polyfunctional cytokine profile, with high IFNγ/IL-4 and IFNγ/IL-10 ratios. Low Treg levels were associated with improved OS (p = 0.037) and a favorable cytokine profile, including higher IFNγ/IL-10 ratios. High CD33+ MDSC levels were associated with poorer immune response rate (p = 0.005). The levels of CD14+ MDSC were significantly higher in patients than in healthy controls (p = 0.012). We conclude that a randomized GV1001 trial in NSCLC is warranted. The findings suggest that Tregs and MDSCs are associated with a tolerogenic cytokine milieu and impaired clinical efficacy of vaccine responses.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-015-1766-5) contains supplementary material, which is available to authorized users.
Keywords: Lung cancer, Myeloid-derived suppressor cells, Regulatory T cells, Cytokines, Cancer vaccine, Survival
Introduction
Lung cancer is the most prevalent form of cancer and the leading cause of cancer-related death worldwide [1]. The finding that PD-1 blockade produces clinical responses in non-small cell lung cancer (NSCLC) [2] has yielded increased interest for cancer vaccines and their possible combination with checkpoint inhibitors [3]. GV1001 is a 16-aa telomerase peptide vaccine designed to activate both CD4+ and CD8+ T cells [4]. We have conducted a series of trials evaluating GV1001 therapy in patients with NSCLC, pancreatic cancer, or melanoma [5–7]. Other investigators have performed GV1001 trials in pancreatic cancer and hepatocellular carcinoma patients [8, 9].
Telomeric DNA provides stability to chromosomes [10], and normal somatic cells can undergo a limited number of cell divisions because the telomeres are shortened at each mitosis. Tumor cells reset this biological clock by expressing the enzyme telomerase that synthesizes new telomere units [11]. Telomerase is expressed in most human cancers, including lung cancer [12, 13], and is considered as an attractive target for a universal cancer vaccine [14–16].
The GV1001 peptide is recognized on multiple HLA class II molecules encoded by DP, DQ, and DR subloci [5, 7, 17, 18]. This promiscuous HLA-binding profile suggests that the GV1001 vaccine may be applicable to the general patient population and may elicit a broad T helper response within each individual. Further, GV1001 includes nested HLA class I epitopes, facilitating recruitment of CD8+ cytotoxic T cells.
In the GV1001 trials, we have observed an association between immune response and survival [5–7]. However, we have also noted a considerable difference in survival within the group of immune responders. This observation highlights the issue that an immune response, though tumor specific, is not necessarily beneficial. We therefore decided to investigate the GV1001 vaccine response with regard to key immunological factors hypothesized to be important for clinical efficacy: myeloid suppressor cells, T regulatory cells, and cytokine profile.
A T helper (Th) 1-dominated cytokine profile with high levels of INFγ and TNFα, but low levels of IL-4 and IL-10, is considered desirable in cancer vaccine therapy. Numerous studies have demonstrated that Th1 cytokines promote cytotoxic cellular responses and anti-tumor activity [19]. By contrast, a Th2-dominated response with high IL-4 and IL-10 levels may inhibit Th1 differentiation [19]. Regulatory T cells (Tregs) with a CD4+CD25+CD127low/−FOXP3+ phenotype have been extensively studied in animal models and are considered to counter anti-tumor immunity even in humans [20]. However, it is not known whether Treg levels can serve as biomarkers for clinical efficacy of cancer vaccination. Myeloid-derived suppressor cells (MDSCs) have also been shown to suppress immunity against tumor [21–23]. In mice, MDSCs are characterized by cell surface markers Gr1 and CD11b and have a granulocytic or monocytic morphology [22, 23]. In humans, MDSCs constitute a diverse group of cells, the common denominator being their suppressive function and myeloid origin [21]. The most extensively studied MDSC phenotype in humans is defined as CD33+CD11b+ HLA-DR− cells lacking expression of markers of mature myeloid and lymphoid cells [22, 24, 25]. Another key MDSC subset has been identified as CD14+HLA-DRlow [26–28].
In the present article, we report long-term clinical data from the two GV1001 vaccine trials (CTN-2000 and CTN-2006) in NSCLC patients. We further report the results of our investigations of cytokine profiles, Tregs, and MDSCs in the vaccinated patients and how these factors relate to clinical outcome.
Materials and methods
Patients, protocols, and PBMC collection
Patient characteristics are given in Tables 1 and 2. The patients were included in protocols CTN-2000 or CTN-2006, which have been reported previously [6, 17]. Briefly, in the CTN-2000 protocol, 27 subjects with advanced NSCLC were vaccinated with two telomerase peptides (GV1001 and I540) [6, 17]. The majority (21/27) had stage IV disease. All CTN-2000 patients were considered incurable and had completed standard therapy before inclusion into the protocol. In the CTN-2006 trial, 23 inoperable stage III patients received radiotherapy (2 Gy × 30) and weekly docetaxel, followed by GV1001 vaccination [6]. These patients were previously untreated. The last update on clinical data was performed June 05, 2015, and is included in the present report.
Table 1.
CTN-2000
| Patienta | Age | Sex | Stage | Tumor responseb | Immune responsec | Progression-free survival (months) | Survival (months) | % Tregs | % CD14+ MDSC | % CD33+ MDSC |
|---|---|---|---|---|---|---|---|---|---|---|
| 701 | 60 | M | IIIB | SD | − | 10 | 11 | nsf | ns | ns |
| 702 | 69 | M | IV | PD | NE | <3 | 4 | ns | ns | ns |
| 703 | 68 | F | IV | PD | + | <3 | 15 | 7.07 | 0.03 | 0.58 |
| 704 | 60 | F | IV | SD | + | 8 | 18 | ns | ns | ns |
| 706 | 60 | M | IV | PD | + | <3 | 3 | ns | ns | ns |
| 707 | 59 | M | IV | PD | − | <3 | 3 | ns | ns | ns |
| 708 | 39 | M | IV | SD | + | 4 | 7 | 10.13 | ns | ns |
| 709 | 63 | F | IV | PD | − | <3 | 3 | ns | ns | ns |
| 710 | 55 | M | IIIA | CRd | + | >141d | >159Alive | 3.98 | 0.27 | 0.25 |
| 711 | 48 | M | IV | PD | + | <3 | 25 | 2.97 | ns | ns |
| 712 | 49 | F | IV | SD | + | 16 | 71 | 3.11 | 0.04 | 0.72 |
| 713 | 76 | F | IIIB | PD | + | <3 | 19 | ns | ns | ns |
| 714 | 68 | M | IV | PD | + | <3 | 32 | 4.01 | 0.08 | 1.00 |
| 715 | 45 | F | IV | PD | NE | <3 | 5 | ns | ns | ns |
| 716 | 72 | M | IV | PD | − | <3 | 2 | ns | ns | ns |
| 717 | 56 | M | IV | PD | − | <3 | 3 | ns | ns | ns |
| 718 | 57 | F | IV | PD | − | <3 | 4 | ns | ns | ns |
| 719 | 44 | M | IV | PD | − | <3 | 4 | ns | ns | ns |
| 720 | 57 | F | IV | NE | NE | <3 | 9 | ns | ns | ns |
| 721 | 63 | F | IV | PD | + | <3 | 6 | 4.27 | ns | ns |
| 722 | 60 | F | IV | PD | − | <3 | 4 | ns | ns | ns |
| 723 | 53 | M | IV | PD | + | <3 | 17 | 3.84 | 0.05 | ns |
| 724 | 60 | F | IV | PD | − | <3 | 10 | 3.15 | 0.6 | 1.05 |
| 725 | 55 | F | IIB | SD | + | 19 | 46 | 4.1 | 0.81 | 0.66 |
| 726 | 49 | M | IV | PD | − | <3 | 4 | ns | ns | ns |
| 727 | 57 | M | IIIB | NEDe | + | >144e | >144Alive | ns | 2.03 | 0.92 |
| 728 | 50 | M | IIIB | PD | − | <3 | 18 | 8.66 | 1.57 | 1.62 |
CR complete response, SD stable disease, PD progressive disease, NE not evaluable, NED no evidence of disease
a12/26 patients did not complete the vaccination period (8 vaccines) prior to first clinical evaluation at week 12, due to early disease progression. Patient 728 was enrolled after the scheduled inclusion period and was not included in the trial paper
bBest tumor response after vaccination (RECIST)
cGV1001-specific immune response, as demonstrated in T cell assays (n = 10) and/or delayed-type hypersensitivity skin reactions (n = 9)
dPatient 710 had a durable complete response at last clinical follow-up (month 141) and is still alive per June 05, 2015 (month 159)
ePatient 727 has residual fibrosis, with no detectable disease (144 months of observation)
fns = no samples available for testing
Table 2.
CTN-2006
| Patient | Age | Sex | T cell response | Relapse-free survival (days) | Survival (days) | % Tregs | % CD14+ MDSC | % CD33+ MDSC |
|---|---|---|---|---|---|---|---|---|
| 101 | 66 | M | POS | 359 | 1147 | 4.36 | ns | ns |
| 102 | 62 | M | NEG | 70 | 288 | 5.26 | 0.13 | 4.74 |
| 104 | 57 | F | POS | 238 | 705 | 6.38 | 0.13 | 1.99 |
| 105 | 50 | M | POS | 267 | 630 | 7.25 | 0.92 | 1.44 |
| 106 | 53 | M | POS | 179 | 446 | 5.16 | 0.19 | 1.00 |
| 107 | 57 | F | POS | >2818b | >2818Alive | 4.82 | 0.04 | ns |
| 108 | 54 | M | POS | 151 | 220 | 6.12 | 2.49 | 0.88 |
| 110a | 49 | F | NEGa | 63 | 144 | nsc | ns | ns |
| 111 | 59 | M | POS | >1819b | 2204 | 5.32 | 0.07 | ns |
| 113 | 63 | F | POS | 63 | 564 | 4.91 | 0.03 | 0.51 |
| 115 | 58 | F | POS | 259 | 1059 | 5.54 | 0.15 | 0.17 |
| 117 | 52 | M | NEG | 357 | 860 | 10.80 | 0.22 | 10.94 |
| 202 | 63 | F | NEG | 1251 | 1304 | 5.32 | 0.06 | 5.03 |
| 203a | 74 | M | NEGa | 93 | 1120 | 3.45 | 0.02 | 3.36 |
| 204a | 63 | M | NEGa | 491 | 766 | ns | ns | ns |
| 205 | 60 | F | POS | 382 | 667 | 7.99 | 0.11 | 1.76 |
| 301 | 70 | F | NEG | 182 | 361 | ns | 0.11 | 3.38 |
| 302 | 51 | M | POS | >2944b | >2944Alive | 3.37 | 0.26 | 0.68 |
| 303 | 78 | M | POS | >2909b | >2909Alive | 11.30 | 0.01 | ns |
| 304 | 56 | M | POS | 1079 | 1143 | 2.69 | 0.04 | ns |
| 305 | 58 | M | POS | 540 | 1155 | 3.35 | ns | 0.70 |
| 306 | 69 | F | POS | 183 | 360 | 5.17 | 0.25 | 0.62 |
| 307 | 46 | F | POS | >2622b | >2622Alive | 3.24 | 0.08 | 1.01 |
aNot evaluable per protocol (included in intention-to-treat population)
bNo relapse per June 05, 2015
cns = no samples available for testing
In both CTN-2000 and CTN-2006, PBMCs were obtained prior to vaccination, week 6, week 10, and at later booster vaccinations. PBMCs were isolated from blood by gradient centrifugation (Lymphoprep, Axis-Shield, Oslo, Norway) and frozen for storage in liquid nitrogen, as previously described [29]. T cell proliferation (3H-thymidine) assays were used to determine the immune response to GV1001. T cell responses were considered Ag specific when the stimulatory index (response with Ag divided by response without Ag) was above 2. Materials and methods for the CTN-2000 and CTN-2006 trials are described in the respective trial articles [6, 17]. In the CTN-2000 trial, 13/24 evaluable subjects developed a GV1001 response. In CTN-2006, a GV1001-specific response developed in 16/20 evaluable patients.
In the extended immunological studies reported below, we included all 33 patients whose samples were available after completion of standard immune-monitoring, i.e., 33/42 patients who completed the first 6 weeks of vaccination. We mostly used patient PBMCs collected between study months 2 and 7. In addition, 15 healthy blood donors were used as controls. These controls were not age/sex matched. The patients available for this immunological follow-up study represented 22/23 subjects in CTN-2006 and 27/30 patients with locally advanced disease (stage IIb/III). Among stage IV patients, samples were only available from 7 patients, and these were not representative for all stage IV subjects regarding clinical outcome. We therefore decided not to perform statistical analyses relating Treg/MDSC values with clinical outcome for stage IV patients.
Ethical concerns
The trials were approved by the Norwegian Medicines Agency, the Regional Committee for Medical Research Ethics and the Hospital Review Board. Signed informed consent was obtained from all patients. The study was performed in compliance with the World Medical Association Declaration of Helsinki.
Peptides
The vaccine peptide GV1001 (EARPALLTSRLRFIPK) was supplied by Pharmexa (Horsholm, Denmark).
Bioplex assay
Thawed PBMCs were plated in 24-well plates (1,5–2 × 106 c/w) and stimulated once in vitro with the GV1001 peptide, as described earlier [5, 17]. The T cells were then restimulated in 96-well plates with irradiated PBMCs ± GV1001 peptide. Bioplex cytokine analyses were performed on supernatants harvested 48 h after T cell stimulation, according to the manufacturer’s protocol (Bio-Rad Laboratories, Hercules, CA, USA). Supernatants were analyzed in duplicate, each parallel kept separate through T cell stimulation and Bioplex assays. Supernatant levels of IL-1b, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12, IL-13, IL-17, G-CSF, GM-CSF, INFγ, MCP-1, MIP-1b, TNFα were measured. The T cells used for Bioplex assays were also tested in T cell proliferation assays, performed in triplicate as previously described [29]. Both proliferation and cytokine responses were considered Ag specific when the stimulatory index was above 2.
Flow cytometry
Analysis of HLA-DRlowCD14+ MDSCs
PBMC from patient samples (n = 28) and healthy controls (n = 11) were obtained as described above. Thawed cells were stained with anti-CD19-APC, anti-CD14-PE (Becton Dickinson, Trondheim, Norway), anti-HLA-DR-e450 (eBioscience, San Diego, CA, USA), or e450-isotype control (eBioscience).
Analysis of Lin−/loHLA-DR−CD33+CD11b+ MDSCs (CD33+ MDSCs)
PBMCs from patient samples (n = 24) and healthy controls (n = 10) were obtained as described above. Thawed cells were stained with anti-lineage antibodies (anti-CD4/CD8/CD19/CD57-e450, eBioscience) and anti-CD14-PerCP-Cy5 (eBioscience). The samples were further stained with either anti-HLA-DR-FITC, anti-CD33-APC, and anti-CD11b (Becton Dickinson) or FITC-isotype, APC-isotype, and PE-isotype (Becton Dickinson).
Analysis of CD4+CD127lowFoxp3highCD25high regulatory T cells
PBMCs from patients samples (n = 31) were obtained as described above. Thawed cells were stained with anti-CD4-e450 (eBioscience), anti-CD25-AF700 (BioLegend, San Diego, CA, USA), anti-CD127-PE, and anti-FoxP3-AF488 (Becton Dickinson). Isotype controls were stained with anti-CD4-e450 (eBioscience), PE-isotype, AF488-isotype (Becton Dickinson), and AF700-isotype (BioLegend). Intracellular staining with anti-FoxP3 or AF488-isotype was done using Foxp3 Staining Buffer Set (eBioscience).
The flow cytometry was performed using a BD LSRII flow cytometer and FlowJo software. The quantities of MDSCs or Tregs were calculated as percentage of total PBMCs or CD4+ T cells, respectively.
Statistical analysis
Descriptive
Data were described with proportions (percentage) for categorical data and with median, mean, and range for continuous data.
Univariate analyses
Mann–Whitney U test was used for comparing patients and healthy controls with regard to distribution of Lin-CD33+ MDSCs or HLA-DRlowCD14+ MDSCs.
Survival analyses
Overall survival (OS) was defined as time from the start of vaccination to death or end of study (June 05, 2015), whichever came first. Progression-free survival (PFS) was defined as time from the start of vaccination to progression, death, or end of study.
OS and PFS were analyzed with Kaplan–Meier analysis and compared between immune responders and non-responders using log-rank test. In accordance with the criteria in protocols CTN-2000 and CTN-2006, the patients were classified as immune responders if and only if they exhibited a GV1001-specific T cell response in proliferation assays with a stimulatory index above 2. Kaplan–Meier analyses with log-rank tests were also performed to compare PFS between the patients with high versus low levels of Tregs or MDSCs. For this purpose, the patients were dichotomized into a “high” and “low” group for Tregs or MDSC, based on the median Treg or MDSC level as cutoff.
All tests were two sided. We consider this study an exploratory analysis. Therefore, we did not perform any correction for multiple testing, and p values <0.05 were considered statistically significant. All analyses were performed using SPSS, version 18 (SPSS Inc., USA).
Results
Long-term clinical follow-up
Tables 1 and 2 show patient characteristics, immune responses, and updated clinical data (June 2015) for CTN-2000 and CTN-2006, respectively. The long-term survivors have been continuously followed at the study hospitals. Serious adverse events have not been observed in either trial. The patients received no further cancer therapy until disease progression. For most CTN-2000 subjects, further therapy was not warranted even after progression, as they had already completed standard therapy. No study subjects from either protocol have received immunological checkpoint inhibitors.
Two patients from the CTN-2000 study are still alive, 12 and 13 years after the start of vaccination, respectively. Neither of them has evidence of cancer relapse. From the CTN-2006 protocol, four patients are still alive, all without evidence of relapse (observation time 7–8 years). All six survivors in the two protocols belong to the immunological vaccine responders and have retained durable GV1001-specific T cell responses as far as monitored. None of the patients have developed evidence of adverse effects. The six survivors have not received any additional NSCLC therapy.
Immune responders had prolonged survival
We observed GV1001-specific T cell responses in 29/45 evaluable subjects in the two protocols taken together [17]. Kaplan–Meier/log-rank analyses on the updated data (June 2015) confirmed our previous observation that immune responders, compared to non-responders, had increased progression-free survival (PFS) and overall survival (OS). All patients tested for immune response were included in the analyses. The OS and PFS advantages applied to both protocol CTN-2000 and CTN-2006 (Tables 1 and 2), though statistically significant only for CTN-2000 when the protocols were analyzed separately (OSCTN2000 p < 0.001, PFSCTN2000 p = 0.026, OSCTN2006 p = 0.10, PFSCTN2006 p = 0.08). Taking both trials together, log-rank test demonstrated a statistically significant advantage for immune responders, both for PFS (p = 0.003) and for OS (p < 0.001) (Fig. 1). The mean PFS advantage was 35 versus 6.7 months, and mean OS advantage was 54 versus 13 months. We also performed Kaplan–Meier analysis on all subjects with locally advanced disease (n = 29). Again, immune responders had significantly extended PFS (p = 0.02) and OS (p = 0.01). There was a 4.6-fold increase in mean PFS (50 vs. 11 months) and a 3.3-fold OS advantage (69 vs. 21 months).
Fig. 1.

Survival versus immune response. Kaplan–Meier analysis comparing progression-free survival (PFS, a) or overall survival (b) between patients with/without a GV1001-specific T cell response. Log-rank p values are displayed
The observed association between immune response and clinical outcome was most evident for the “tail” of the PFS/OS curves (Fig. 1), representing long-term survivors. All six patients still alive were immune responders. On the other hand, a considerable subpopulation of immune responders had short survival. This variability points to the need to study factors that influence whether an immune response translates to clinical benefit. In the studies of Tregs and MDSCs reported below, we investigated whether these regulatory cells represent biomarkers of clinical outcome.
Immune responders with low Treg levels recorded prolonged PFS
Regulatory T cells are hypothesized to impair the development of anti-cancer vaccine responses and to counter their clinical effectiveness, but there is sparse evidence from clinical studies. Here, we performed Treg flow cytometry analysis on PBMCs from all 31 patients with available samples. Tregs were defined as CD4+CD127lowFoxp3highCD25high cells (Suppl Fig. 1). The patients were divided into a Treghigh or Treglowgroup, using as cutoff the median Treg value (4.91 %) for all 31 patients. There was no significant difference between the Treglow and Treghigh group with regard to the frequency of immune responders.
We addressed whether Tregs may counter the clinical effectiveness of vaccine responses by comparing OS and PFS for the two Treg groups. The statistical analysis was performed on the CTN-2006 population, as expected survival was for other reasons (stage and pre-treatment) substantially different in CTN-2000 patients. Interestingly, immune responders with low Treg levels recorded significantly increased OS (p = 0.037, Fig. 2a) and PFS (p = 0.046). The median PFS for immune responders in CTN-2006 was 61 months in the Treglow group compared to 8.5 months in the Treghigh group.
Fig. 2.

a Treg level versus survival. PBMCs from 31 GV1001 trial patients were analyzed by flow cytometry for T regulatory cells (Tregs, CD4+CD25+CD127low/−FOXP3+), and the percentage of Tregs in CD4+ T cells was calculated. The patients were dichotomized into a Treghigh and a Treglow group, based on the median Treg percentage as cutoff. Overall survival (OS) for CTN-2006 immune responders was compared between the Treghigh and Treglow groups by Kaplan–Meier analysis. The log-rank p value is displayed. b CD33+ MDSC levels in NSCLC patients compared to healthy controls. PBMCs from 24 trial patients plus 10 healthy controls were analyzed by flow cytometry for CD33+ MDSCs (Lin−/loHLA-DR−CD33+CD11b+). The percentage of CD33+ MDSCs in PBMCs was calculated. The CD33+ MDSC levels were not increased in NSCLC patients compared to healthy controls (median 1.0 versus 0.95 %, p = 0.35, Mann–Whitney U test). c CD33+ MDSC levels versus immune response. The figure shows the CD33+ MDSC percentage in PBMCs among immune responders and non-immune responders, indicating median (line), 25/75 % quartiles (boxes), and range. The level of CD33+ MDSCs was significantly lower among immune responders (Mann–Whitney U test, p = 0.001). d CD14+ MDSC levels in NSCLC patients compared to healthy controls. PBMCs from 28 trial patients and 11 healthy controls were analyzed for the presence of CD14+ MDSCs by flow cytometry. The percentage of CD14+ MDSCs in PBMCs was calculated. The CD14+ MDSC levels were increased in NSCLC patients compared to healthy controls (p = 0.012, Mann–Whitney U test). e CD14+ MDSC level versus survival. The patients were divided into a CD14+ MDSChigh and a CD14+ MDSClow level group, based on the median value as cutoff. Overall survival (OS) for CTN-2006 immune responders was compared between the MDSChigh and MDSClow level groups by Kaplan–Meier analysis. Log-rank p value is displayed
Lin−CD33+ MDSCs were associated with immune response rate
To determine the level of Lin−/loHLA-DR−CD33+CD11b+ MDSCs (CD33+ MDSCs), PBMCs from NSCLC patients (n = 24) and healthy controls (n = 10) were stained for expression of CD14, HLA-DR, CD33, CD11b, and lineage markers (CD4/CD8/CD19/CD57). The gating strategy is shown in Suppl. Figure 2. We found no significant difference between NSCLC patients and healthy controls in levels of CD33+ MDSCs (Fig. 2b).
Next, we investigated whether levels of CD33+ MDSCs were associated with PFS or immune response rate. The patients were divided into a high or a low CD33+ MDSC level group using the median value (1.00 %). We observed no association between PFS and CD33+ MDSC level (not shown). However, there was a striking difference in immune response rate between the CD33+ MDSClow and the CD33+ MDSChigh group. Whereas all (12/12) CD33+ MDSClow patients were immune responders, only 5/12 CD33+ MDSChigh patients developed an immune response (p = 0.005, Fisher’s exact test). Figure 2c shows the recorded CD33+ MDSC distribution among immune responders and non-responders.
CD14+ MDSCs were present only in NSCLC patients and associated with poorer PFS
To investigate the levels of HLA-DRlowCD14+ MDSCs (CD14+ MDSCs), we stained PBMCs from 28 NSCLC patients and 11 healthy controls (Suppl. Figure 3). The measured values among patients ranged from 0.01 to 2.49 % (median 0.12), compared to 0.01–0.17 for healthy controls (median 0.03) (Fig. 2d). This difference was statistically significant (Mann–Whitney U test, p = 0.012). Finally, we asked whether the level of CD14+ MDSCs may influence clinical outcome among immunological vaccine responders. To address this question, we used the median CD14+ MDSC level as cutoff and compared OS and PFS for CTN-2006 immune responders with low versus high CD14+ MDSC levels. The median PFS was 60 versus 7.8 months, in favor of the group with low CD14+ MDSC levels (p = 0.08). Similarly, OS was extended in the CD14+ MDSClow group (median 73 versus 21 months, Fig. 2e).
Polyfunctional cytokine profiles with high IFNγ/IL-4 and IFNγ/IL-10 ratios
Samples from 15 NSCLC patients (8 from CTN-2006 and 7 from CTN-2000) were tested in Bioplex cytokine assays. These patients represented all immune responders with available samples for Bioplex assays. We demonstrated a GV1001-specific cytokine response in 15/15 subjects (stimulatory index >2, Suppl. Table 1). Moreover, GV1001-specific T cell proliferation was observed for all samples in parallel 3H-thymidine assays (stimulatory index >2, data not shown).
A polyfunctional GV1001-specific cytokine pattern was detected for most patients (Fig. 3a). Applying an arbitrary cutoff value of 30 pg/ml, a majority of patients showed substantial secretion of at least 7 cytokines. There is an increasing evidence that Th17-cells, secreting IL-17, is a key player in shaping immune responses and may enhance anti-tumor activity [30, 31]. Here, we demonstrated secretion of IL-17 in cultures from 9/15 patients (Fig. 3b).
Fig. 3.

Cytokine profile. The cytokine profile of T cell cultures from 15 NSCLC patients was determined in Bioplex assays. T cells were stimulated with irradiated PBMCs ± peptide GV1001. Supernatants were harvested after 48 h and analyzed as duplicates in Bioplex assays, measuring 17 cytokines. The duplicates were kept separate from start of T cell stimulation and throughout the Bioplex assays. a Polyfunctionality. The sectors represent the fraction of patients producing the given number of cytokines, at concentrations above the chosen cutoff value (>30 pg/ml). b IL-17 secretion was demonstrated in cultures form 9/15 patients, at concentrations above the chosen cutoff value (>30 pg/ml). Columns represent mean concentration (pg/ml) from duplicates. c Mixed Th1/Th2 cytokine profile (patient 307). The diagram depicts a representative cytokine pattern, with high leves of IFNγ (Th1), TNFα (Th1), IL-5 (Th2), and IL-13 (Th2), amid low levels of IL-4 (Th2) and IL-10 (Tr1/Th2). Columns represent mean concentration (pg/ml) from duplicates. d–g Secretion of IFNγ, IL-4, IL-10, IL-5, and IL-13. T cells from all patients tested secreted IL-5 and IL-13, while IFNγ was detected for all except patient 205. Most patients showed high IFNγ/IL-4 and IFNγ/IL-10 ratios (d, e). The levels of Th2 cytokines IL-5 (f) and IL-13 (g) were still generally high, suggesting that the cytokine patterns did not follow a Th1/Th2 delineation. Patient 205 was the only subject not secreting IFNγ, IL-4, or IL-10
The Th1/Th2 cytokine pattern for a representative patient is shown in Fig. 3c. In general, the levels of Th1 cytokines IFNγ and TNFα were high, while levels of IL-4 and IL-10 were low. As shown in Fig. 3d, e, the IFNγ/IL-4 and INFγ/IL-10 ratios were high across both protocols. This pattern may suggest a favorable balance between immunity and tolerance. The high IFNγ/IL-4 ratios, however, did not reflect a classic Th1 profile as the key Th2 cytokines IL-5 and IL-13 were both high (Fig. 3f, g). We have previously observed a similar breach of Th1/Th2 delineation at a clonal level [18, 32].
Patients with low Treg levels recorded higher IFNγ/IL-10-ratios
IL-10 is known to be secreted by tolerogenic cells including type 2 macrophages and T regulatory type 1 (Tr1) cells and is considered to promote tumor tolerance [33]. We hypothesized that high levels of Foxp3+ Tregs, presumably reflecting a tolerogenic environment, may correlate with elevated IL-10 levels as compared to IFNγ. This hypothesis was assessed by comparing the Treghigh and Treglow groups (defined above) with regard to IFNγ/IL-10 ratio (n = 13). Interestingly, we found that the Treghigh group had markedly decreased IFNγ/IL-10 ratios (Fig. 4a). We next asked whether the Treglow group had a more Th1-driven response. Indeed, the IFNγ/IL-5 and IFNγ/IL-13 ratios were substantially higher in the Treglow group, suggesting a more Th1-weighted pattern (Fig. 4b). The mean IFNγ/IL-5 ratio was increased 16-fold (22 vs. 1.33).
Fig. 4.
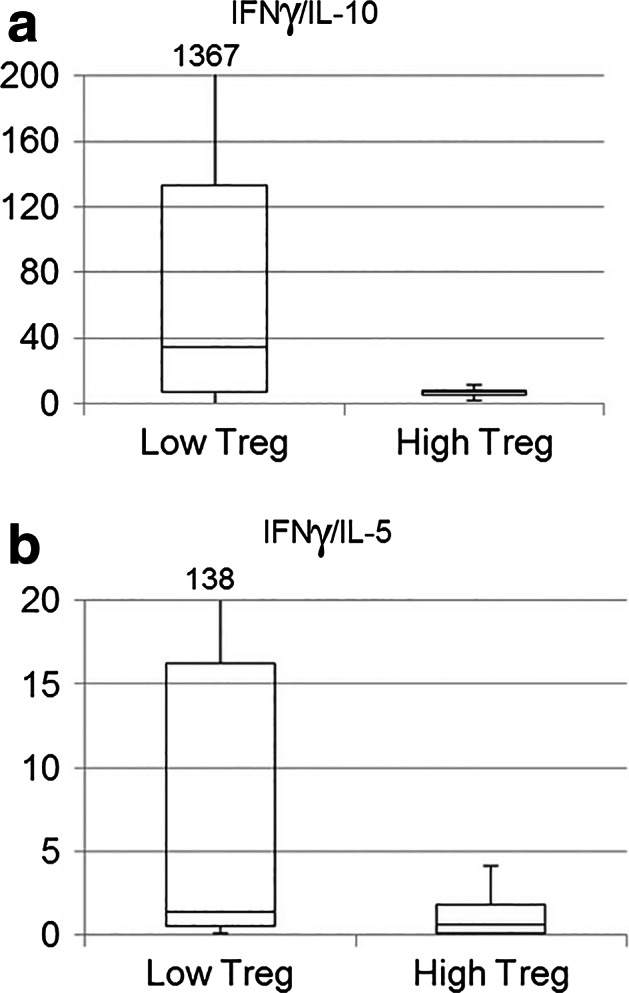
Treg level versus cytokine profile. The cytokine profile of samples from 15 NSCLC patients was determined in Bioplex assays. Their Treg levels were determined by flow cytometry analysis (Tregs: CD4+CD25+CD127low/−FOXP3+). The patients were dichotomized into a Treghigh and a Treglow group, based on the median Treg value as cutoff. The box plots in a and b indicate median (line), 25/75 % quartiles (boxes), and range. a Lower Treg levels in patients with high IFNγ/IL-10 ratios. The Treghigh group had decreased IFNγ/IL-10 ratios compared to the Treglow group (median 7.0 vs 34.4). b) Th1-like response in Treglow group. The IFNγ/IL-5 (b) and IFNγ/IL-13 (not shown) ratios were higher in the Treglow group, suggesting a more Th1-weighted pattern
CTN-2000 patients had a more Th1 - weighted cytokine profile compared to CTN-2006
The patients in protocol CTN-2006 differed considerably from protocol CTN-2000 in terms of disease stage and prior treatment. Further, the CTN-2006 group received vaccination as adjuvant therapy, at a point where the immune system would be strongly influenced after 6 weeks of chemoradiation. We investigated whether these differences resulted in different cytokine profiles between patients in the CTN-2000 versus the CTN-2006 protocol. The results showed that the levels of IFNγ as well as the IFNγ/IL-13 and IFNγ/IL-5 ratios were considerably higher in CTN-2000, suggesting a more Th1-like profile (Suppl. Figure 4). This observation may reflect how chemoradiation influences the cytokine profile, but needs to be confirmed in a new study.
Discussion
We observed a statistically significant association between survival and immune response in the GV1001 trials. We also noted a considerable diversity in survival within the group of immune responders and investigated what separates the clinically useful from the pointless immune response. This question is as challenging as it is important. Cytokine profiles, Tregs, and MDSCs may be decisive factors. However, it has proven difficult to establish whether the clear effects of these factors reported in animal models apply in a complex human setting. Further, we performed in-depth cytokine profiling of the GV1001 response. The results support the notion that high levels of Tregs and MDSCs impair cancer vaccine responses and suggest that these cell types are associated with lower IFNg/IL-10 ratios and reduced progression-free survival among immune responders.
A randomized trial would be needed to determine whether the superior survival for immune responders is caused by the vaccine, but the observed strong association is of interest. The clinical follow-up data reported herein suggest that the OS and PFS advantages for immune responders are maintained long term, without development of side effects. One may argue that patients with less advanced disease may be more likely to respond immunologically to vaccination. However, the survival advantage appeared not to be explained by disease stage and held firm in separate analyses on stage III patients. These observations support the rationale for further development of the GV1001 vaccine.
Increased levels of Tregs are found in many cancer types [20, 34], including NSCLC [35, 36]. The detrimental effect of increased numbers of Tregs is supported in this study. We find that immune responders with low levels of Foxp3+ Tregs have increased PFS and a more favorable cytokine profile, including higher IFNγ/IL-10 ratios, compared to those with high Treg levels. These findings are in line with compelling evidence from animal studies indicating that Tregs counter anti-tumor immune responses, as well as some clinical studies suggesting that increased Treg levels are negative prognostic factors [20, 37, 38].
In the CTN-2006 trial, we combined vaccination with chemoradiation. The observations in our study may reflect that a Treg-/IL-10-dominated tumor milieu counters the efficacy of T cell responses induced by chemoradiation. This hypothesis is in line with current understanding of immunogenic cell death [33] and of particular relevance for regimes combining vaccination with chemotherapy or radiation. Our data further suggest that Treg levels influence the quality of the immune response, but not the frequency of immune responders. Possibly, only patients with low Treg levels are able to benefit from a GV1001 vaccine response. This represents a possible explanation as to why some immune responders experience long-term survival, while others do not. The mechanism may be related to epitope spreading. We have previously observed that long-term survivors after GV1001 vaccination exhibit increased epitope spreading compared to immune responders without evidence of clinical benefit [39].
We observed differences in cytokine profiles both between patients with different Treg levels and between the CTN-2000 and CTN-2006 protocols. The limited number of patients did not allow for statistical analysis of whether these factors were independent or influenced each other. This point illustrates the exploratory nature of our study and the need to confirm the findings in new studies.
There is increasing evidence that MDSCs are potent suppressors of anti-tumor T cell responses. The phenotypes of these cells are heterogenous in humans. We evaluated two categories of MDSCs considered to be highly suppressive of human anti-tumor responses [22, 24, 26–28]. The levels of Lin−/loHLA-DR−CD33+CD11b+ MDSCs have been reported to be increased in many forms of cancer [25, 40], including one study of NSCLC [41]. However, we found no significant increase in NSCLC patients, compared to healthy controls. Moreover, the levels did not correlate with progression-free survival. We did not have sufficient material to perform functional studies with sorted MDSCs, and circulating levels of MDSCs may not reflect the MDSCs activity in lymph nodes and tumor. The results should therefore be interpreted with caution. However, biomarker analyses in clinical practice can hardly rely on functional assays or tumor-derived MDSCs. Hence, our data suggest that Lin−/loHLA-DR−CD33+CD11b+ MDSCs may not be a suitable biomarker in NSCLC.
We identify substantial levels of HLA-DRlowCD14+ MDSCs only in NSCLC patients. Cells with this phenotype have also been detected in cancer patients in other studies [26–28, 38]. In our study, the minimal levels of such cells (0.01–0.17 %) in healthy controls suggest that the HLA-DRlowCD14+ cells are associated with NSCLC development and may be a candidate biomarker. Our observation that OS and PFS were inferior in subjects with HLA-DRlowCD14+ MDSCs is in line with other studies, including data from NSCLC patients who had received chemotherapy [42] and the IMA901 peptide vaccine trial in renal carcinoma patients [38]. Interestingly, an experimental agent for countering MDSC has been reported to enhance the immune response after vaccination in NSCLC patients [43].
Cancer vaccines do not enter a virgin environment but need to overcome established tumor tolerance. Compelling evidence from in vitro studies and animal models suggests that Foxp3+ Tregs interact with other regulatory cell types, including tolerogenic DCs, type 2 macrophages, and Tr1 cells [20]. This network theory is supported by our observation that patients with low levels of Foxp3+ Tregs develop vaccine responses with decreased IFNγ/IL-10 ratios. Moreover, the association with lower IFNγ/IL-5 and INFγ/IL-13 ratios supports the notion that a Treg-/Tr1-driven environment counters Th1 differentiation, yielding a muted and more Th2-like response (Fig. 4). Our data support the notion that GV1001 and other cancer vaccines may need to be combined with drugs countering established tolerance. We have previously observed that GV1001 can safely be combined with temozolomide [7], which has been shown to reduce Treg levels in several studies [44]. Regarding the clinical importance of Tregs, it is of interest that the melanoma breakthrough drug ipilimumab, which was until recently considered mainly to work on effector T cells, has been shown to efficiently deplete Tregs from tumor [45, 46].
The Th1/Th2 paradigm was established based on elegant studies in mouse models [47] and has led to widespread use of INFγ/IL-4 assays to determine a Th1 or Th2 profile. Our data from the present and previous studies suggest that this practice may be misleading, as an IFNγ/IL-4 response may include high levels of other Th2 cytokines (IL-5, IL-13) as well as IL-10. Here, we have demonstrated this in T cell bulk cultures. Previously, we have found this in T cell clones [18, 32]. In our experience, the breach of Th1/Th2 dichotomy is observed across different cancer forms, disease stages, and vaccine strategies [7, 18, 48]. These observations suggest that it may be wise to avoid Th1/Th2-lables and rather focus on polyfunctionality or the ratio between key cytokines, like IFNγ/IL-10.
The GV1001 cancer vaccine has been approved in Korea for treatment of pancreatic cancer (personal communication, Dr. M Kim, CEO, Kael-Gemvax). A randomized GV1001 trial in NSCLC patients has been prepared (NCT01579188). Such a trial may present an opportunity to follow up the leads from the present study, in particular by comparing cytokine patterns and levels of Tregs and MDSCs with clinical outcome. The extensive cytokine panel used in the present study was useful for our purpose, but in a large-scale trial, it is important to limit the panel. Based on the close association between IL-5 and IL-13, one may omit one of these cytokines. It is probably more important to retain IL-17 and chemokines like MIP-1a and MIP-1b to assess polyfunctionality. The polyfunctional profile observed in most patient samples in the present study may be beneficial, as suggested from animal studies and data from humans with HIV and other chronic infections [49, 50]. We have not studied the cytokine pattern at single-cell level, but have previously observed that the polyfunctionality is retained in GV1001-specific T cell clones confirmed to be monoclonal by TCR mapping [18].
We conclude that a randomized GV1001 trial in NSCLC patients is warranted, based on a high immune response rate, favorable cytokine profiles, low toxicity after long-term observation, and superior survival for immune responders. The development of effective cancer vaccines depends on identifying factors influencing whether an immune response is clinically effective. Putative biomarkers like cytokine profiles, Tregs, or MDSCs can be addressed one by one in elegant animal studies, but in patients, the role of each factor is difficult to clarify. Because large patient cohorts are rarely available, we consider it important to perform in-depth studies of material from small-scale trials and accumulate data from different studies. The present investigation contributes to this collective bank of knowledge and provides leads as to which factors may be important. Our findings need to be confirmed in new patient cohorts, but suggest that Tregs, CD33+ MDSCs, and CD14+ MDSCs may represent biomarkers for prognosis after cancer vaccination and serve to identify patients not benefitting from vaccine therapy. The results also support the notion that vaccine responses with an IFNγhigh/IL-10low/IL-4low and polyfunctional cytokine profile may counter cancer. Finally, the data indicate that HLA-DRlowCD14+ MDSCs are associated with development of NSCLC.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
The work was supported by the Norwegian Health Region South-East and the Faculty of Medicine, University of Oslo. Pharmexa (Denmark) supplied GV1001 free of charge. The authors thank Sissel Trachsel and Mira Roness for valuable technical assistance. We also thank doctors and nurses at the study hospitals for excellent clinical follow-up and patient care. Special thanks to Steinar Aamdal, Christian Kersten, Stein Sundstrøm, Else Marit Suso, Svein Dueland, Odd Terje Brustugun, Kristin Øwre, and Ingun Sve.
Abbreviations
- MCP
Monocyte chemoattractant protein
- MDSC
Myeloid-derived suppressor cell
- MIP
Macrophage inflammatory protein
- NSCLC
Non-small cell lung cancer
- OS
Overall survival
- PFS
Progression-free survival
- Th
T helper
- Treg
T regulatory cell
Compliance with ethical standards
Conflict of interest
J.A. Kyte has received research support (≈10,000 Euro) from Kael-Gemvax. G. Gaudernack is a member of Kael-Gemvax advisory board. The other authors declare that they have no conflict of interest.
References
- 1.Brambilla E, Travis WD. World cancer report. World Health Organization, Lyon: Lung cancer; 2014. [Google Scholar]
- 2.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 3.Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, Thatcher N, Wagstaff J, Zielinski C, Faulkner I, et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol. 2014;11(9):509–524. doi: 10.1038/nrclinonc.2014.111. [DOI] [PubMed] [Google Scholar]
- 4.Kyte JA. Cancer vaccination with telomerase peptide GV1001. Expert Opin Investig Drugs. 2009;18(5):687–694. doi: 10.1517/13543780902897631. [DOI] [PubMed] [Google Scholar]
- 5.Bernhardt SL, Gjertsen MK, Trachsel S, Moller M, Eriksen JA, Meo M, Buanes T, Gaudernack G. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: a dose escalating phase I/II study. Br J Cancer. 2006;95(11):1474–1482. doi: 10.1038/sj.bjc.6603437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brunsvig PF, Kyte JA, Kersten C, Sundstrom S, Moller M, Nyakas M, Hansen GL, Gaudernack G, Aamdal S. Telomerase peptide vaccination in NSCLC: a phase II trial in stage III patients vaccinated after chemoradiotherapy and an 8-year update on a phase I/II trial. Clin Cancer Res. 2011;17(21):6847–6857. doi: 10.1158/1078-0432.CCR-11-1385. [DOI] [PubMed] [Google Scholar]
- 7.Kyte JA, Gaudernack G, Dueland S, Trachsel S, Julsrud L, Aamdal S. Telomerase peptide vaccination combined with temozolomide: a clinical trial in stage IV melanoma patients. Clin Cancer Res. 2011;17(13):4568–4580. doi: 10.1158/1078-0432.CCR-11-0184. [DOI] [PubMed] [Google Scholar]
- 8.Greten TF, Forner A, Korangy F, N’Kontchou G, Barget N, Ayuso C, Ormandy LA, Manns MP, Beaugrand M, Bruix J. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer. 2010;10:209. doi: 10.1186/1471-2407-10-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Middleton G, Silcocks P, Cox T, Valle J, Wadsley J, Propper D, Coxon F, Ross P, Madhusudan S, Roques T, et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): an open-label, randomised, phase 3 trial. Lancet Oncol. 2014;15(8):829–840. doi: 10.1016/S1470-2045(14)70236-0. [DOI] [PubMed] [Google Scholar]
- 10.Szostak JW, Blackburn EH. Cloning yeast telomeres on linear plasmid vectors. Cell. 1982;29(1):245–255. doi: 10.1016/0092-8674(82)90109-X. [DOI] [PubMed] [Google Scholar]
- 11.Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43(2 Pt 1):405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- 12.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266(5193):2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 13.Fernandez-Garcia I, Ortiz-de-Solorzano C, Montuenga LM. Telomeres and telomerase in lung cancer. J Thorac Oncol. 2008;3(10):1085–1088. doi: 10.1097/JTO.0b013e3181886713. [DOI] [PubMed] [Google Scholar]
- 14.Parkhurst MR, Riley JP, Igarashi T, Li Y, Robbins PF, Rosenberg SA. Immunization of patients with the hTERT: 540–548 peptide induces peptide-reactive T lymphocytes that do not recognize tumors endogenously expressing telomerase. Clin Cancer Res. 2004;10(14):4688–4698. doi: 10.1158/1078-0432.CCR-04-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolonaki I, Kotsakis A, Papadimitraki E, Aggouraki D, Konsolakis G, Vagia A, Christophylakis C, Nikoloudi I, Magganas E, Galanis A, et al. Vaccination of patients with advanced non-small-cell lung cancer with an optimized cryptic human telomerase reverse transcriptase peptide. J Clin Oncol. 2007;25(19):2727–2734. doi: 10.1200/JCO.2006.10.3465. [DOI] [PubMed] [Google Scholar]
- 16.Vonderheide RH, Hahn WC, Schultze JL, Nadler LM. The telomerase catalytic subunit is a widely expressed tumor-associated antigen recognized by cytotoxic T lymphocytes. Immunity. 1999;10(6):673–679. doi: 10.1016/S1074-7613(00)80066-7. [DOI] [PubMed] [Google Scholar]
- 17.Brunsvig P, Aamdal S, Gjertsen MK, Kvalheim G, Markowski-Grimsrud CJ, Sve I, Dyrhaug M, Trachsel S, Moeller M, Eriksen JA, et al. Telomerase peptide vaccination: a phase I/II study in patients with non-small cell lung cancer. Cancer Immunol Immunother. 2006;55(12):1553–1564. doi: 10.1007/s00262-006-0145-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kyte JA, Trachsel S, Risberg B, thor Straten P, Lislerud K, Gaudernack G. Unconventional cytokine profiles and development of T cell memory in long-term survivors after cancer vaccination. Cancer Immunol Immunother. 2009;58(10):1609–1626. doi: 10.1007/s00262-009-0670-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54(8):721–728. doi: 10.1007/s00262-004-0653-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mougiakakos D, Choudhury A, Lladser A, Kiessling R, Johansson CC. Regulatory T cells in cancer. Adv Cancer Res. 2010;107:57–117. doi: 10.1016/S0065-230X(10)07003-X. [DOI] [PubMed] [Google Scholar]
- 21.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182(8):4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166(1):678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 24.Lechner MG, Megiel C, Russell SM, Bingham B, Arger N, Woo T, Epstein AL. Functional characterization of human Cd33+ and Cd11b+ myeloid-derived suppressor cell subsets induced from peripheral blood mononuclear cells co-cultured with a diverse set of human tumor cell lines. J Transl Med. 2011;9:90. doi: 10.1186/1479-5876-9-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. 2011;60(10):1419–1430. doi: 10.1007/s00262-011-1028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Filipazzi P, Valenti R, Huber V, Pilla L, Canese P, Iero M, Castelli C, Mariani L, Parmiani G, Rivoltini L. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol. 2007;25(18):2546–2553. doi: 10.1200/JCO.2006.08.5829. [DOI] [PubMed] [Google Scholar]
- 27.Brimnes MK, Vangsted AJ, Knudsen LM, Gimsing P, Gang AO, Johnsen HE, Svane IM. Increased level of both CD4+FOXP3+ regulatory T cells and CD14+HLA-DR(−)/low myeloid-derived suppressor cells and decreased level of dendritic cells in patients with multiple myeloma. Scand J Immunol. 2010;72(6):540–547. doi: 10.1111/j.1365-3083.2010.02463.x. [DOI] [PubMed] [Google Scholar]
- 28.Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, Manns MP, Greten TF, Korangy F. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. 2008;135(1):234–243. doi: 10.1053/j.gastro.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 29.Kyte JA, Kvalheim G, Aamdal S, Saebøe-Larssen S, Gaudernack G. Preclinical full-scale evaluation of dendritic cells transfected with autologous tumor-mRNA for melanoma vaccination. Cancer Gene Ther. 2005;12(6):579–591. doi: 10.1038/sj.cgt.7700837. [DOI] [PubMed] [Google Scholar]
- 30.Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Fili L, Ferri S, Frosali F, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204(8):1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, Hwu P, Restifo NP, Overwijk WW, Dong C. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity. 2009;31(5):787–798. doi: 10.1016/j.immuni.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kyte JA, Kvalheim G, Lislerud K, Thor Straten P, Dueland S, Aamdal S, Gaudernack G. T cell responses in melanoma patients after vaccination with tumor-mRNA transfected dendritic cells. Cancer Immunol Immunother. 2007;56(5):659–675. doi: 10.1007/s00262-006-0222-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, Daniel D, Hwang ES, Rugo HS, Coussens LM. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26(5):623–637. doi: 10.1016/j.ccell.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6(4):295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 35.Woo EY, Yeh H, Chu CS, Schlienger K, Carroll RG, Riley JL, Kaiser LR, June CH. Cutting edge: regulatory T cells from lung cancer patients directly inhibit autologous T cell proliferation. J Immunol. 2002;168(9):4272–4276. doi: 10.4049/jimmunol.168.9.4272. [DOI] [PubMed] [Google Scholar]
- 36.Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G, Rubin SC, Kaiser LR, June CH. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001;61(12):4766–4772. [PubMed] [Google Scholar]
- 37.Elkord E, Alcantar-Orozco EM, Dovedi SJ, Tran DQ, Hawkins RE, Gilham DE. T regulatory cells in cancer: recent advances and therapeutic potential. Expert Opin Biol Ther. 2010;10(11):1573–1586. doi: 10.1517/14712598.2010.529126. [DOI] [PubMed] [Google Scholar]
- 38.Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, Staehler M, Brugger W, Dietrich PY, Mendrzyk R, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med. 2012;18(8):1254–1261. doi: 10.1038/nm.2883. [DOI] [PubMed] [Google Scholar]
- 39.Inderberg-Suso EM, Trachsel S, Lislerud K, Rasmussen AM, Gaudernack G. Widespread CD4+ T-cell reactivity to novel hTERT epitopes following vaccination of cancer patients with a single hTERT peptide GV1001. Oncoimmunology. 2012;1(5):670–686. doi: 10.4161/onci.20426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin–cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58(1):49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu CY, Wang YM, Wang CL, Feng PH, Ko HW, Liu YH, Wu YC, Chu Y, Chung FT, Kuo CH, et al. Population alterations of L-arginase- and inducible nitric oxide synthase-expressed CD11b+/CD14(−)/CD15+/CD33+ myeloid-derived suppressor cells and CD8+ T lymphocytes in patients with advanced-stage non-small cell lung cancer. J Cancer Res Clin Oncol. 2010;136(1):35–45. doi: 10.1007/s00432-009-0634-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang A, Zhang B, Wang B, Zhang F, Fan KX, Guo YJ. Increased CD14(+)HLA-DR (-/low) myeloid-derived suppressor cells correlate with extrathoracic metastasis and poor response to chemotherapy in non-small cell lung cancer patients. Cancer Immunol Immunother. 2013;62(9):1439–1451. doi: 10.1007/s00262-013-1450-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iclozan C, Antonia S, Chiappori A, Chen DT, Gabrilovich D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol Immunother. 2013;62(5):909–918. doi: 10.1007/s00262-013-1396-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ridolfi L, Petrini M, Granato AM, Gentilcore G, Simeone E, Ascierto PA, Pancisi E, Ancarani V, Fiammenghi L, Guidoboni M, et al. Low-dose temozolomide before dendritic-cell vaccination reduces (specifically) CD4+ CD25++ Foxp3+ regulatory T-cells in advanced melanoma patients. J Transl Med. 2013;11:135. doi: 10.1186/1479-5876-11-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210(9):1695–1710. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, Rajapaksa R, Green MR, Torchia J, Brody J, et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest. 2013;123(6):2447–2463. doi: 10.1172/JCI64859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136(7):2348–2357. [PubMed] [Google Scholar]
- 48.Kyte JA, Mu L, Aamdal S, Kvalheim G, Dueland S, Hauser M, Gullestad HP, Ryder T, Lislerud K, Hammerstad H, et al. Phase I/II trial of melanoma therapy with dendritic cells transfected with autologous tumor-mRNA. Cancer Gene Ther. 2006;13(10):905–918. doi: 10.1038/sj.cgt.7700961. [DOI] [PubMed] [Google Scholar]
- 49.Duvall MG, Precopio ML, Ambrozak DA, Jaye A, McMichael AJ, Whittle HC, Roederer M, Rowland-Jones SL, Koup RA. Polyfunctional T cell responses are a hallmark of HIV-2 infection. Eur J Immunol. 2008;38(2):350–363. doi: 10.1002/eji.200737768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Darrah PA, Patel DT, De Luca PM, Lindsay RW, Davey DF, Flynn BJ, Hoff ST, Andersen P, Reed SG, Morris SL, et al. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med. 2007;13(7):843–850. doi: 10.1038/nm1592. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.