

Abstract
CD4+ T cells contribute importantly to the antitumor T cell response, and thus, long peptides comprising CD4 and CD8 epitopes may be efficient cancer vaccines. We have previously identified an overexpressed antigen in melanoma, MELOE-1, presenting a CD8+ T cell epitope, MELOE-136–44, in the HLA-A*0201 context. A T cell repertoire against this epitope is present in HLA-A*0201+ healthy subjects and melanoma patients and the adjuvant injection of TIL containing MELOE-1 specific CD8+ T cells to melanoma patients was shown to be beneficial. In this study, we looked for CD4+ T cell epitopes in the vicinity of the HLA-A*0201 epitope. Stimulation of PBMC from healthy subjects with MELOE-126–46 revealed CD4 responses in multiple HLA contexts and by cloning responsive CD4+ T cells, we identified one HLA-DRβ1*1101-restricted and one HLA-DQβ1*0603-restricted epitope. We showed that the two epitopes could be efficiently presented to CD4+ T cells by MELOE-1-loaded dendritic cells but not by MELOE-1+ melanoma cell-lines. Finally, we showed that the long peptide MELOE-122–46, containing the two optimal class II epitopes and the HLA-A*0201 epitope, was efficiently processed by DC to stimulate CD4+ and CD8+ T cell responses in vitro, making it a potential candidate for melanoma vaccination.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-010-0938-6) contains supplementary material, which is available to authorized users.
Keywords: T cell epitope, CD4+ T cells, Melanoma, Vaccination
Introduction
Since the identification of multiple melanoma antigens and T cell epitopes, many attempts at therapeutic vaccination against melanoma have been made in recent years using short peptides corresponding to HLA class I-restricted epitopes, delivered either alone, with immunostimulants or in viruses. However, as reviewed by Rosenberg et al. [1], the results of these clinical trials have been disappointing so far with an objective response rate around 3–4%. A combination of factors was proposed by Melief and van der Burg to account for the poor efficacy of vaccinations with short peptides. According to the authors, the most important drawbacks of short peptide vaccination would be the inability of a short peptide to evoke a CD4+ T cell response and the possibility for the peptide to load onto non professional APC and thus induce tolerance of specific CD8+ T cells [2]. In support of this hypothesis, Melief’s group and others have shown in mice models that, unlike short peptide vaccination, vaccination with long peptides comprising class II epitopes in addition to class I epitopes could efficiently induce CD4+ T cell responses that are necessary to support strong CD8+ T cell responses [3, 4]. Vaccination-induced CD4+ T cells may even be able to counteract the action of CD4+ Tregs present in the tumor environment [5]. Recently, Melief’s team demonstrated the clinical efficacy of vaccination with long peptides from HPV-16 in vulvar intraepithelial neoplasia [6].
Considering the above, we are eager to design and test similar long peptides for therapeutic vaccination of metastatic melanoma. We have recently characterized two new antigens, MELOE-1 [7] and MELOE-2 [8], encoded by distinct ORFs from the same unspliced mRNA, coined meloe for “melanoma overexpressed”. We have shown that the presence of CD8+ T cells specific for a HLA-A*0201-restricted epitope derived from MELOE-1 among TIL re-injected to melanoma patients in an adjuvant setting was strongly correlated with prolonged relapse-free survival [7] and we have documented the existence of a frequent CD8+ T cell repertoire against MELOE-1 in HLA-A*0201 healthy individuals and melanoma patients strongly suggesting the immunogenicity of this antigen [9]. In the present work, we aimed at defining a long peptide comprising the previously identified HLA-A*0201-restricted MELOE-136–44 epitope that would elicit significant CD4+ T cell responses in healthy HLA-A*0201 individuals in whom a CD8+ T cell response against MELOE-1 was present. In addition, we characterized some of the HLA class II-restricted epitopes to design a peptide of optimal length that could be processed by dendritic cells and thus used for vaccination.
Materials and methods
Cells
Blood samples from healthy subjects were obtained from Etablissement Français du Sang, Nantes, France. All cell-lines were maintained in RPMI 1640 containing 10% fetal calf serum (FCS) or 8% pooled human serum (HS). Lymphocytes were grown in RPMI 1640 8% HS with 50 or 150 IU/ml of interleukin-2 (IL-2, Chiron, France). For some experiments including dendritic cells, serum-free AIM-V medium (Gibco) was used to avoid peptide degradation by serum proteases. Melanoma cell-lines were established in our laboratory from fragments of metastatic tumors. Mouse fibrosarcoma WEHI 164 clone 13 was obtained from Pr Boon (Ludwig Institute for Cancer Research, Brussels, Belgium). The different EBV-B cell-lines were obtained in our laboratory.
Reagents
All antibodies were purchased from BD Biosciences-France. Purified cytokines were purchased from Abcys, France. The different peptides (Eurogentec, Belgium, purity >80%) used in this study were: MELOE-122–46 (AACPPWHPSERISSTLNDECWPASL), MELOE-126-46 (PWHPSERISSTLNDECWPASL), MELOE-126–40 (PWHPSERISSTLNDE), MELOE-132–46 (RISSTLNDECWPASL), MELOE-136–44 (TLNDECWPA) and the modified Melan-A16–40 A27L (GHGHSYTTAEELAGIGILTVILGVL).The MELOE-1 whole protein (purity >90%) was produced by Millegen (Labege, France). HLA-A*0201/MELOE-136-44 monomers were generated by the local recombinant protein facility. Chemicals were purchased from Sigma–Aldrich Co.
Dendritic cells generation and loading
Monocytes were purified from PBMC of HLA-A*0201, HLA-DRβ1*1101 and/or HLA-DQβ1*0603 healthy donors by counter-flow centrifugal elutriation (Clinical Transfer Facility, Dr Gregoire, Nantes). Immature dendritic cells were generated by culturing monocytes in AIM-V supplemented with 1000 IU/mL of GM-CSF and 200 IU/mL of IL-4 for 5 days. Then, DC were pulsed with the whole MELOE-1 protein or the modified Melan-A16–40 A27L as negative control at 10 μM and matured with 20 ng/mL of TNF-α and 50 μg/mL of PolyI:C for 4 h at 37°C. Finally they were fixed for 1 min with PBS/0.02% glutaraldehyde. Alternatively, DC were first matured, fixed and then pulsed with antigen. For PBL priming against MELOE-122–46, DC were loaded for 4 h with 10 μΜ MELOE-122–46, washed and matured with TNF-α and PolyI:C for 12 h. DC loading with lysate from the meloe-1 positive M117 melanoma cell-line or with the meloe-1 negative colon carcinoma cell line SW480 was performed as follows: M117 or SW480 cells were lysed by five cycles of rapid freeze/thawing, cell debris were spun down at 13,000 rpm for 10 min and supernatants were mixed with immature DC at a ratio of two tumor cell equivalents to 1 DC for 8 h, before adding maturation agents for 36 h.
Stimulation of MELOE-1 specific T cells
PBMC from healthy donors (2 × 105 cells/well) were cultured for 14 days with 10 μM of peptide alone (MELOE-126–46 or MELOE-132–46) in RPMI/8% HS/50 IU/ml of IL-2. Cultures were then restimulated with the specific peptide in the presence of 10 μg/mL brefeldin A for 5 h and the percentage of CD4+ specific T cells was assessed by IFN-γ intracellular staining. Alternatively, PBL were stimulated by autologous MELOE-122–46-loaded DC at a 10:1 ratio. Two culture conditions were evaluated: a 14-day culture with peptide-loaded DC in RPMI 8% HS and 50 IU/mL of IL-2 or a 7-day culture with peptide-loaded DC in RPMI 8% HS with 5 ng/mL of IL-6 and 5 ng/mL of IL-12, followed by a second stimulation with loaded DC for another 7 day culture with 10 IU/mL of IL-2. The presence of CD8+ and CD4+ specific T cells was assessed by restimulating cultures with, respectively, 10 μM of MELOE-136–44 or MELOE-124–37 for 5 h before IFN-γ intracellular staining. A negative control without peptide was included in all experiments.
T cell cloning and TCR characterization
Polyclonal cultures containing specific CD4+ T cells were cloned by limiting dilution as previously described [10]. After 2 weeks, each clone was checked for peptide specificity by IFN-γ intracellular staining. For TCR sequencing, RNA from 5 × 106 T cell clones was extracted with RNable reagent (Eurobio, France) according to the supplier’s instructions. Reverse transcriptions, PCR amplifications and sequencing were performed as described [11]. We used the TCR nomenclature established by Arden et al. [12].
TNF production assay
CD4+ T cell clones were cultured for 6 h at 37°C with EBV-B cells loaded or not for 1 h with antigenic peptide. Culture supernatants were harvested and TNF was measured in a biological assay using cytotoxicity against WEHI 164 clone 13 [13].
Cytokine intracellular staining
Lymphocytes were stimulated for 5 h in the presence of brefeldin A (10 μg/mL) either with peptide alone (10 μM) in an autopresentation assay or with peptide-loaded melanoma cells pre-treated or not with IFN-γ (500 IU/mL for 48 h), or with peptide-loaded EBV-B cell-lines or with peptide- or lysate-loaded DC. In some experiments, blocking mAb against HLA-DP (clone B7.21 from Dr Charron, UMR940, Paris), HLA-DQ (clone SK10, BD Biosciences) or HLA-DR (clone L243, BD Biosciences) were added. Cells were then stained with APC-conjugated anti-CD8 or anti-CD4 mAb, fixed with 4% paraformaldehyde, labelled with PE-conjugated anti-IFNγ or anti-TNF-α mAb and analyzed by flow cytometry. To determine their cytokine profile, clones were stimulated for 5 h with either OKT-3 coated at 1 μg/mL or with 0.1 μg/mL phorbol-myristate-acetate (PMA) and 1 μg/mL calcium ionophore (CaI).
Cytotoxicity assay
Cytotoxicity was assessed in a standard chromium release assay. Target cells (melanoma or EBV-B cell-lines) were pulsed for 1 h with Na512CrO4 (NEN Life Science, France) and then washed. EBV-B cell-lines were incubated or not with 10 μM MELOE-1 peptides for 1 h and 103 target cells/well were mixed with effector cells at different effector/target ratios for 4 h at 37°C. The percentage of lysis was calculated as follows: (sample release−spontaneous release)/(maximum release−spontaneous release) × 100.
Results
Peptide stimulation, expansion and cloning of MELOE-1 specific CD4+ T cells
Our purpose was to look for the existence of a CD4+ T cell repertoire against a MELOE-1 long peptide that would include the previously identified HLA-A*0201-restricted MELOE-136–44 epitope. We arbitrarily chose a 21 aa long peptide, MELOE-126–46 to test for CD4 responses in seven healthy HLA-A*0201 donors in whom a CD8 response against the nonapeptide MELOE-136–44 had been detected after in vitro peptide stimulation by specific tetramer labelling (an example is shown in Online Resource 1a). We stimulated their PBMC (2 × 105cells/well) with the peptide MELOE-126–46 at 10 μM for 14 days. Wells were screened for CD4 reactivity by restimulating cells with MELOE-126–46 in an autopresentation assay and by analyzing the percentage of IFN-γ-secreting CD4+ T cells (Online Resource 1b). As shown in Table 1, all seven donors presented CD4 responses against the 21 mer peptide despite marked differences in the frequencies of positive responses among donors. Considering that no HLA class II isotype was shared by all donors, we reasoned that either the 21 mer peptide contained a single epitope that could be presented in multiple HLA contexts or that distinct epitopes were present in this long peptide.
Table 1.
Assessment of MELOE-126–46 CD4+ T cell responses in PBMC from healthy donors
| Donor | DPβ1 | DRβ1 | DQβ1 | Wells containing MELOE-126–46 specific CD4+ T cells (% positive wells) |
|---|---|---|---|---|
| DS1 | 0201/0402 | 0401/1201 | 0301/0302 | 1/96 (1) |
| DS2 | 0401/0401 | 0102/0801 | 04/05 | 2/60 (3.3) |
| DS6 | 0401/1101 | 0301/1601 | 0201/0502 | 5/96 (5.2) |
| DC1 | 0201/0401 | 0404/1101 | 0301/0302 | 8/96 (8.3) |
| DA3 | 0301/0401 | 1101/1301 | 0301/0603 | 9/60 (15) |
| DS8 | 0401/1101 | 0701/1401 | 0202/0503 | 29/48 (60) |
| DS7 | 0201/0401 | 0401/1101 | 0301/0301 | 43/48 (89.6) |
PBMC from healthy donors were stimulated with MELOE-126–46 at 10 μM. After 14 days, presence of MELOE-26–46 specific CD4+ T cells was assessed by restimulating cells with MELOE-126–46, followed by CD4/TNF-α double staining and flow cytometry analysis
To explore these hypotheses, we reproduced the same stimulation experiments with a shorter peptide MELOE-132–46 on donor DC1 and DA3. Donor DA3 (Fig. 1a) displayed CD4 responses against this shorter peptide but not donor DC1 (not shown). This suggested that at least two distinct epitopes were present in MELOE-126–46 and we decided to identify them to confirm this hypothesis. We started with the characterization of the response toward the short peptide MELOE-132–46 in donor DA3. Out of the 96 wells initially seeded, 6 showed a percentage of IFN-γ-secreting CD4+ T cells above 2.5%. The well with the highest percentage of reactive CD4+ T cells (8.9%) (Fig. 1a, left panel) was cloned. After expansion, clones were screened in an autopresentation assay with MELOE-132–46 peptide (Fig. 1a, right panel). Nine positive clones were expanded and their TCR sequences were characterized. We identified three distinct clonotypes (Fig. 1b) with the predominance (7/9) of the AV20.1/BV13.3 T cell clone. One such clone, 1A3, that showed both a consistent response to the peptide and a robust proliferation was used for further experiments.
Fig. 1.
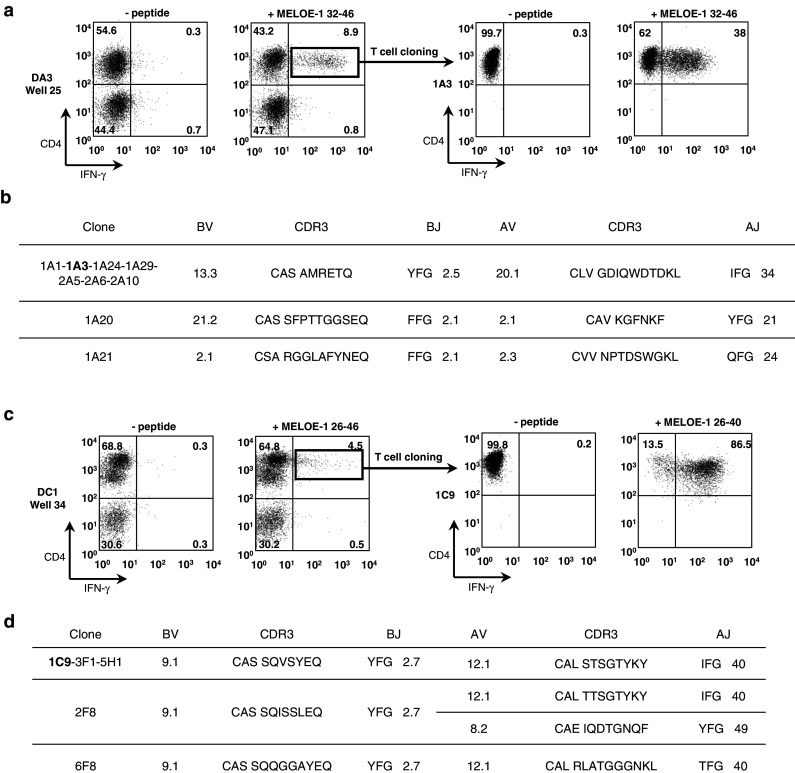
Expansion and TCR characterization of MELOE-132–46 and MELOE-126–40 specific CD4+ clones. a PBMC from healthy donor DA3 were cultured for 14 days with 10 μM MELOE-132–46 and the presence of specific CD4+ T cells was assessed by CD4/IFN-γ double staining after restimulation with peptide (left panel). Specific T cells were cloned by limiting dilution and specificity of the clones was also assessed by CD4/IFN-γ double staining (right panel clone 1A3). b TCR sequences of the MELOE-132–46 specific CD4+ T clones. c PBMC from donor DC1 were stimulated as in a with MELOE-126–46 (left panel) and cloned. Specificity of the clones was assessed after restimulation with MELOE-126–40 (right panel clone 1C9). d TCR sequences of MELOE-126–40 specific CD4+ T clones
Since donor DC1 showed CD4 reponses to MELOE-126–46 but not to the shorter MELOE-132–46 peptide, it suggested that donor DC1 reacted against an epitope distinct from that recognized by donor DA3 and it was thus worth deriving CD4+ T cell clones from this donor to characterize the new epitope. The highest positive well (4.5% of reactive CD4+ T cells, Fig. 1c, left panel) was cloned. Five positive clones were identified by IFN-γ staining and their TCR chains characterized (Fig. 1d). It turned out that all five clones used the same AV/BV combination, namely AV12.1/BV9.1 among which three distinct clonotypes were identified. All clones showed strong reactivity against the 15 aa-peptide MELOE-126–40 thus confirming that the recognized epitope was distinct from that of donor DA3 (Fig. 1c, right panel). Clone 1C9 was used for further experiments.
DC processing of the whole MELOE-1 protein generates the recognized epitopes
We needed to confirm that the peptides recognized by our clones were naturally generated after processing of the whole MELOE-1 protein by DC and that we did not artificially expand irrelevant clones with our in vitro peptide stimulation. To this end, we loaded immature DC with the whole purified MELOE-1 protein or Melan-A16–40 as negative control, matured them and fixed them before presenting them to the clones. To ensure that in our culture conditions the antigen MELOE-1 could not be degraded into peptides that could then load externally onto MHC class II molecules, we also perform the following control experiment: we matured DC, fixed them and then added the MELOE-1 whole protein to evaluate external loading, as previously described by Faure et al. [14]. When DC were fixed prior to incubation with MELOE-1, no response of clone 1A3 was observed indicating that MELOE-1 was not externally degraded into peptides (Fig. 2a). In contrast, both clones 1A3 and 1C9 produced TNF-α and IFN-γ in response to DC that had internalized and processed the whole MELOE-1 protein (but not Melan-A16–40) before fixation thus demonstrating that their specific epitopes were naturally presented by DC (Fig. 2b).
Fig. 2.

Recognition of DC loaded with MELOE-1 whole protein by the CD4+ T cell clones. a Negative control. DC were matured with TNF-α and PolyI:C for 4 h, fixed and then incubated in serum-free medium with MELOE-1 for 4 h, to verify the absence of exogenous loading on class II HLA molecules. b DC were loaded with MELOE-1 whole protein or with the control peptide Melan-A16–40 A27L, for 4 h in serum-free medium with TNF-α and PolyI:C prior to fixation. Co-cultures with clones 1C9 or 1A3 were performed at a 1:1 clone:DC ratio and responses of each clone was assessed by TNF-α and IFN-γ intracellular staining and flow cytometry analysis. A representative experiment is shown out of six performed. ND not done
HLA restriction and epitope characterization
To determine HLA class II restriction of the clones, we first measured TNF-α production following autopresentation of the relevant peptides in the presence of ranging concentrations of anti HLA-DP, DR or DQ mAbs. Reactivity of clone 1A3 was inhibited by the anti-HLA-DQ mAb while clone 1C9 was partially inhibited only by the anti-HLA-DR mAb (Fig. 3a). To identify the restricting allele in each case, we used different EBV-transformed cell-lines (BLCL) as presenting cells. Clone 1A3 (from a HLA-DQβ1*0301/0603 donor) recognized the MELOE-132–46 peptide presented by the BLCL BL30/95 (DQβ1*0201/0603) but not the BLCL LAZ (DQβ1*0301/0501) thus its response was restricted by DQβ1*0603 (Fig. 3b). Likewise, clone 1C9 (from a HLA-DRβ1*0404/1101 donor) recognized the MELOE-126–40 peptide presented by BLCL C3 (DRβ1*1101/1601) but not BLCL BOI (DRβ1*0404/1501) and was thus restricted by DRβ1*1101 (Fig. 3b).
Fig. 3.
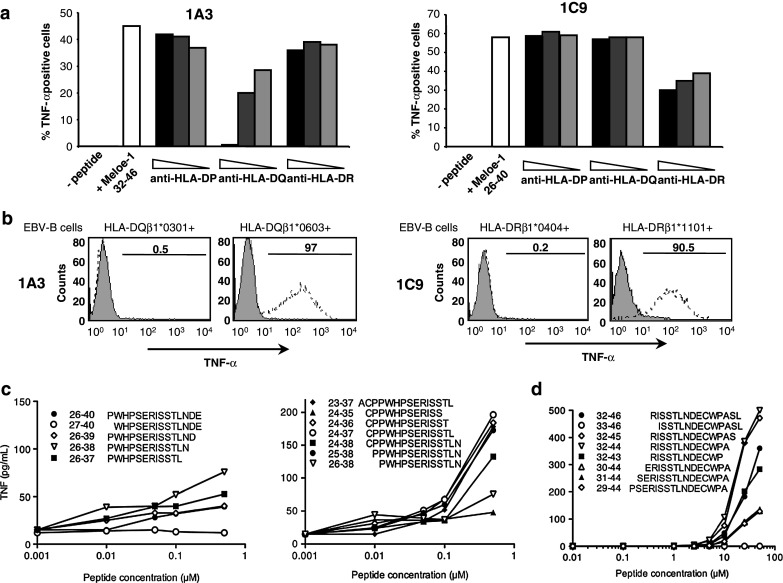
HLA class II restrictions and optimal epitopes recognized by the two clones. a Clones 1A3 and 1C9 were stimulated by MELOE-132–46 and MELOE-126–40, respectively, in the presence or not of decreasing concentrations of anti-HLA-DP, -DQ or -DR mAbs. b The HLA class II-restricting alleles were determined by assessing 1A3 and 1C9 TNF- α responses against unpulsed (grey histogram) or peptide-pulsed (dotted line) EBV-B cells expressing HLA-DQ or -DR alleles from the donors DA3 and DC1. c Clone 1C9 was incubated at a 1:1 ratio with the HLA-DRβ1*1101+ C3 EBV-B cell-line loaded with various concentrations of the indicated peptides and TNF production was measured in the WEHI biological assay. d As in c with clone 1A3 and the HLA-DQβ1*0603+ BL30/95 EBV-B cell-line
Since MELOE-126–40 and MELOE-132–46 peptides were arbitrarily chosen, we tested a series of overlapping peptides on the two clones to define the optimal epitopes. Concerning the optimal epitope(s) recognized by clone 1C9 in the DRβ1*1101 context, we started by testing peptides shortened from the COOH end. Peptide MELOE-126–38 evoked the highest TNF response (Fig. 3c, left). We then tested peptides elongated on the NH2 terminus and observed a significant increase in reactivity up until residue 24 (Fig. 3c, right). Finally, peptides that consistently gave the highest responses were MELOE-124–36 and MELOE-124–37.
Likewise, we tested overlapping peptides for recognition by clone 1A3. The optimal peptide identified was either MELOE-132–45 or MELOE-132–44 since both gave identical responses in terms of TNF production (Fig. 3d). It should be noted that reactivity of clone 1A3 required much higher concentrations of peptide than that required for reactivity of clone 1C9. This probably resulted in part from a much lower expression of HLA-DQ molecules as compared to HLA-DR at the cell surface of the presenting EBV-B cells (data not shown) but could also reflect differences in TCR affinities between the two clones.
Phenotypic and functional characterization of the CD4+ T cell clones
Clones 1C9 and 1A3 were stimulated for 5 h with immobilized anti-CD3 mAb (or PMA/ionomycin for IL-17 production) to identify their T helper profile (Th1, Th2, Th17 or Treg). Both clones expressed Th1 cytokines (TNF-α, IFN-γ, IL-2 and GM-CSF) and some Th2 cytokines (Il-4 and IL-13 but no IL-5) but produced neither IL-10, TGF-β nor IL-17 (Online Resource 2). Expression of chemokine receptors confirmed the Th1 phenotype of both clones with the expression of CCR5 and CXCR3 but not CCR3 (Online Resource 3). Concerning CCR6, it was expressed by clone 1A3 but not clone 1C9 and although it is generally considered as a Th17 marker, it has also been reported on regular Th1 [15]. Otherwise, both clones expressed a phenotypic profile associated with antigen-experienced T cells (CCR7−, CD11ahigh, CD45RAlow, CD27−) (Online Resource 3).
We then tested the cytotoxic potential of both clones on HLA-matched peptide-loaded EBV-B cell-lines. Clone 1C9 was cytotoxic whereas clone 1A3 was not (Fig. 4a). We also tested the reactivity of both clones against HLA-matched melanoma cell-lines treated or not with 500 IU/mL of IFN-γ for 48 h to enhance HLA expression. All melanoma cell-lines expressed the meloe mRNA. The melanoma cell-line M204 expressed a high basal level of HLA-DR that was further increased by IFN-γ treatment whereas untreated M301 expressed a very low level of HLA-DQ that was significantly increased by IFN-γ treatment (Fig. 4b). Neither clones showed any cytokine response when challenged with the relevant melanoma target, even after IFN-γ enhancement of HLA expression. In contrast, clones responded well after peptide loading of the same melanoma cell-lines (Fig. 4c). This absence of reactivity was confirmed on three different HLA-DQβ1*0603-expressing melanoma cell-lines for clone 1A3 and on five different DRβ1*1101-expressing melanoma cell-lines for clone 1C9. Furthermore, clone 1C9 showed no cytotoxicity against the relevant melanoma cell-lines (not shown). Thus, we could not evidence any direct recognition of melanoma cell-lines by our two CD4+ MELOE-1 specific T cell clones. Finally, we tested the ability of DC to present the HLA-DQβ1*0603-restricted MELOE-1 epitope following exposure to MELOE-1+ melanoma or MELOE-1 negative SW480 cell lysates. To this end, we loaded immature DC with M117 or SW480 cell lysates for 8 h, matured them for 36 h, co-cultured them with clone 1A3 for 5 h and tested IFN-γ production by the clone. We used DC loaded with MELOE-132–46 as positive control. As shown in Fig. 4d, a small but significant percentage of clone 1A3 (1.1%) responded to DC loaded with M117 lysate but not with SW480 lysate, demonstrating that DC could indeed process and present the MELOE-1 whole protein from melanoma cell lysates.
Fig. 4.

Functional characterization of the CD4+ T cell clones. a Cytotoxicity of CD4+ T cell clones 1A3 and 1C9 against peptide-pulsed EBV-B cells was assessed by a standard chromium release assay. b Representative examples of HLA-DQ or HLA-DR expression by melanoma cell-lines after incubation with (bold line) or without (thick line) 500 IU/ml IFN-γ for 48 h. c IFN-γ production by CD4+ T cell clones 1A3 and 1C9 in response to HLA class II-matched melanoma cell-lines, treated or not with IFN-γ and loaded or not with MELOE-1 peptides. Shown are representative responses of 1A3 and 1C9 T cell clones toward one out of three HLA-DQβ1*0603+ and one out of five HLA-DRβ1*1101+ MELOE-1-expressing melanoma cell-lines tested, respectively. d IFN-γ production by clone 1A3 in response to HLA-matched DC loaded with M117 or SW480 cell lysate or the MELOE-132–46 peptide as positive control (n = 2)
In vitro priming with MELOE-122–46-loaded DC
Since we showed that the optimal HLA-DRβ1*1101-restricted epitope started at residue 24 of MELOE-1, we decided to test the stimulation potential of a long peptide, MELOE-122–46, that comprises the optimal epitope (24–37) and yet requires DC processing to generate it. We first tested whether peptide MELOE-122–46 could in fact be processed by DC and stimulate both the HLA-DRβ1*1101-restricted response of clone 1C9 and the HLA-A*0201-restricted response of the MELOE-136–44-specific CD8+ T cell clone M170.48. DC loaded with MELOE-122–46 very efficiently stimulated clone 1C9 and also cross-presented MELOE-136–44 to the CD8 clone M170.48 (Fig. 5a). Next, we used DC loaded with MELOE-122–46 to stimulate PBL from donor DS7 (Table 1) using two different protocols. The first protocol used a single stimulation for 14 days of PBL in 96 wells with peptide-loaded DC while the second protocol used a 7 day stimulation with IL-6 and IL-12 followed by a second stimulation with DC (see material and methods). In both cases, T cells in each well were challenged with MELOE-124–37 to reveal activation of specific CD4+ T lymphocytes and with MELOE-136–44 to detect CD8 responses. Typical examples of the results obtained with the first and the second protocol are presented in Fig. 5b and 5c, respectively. With the first protocol we detected CD4 positive responses in 6/96 wells (range of IFN-γ-positive cells from 0.2 to 0.5% among CD4+ T cells) and CD8 positive responses in 4/96 wells (range of IFN-γ-positive cells from 1.6 to 12.2% among CD8+ T cells). With the second protocol, it was not significantly different with CD4 responses in 4/96 wells (range of IFN-γ-positive cells from 0.25 to 2.1% among CD4+ T cells) and CD8 responses in 6/96 wells (range of IFN-γ-positive cells from 1.5 to 16.3% among CD8+ T cells). Thus, regardless of the protocol used, DC loaded with MELOE-122–46 peptide could efficiently induce both HLA-DRβ1*1101-restricted CD4 responses and HLA-A*0201-restricted responses among PBL from this healthy donor. In some rare instances, we even found both CD4 and CD8 responses in the same well as shown on Fig. 5b.
Fig. 5.

Stimulation of PBL from a HLA-A*0201+ HLA-DRβ1*1101+ donor with MELOE-122–46-loaded autologous DC. a Immature DC were loaded with MELOE-122–46 for 4 h, washed and matured with TNF-α and PolyI:C for 12 h. Efficiency of MELOE-122–46 processing and presentation or cross- presentation was assessed by coculturing loaded DC with the CD4+ T cell clone 1C9 or with the MELOE-136–44-specific CD8+ T cell clone M170.48 at a 1:1 ratio for 5 h. b PBL of a HLA-A*0201+ HLA-DRβ1*1101+ healthy donor (DS7) were stimulated by MELOE-122–46–loaded DC at a 10:1 PBL:DC ratio and cultured for 14 days in the presence of IL-2. c PBL from donor DS7 were stimulated by MELOE-122–46–loaded DC at a 10:1 PBL:DC ratio in the presence of IL-6 and IL-12 for 7 days, restimulated with loaded DC and cultured for 7 more days in the presence of IL-2. In both cases, the presence of MELOE-124–37 specific CD4+ T cells and MELOE-136–44 specific CD8+ T cells was assessed by challenging T cells with MELOE-124–37 or MELOE-136–44 peptides, respectively, followed by CD4/IFN-γ or CD8/IFN-γ double staining and flow cytometry analysis. Examples of wells with MELOE-124–37 (left panels) and/or MELOE-136–44 (right panels) specific T cells expansions are shown. Percentage of specific T cells among CD8+ or among CD4+ T cells are indicated
Discussion
Increasing evidence suggests that efficient therapeutic vaccination against cancer will require the stimulation of CD4+ T cell to buttress cytotoxic CD8+ T cell responses [2] or to generate CD4 effector cells [16, 17]. Thus, there is a growing interest in defining long peptides from relevant tumor antigens that contain both CD4+ and CD8+ T cell epitopes.
In the present study, we looked for CD4+ T cell responses against this antigen in healthy subjects by simply adding different peptides from MELOE-1 to PBMC in vitro and measuring cytokine production following restimulation with the same peptide. A similar strategy (using the APC present within PBMC) was previously used to successfully obtain Melan-A-specific CD4+ T cell clones from healthy donors [18]. In contrast, many research groups use multiple rounds of in vitro stimulations with peptide- or antigen-loaded DC to expand specific CD4+ T cells [19–21]. In the latter cases, both recall of antigen-experienced T cells and T cell priming of naive lymphocytes may occur. We reckon that in our stimulation conditions, (1) APC were monocytes/macrophages and possibly B lymphocytes and (2) these APC are unlikely to prime naive T cells in short-time cultures. Therefore, we probably restimulated MELOE-1-experienced CD4+ T cells in our healthy donors with this stimulation procedure, which would be consistent with our previous detection of CD8 memory T cells directed against MELOE-1 in healthy individuals [9].
This stimulation procedure could efficiently expand MELOE-126–46-specific CD4+ precursors present in the blood of all seven healthy donors tested, despite their broad diversity of HLA class II haplotypes. We wondered whether MELOE-126–46 contained a single epitope presented in multiple HLA class II contexts as previously reported for peptides from MAGE-A3 [22], gp100 [23] or survivin [21] or whether MELOE-126–46 contained distinct epitopes. By testing shorter peptides, we demonstrated the presence of at least two distinct epitopes in this long peptide presented in the HLA-DRβ1*1101 and the HLA-DQβ1*0603 contexts. Cloning of the CD4 reactive populations led to the identification of three distinct clonotypes in each context with a somewhat greater diversity of HLA-DQβ1*0603-restricted clones that used three distinct AV and BV regions while HLA-DRβ1*1101-restricted clones differed only in their CDR3 regions.
Because our in vitro stimulation protocol may lead to the expansion of irrelevant crossreactive CD4+ T cells, we needed to confirm that our clones could recognize peptides from naturally processed MELOE-1 whole protein. To this end, we loaded HLA-matched DC with the whole MELOE-1 protein and tested their ability to stimulate the clones. We ensured that MELOE-1 internalization and processing was necessary for peptide presentation (1) by using serum-free medium to avoid extracellular proteolytic cleavage by serum proteases and possible exogenous loading on class II molecules and (2) by fixing DC prior to DC loading and verifying that presentation was abrogated [14]. We demonstrated that both clones could be efficiently stimulated by MELOE-1-loaded DC, thus supporting their immunological relevance.
Since MELOE-126–46 was arbitrarily chosen, we next ask whether it contained the optimal epitope for each clone and especially for the HLA-DRβ1*1101-restricted clone 1C9 that recognized MELOE-126–40. By using overlapping synthetic peptides we indeed demonstrated that elongation toward the NH2 terminus of MELOE-1 increased clone 1C9 reactivity. This prompted us to design a longer peptide for vaccination purposes, MELOE-122–46, that would contain the two optimal epitopes, namely MELOE-124–37 for clone 1C9 and MELOE-132–44 for clone 1A3. It should be stressed, however, that the optimal epitopes thus defined may not represented the true epitopes presented in vivo, which would only be formally identified by elution from class II HLA/peptide complexes at the cell surface of APCs as previously described [24].
Considering that the aim of vaccination with class II-restricted peptides is to induce Th1 responses that have been shown to play a major role in anti-tumor immunity [25], we have used production of Th1 cytokines (TNF-α, IFN-γ…) to screen for CD4 responses. Indeed, the two MELOE-1-specific CD4 clones that we isolated displayed a Th1 profile, confirmed by analysis of chemokine receptors. This does not exclude the possibility that other CD4+ T cell subtypes may have been stimulated but undetected in our conditions, such as Th2, Th17 or Treg. We reckon, however, that Treg were probably not majorly stimulated in our culture conditions since they would have inhibited Th1 lymphocyte expansion.
Although many authors described direct recognition of melanoma cell-lines by tumor-antigen specific CD4+ T cells [20, 26, 27], others reported only indirect recognition through processing of the tumor antigen by DC [28], which is expected to be the main pathway of presentation of tumor antigens to CD4+ T lymphocytes. In our study, we showed that MELOE-1 could be processed and presented by DC exposed to melanoma cell lysates but we could not evidence any response of the two MELOE-1 specific CD4+ T cell clones toward HLA class II-matched and MELOE-1-expressing melanoma cell-lines. One first explanation could be that direct tumor recognition by our clones may require higher antigen expression levels and/or HLA class II expression (notably in the case of HLA-DQ expression). However, (1) IFN-γ treatment significantly enhanced HLA class II expression on melanoma cells but did not trigger direct recognition by our CD4 clones although it allowed presentation of exogenously added peptide and (2) these melanoma cell-lines express sufficient levels of MELOE-1 to present MELOE-136–44 to CD8+ T lymphocytes. A second explanation would be the inability of melanoma cell-lines to process and present the two class II-restricted MELOE-124–37 and MELOE-132–44 epitopes. The MELOE-1 antigen in melanoma cell is predicted as a non secretory polypeptide and we have no further information to date on its sub-cellular localization. It thus remains to be investigated whether the MELOE-1 whole protein is not processed at all within the MHC II loading compartments in melanoma cells or whether only some epitopes can be generated and presented by melanoma cells as was reported in the case of MAGE-A3 [20].
Nevertheless, our results demonstrate that a long peptide, MELOE-122–46, that contains the two optimal class II epitopes and the HLA-A*0201-restricted epitope was processed and presented by DC to the HLA-DRβ1*1101-restricted clone 1C9. Moreover, we provide evidence for the first time that this long peptide from MELOE-1 can also be efficiently cross-presented to the HLA-A*0201/MELOE-136–44 specific CD8 clone M170.48.
Moreover, we showed that HLA-matched dendritic cells loaded with MELOE-122–46 induced both specific CD8+ and CD4+ T cell responses among PBL from healthy donor DS7, with no significant difference between the two protocols used (one vs. two rounds of stimulation).
Altogether, our data suggest that this long peptide MELOE-122-46 could represent a good candidate for efficient vaccination against MELOE-1 since it can trigger both CD4 and CD8 responses in healthy individuals. Finally, HLA class II prediction algorithms (http://www.syfpeithi.de) [29] predict that this long peptide may generate multiple epitopes with good binding scores in HLA-DRβ1*0101, 0301, 0401, 0701, 1101 and 1501. This remains to be confirmed not only in healthy subjects but also in melanoma patients before considering the future use of this long peptide in melanoma vaccination.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by a grant from “Association pour la Recherche sur le Cancer” (ARC1074XA0830F). Anne Rogel was supported by a grant from INSERM and “Région Pays de La Loire”. We thank Delphine Coulais from the Clinical Transfer Facility for expert technical assistance.
References
- 1.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Melief CJ, van der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer. 2008;8:351–360. doi: 10.1038/nrc2373. [DOI] [PubMed] [Google Scholar]
- 3.Fayolle C, Deriaud E, Leclerc C. In vivo induction of cytotoxic t cell response by a free synthetic peptide requires CD4+ T cell help. J Immunol. 1991;147:4069–4073. [PubMed] [Google Scholar]
- 4.Bijker MS, van den Eeden SJ, Franken KL, Melief CJ, Offringa R, van der Burg SH. CD8+ CTL priming by exact peptide epitopes in incomplete freund’s adjuvant induces a vanishing CTL response, whereas long peptides induce sustained CTL reactivity. J Immunol. 2007;179:5033–5040. doi: 10.4049/jimmunol.179.8.5033. [DOI] [PubMed] [Google Scholar]
- 5.Gritzapis AD, Voutsas IF, Lekka E, Papamichail M, Baxevanis CN. Peptide vaccination breaks tolerance to HER-2/neu by generating vaccine-specific fasL+ CD4+ T cells: first evidence for intratumor apoptotic regulatory T cells. Cancer Res. 2010;70:2686–2696. doi: 10.1158/0008-5472.CAN-09-2517. [DOI] [PubMed] [Google Scholar]
- 6.Kenter GG, Welters MJ, Valentijn AR, Lowik MJ, Berends-van der Meer DM, Vloon AP, Essahsah F, Fathers LM, Offringa R, Drijfhout JW, Wafelman AR, Oostendorp J, Fleuren GJ, van der Burg SH, Melief CJ. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med. 2009;361:1838–1847. doi: 10.1056/NEJMoa0810097. [DOI] [PubMed] [Google Scholar]
- 7.Godet Y, Moreau-Aubry A, Guilloux Y, Vignard V, Khammari A, Dreno B, Jotereau F, Labarriere N. Meloe-1 is a new antigen overexpressed in melanomas and involved in adoptive T cell transfer efficiency. J Exp Med. 2008;205:2673–2682. doi: 10.1084/jem.20081356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Godet Y, Moreau-Aubry A, Mompelat D, Vignard V, Khammari A, Dreno B, Lang F, Jotereau F, Labarriere N. An additional ORF on meloe cDNA encodes a new melanoma antigen, MELOE-2, recognized by melanoma-specific T cells in the HLA-A2 context. Cancer Immunol Immunother. 2010;59:431–439. doi: 10.1007/s00262-009-0762-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Godet Y, Desfrancois J, Vignard V, Schadendorf D, Khammari A, Dreno B, Jotereau F, Labarriere N. Frequent occurrence of high affinity t cells against meloe-1 makes this antigen an attractive target for melanoma immunotherapy. Eur J Immunol. 2010;40:1786–1794. doi: 10.1002/eji.200940132. [DOI] [PubMed] [Google Scholar]
- 10.Gervois N, Labarriere N, Le Guiner S, Pandolfino MC, Fonteneau JF, Guilloux Y, Diez E, Dreno B, Jotereau F. High avidity melanoma-reactive cytotoxic T lymphocytes are efficiently induced from peripheral blood lymphocytes on stimulation by peptide-pulsed melanoma cells. Clin Cancer Res. 2000;6:1459–1467. [PubMed] [Google Scholar]
- 11.Davodeau F, Difilippantonio M, Roldan E, Malissen M, Casanova JL, Couedel C, Morcet JF, Merkenschlager M, Nussenzweig A, Bonneville M, Malissen B. The tight interallelic positional coincidence that distinguishes t-cell receptor jalpha usage does not result from homologous chromosomal pairing during valphajalpha rearrangement. EMBO J. 2001;20:4717–4729. doi: 10.1093/emboj/20.17.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arden B, Clark SP, Kabelitz D, Mak TW. Human t-cell receptor variable gene segment families. Immunogenetics. 1995;42:455–500. doi: 10.1007/BF00172176. [DOI] [PubMed] [Google Scholar]
- 13.Espevik T, Nissen-Meyer J. A highly sensitive cell line, WEHI 164 clone 13, for measuring cytotoxic factor/tumor necrosis factor from human monocytes. J Immunol Methods. 1986;95:99–105. doi: 10.1016/0022-1759(86)90322-4. [DOI] [PubMed] [Google Scholar]
- 14.Faure F, Mantegazza A, Sadaka C, Sedlik C, Jotereau F, Amigorena S. Long-lasting cross-presentation of tumor antigen in human dc. Eur J Immunol. 2009;39:380–390. doi: 10.1002/eji.200838669. [DOI] [PubMed] [Google Scholar]
- 15.Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, Napolitani G. Surface phenotype and antigenic specificity of human interleukin 17-producing t helper memory cells. Nat Immunol. 2007;8:639–646. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 16.Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, Jungbluth A, Gnjatic S, Thompson JA, Yee C. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358:2698–2703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perez-Diez A, Joncker NT, Choi K, Chan WF, Anderson CC, Lantz O, Matzinger P. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood. 2007;109:5346–5354. doi: 10.1182/blood-2006-10-051318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Godefroy E, Scotto L, Souleimanian NE, Ritter G, Old LJ, Jotereau F, Valmori D, Ayyoub M. Identification of two melan-a CD4+ T cell epitopes presented by frequently expressed MHC class II alleles. Clin Immunol. 2006;121:54–62. doi: 10.1016/j.clim.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 19.Jager E, Jager D, Karbach J, Chen YT, Ritter G, Nagata Y, Gnjatic S, Stockert E, Arand M, Old LJ, Knuth A. Identification of NY-ESO-1 epitopes presented by human histocompatibility antigen (HLA)-DRB4*0101–0103 and recognized by CD4(+) T lymphocytes of patients with NY-ESO-1-expressing melanoma. J Exp Med. 2000;191:625–630. doi: 10.1084/jem.191.4.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schultz ES, Lethe B, Cambiaso CL, Van Snick J, Chaux P, Corthals J, Heirman C, Thielemans K, Boon T, van der Bruggen P. A MAGE-A3 peptide presented by HLA-DP4 is recognized on tumor cells by CD4+ cytolytic t lymphocytes. Cancer Res. 2000;60:6272–6275. [PubMed] [Google Scholar]
- 21.Wang XF, Kerzerho J, Adotevi O, Nuyttens H, Badoual C, Munier G, Oudard S, Tu S, Tartour E, Maillere B. Comprehensive analysis of HLA-DR- and HLA-DP4-restricted CD4+ T cell response specific for the tumor-shared antigen survivin in healthy donors and cancer patients. J Immunol. 2008;181:431–439. doi: 10.4049/jimmunol.181.1.431. [DOI] [PubMed] [Google Scholar]
- 22.Kobayashi H, Song Y, Hoon DS, Appella E, Celis E. Tumor-reactive t helper lymphocytes recognize a promiscuous MAGE-A3 epitope presented by various major histocompatibility complex class II alleles. Cancer Res. 2001;61:4773–4778. [PubMed] [Google Scholar]
- 23.Kobayashi H, Lu J, Celis E. Identification of helper t-cell epitopes that encompass or lie proximal to cytotoxic t-cell epitopes in the gp100 melanoma tumor antigen. Cancer Res. 2001;61:7577–7584. [PubMed] [Google Scholar]
- 24.Chicz RM, Urban RG, Gorga JC, Vignali DA, Lane WS, Strominger JL. Specificity and promiscuity among naturally processed peptides bound to HLA-DR alleles. J Exp Med. 1993;178:27–47. doi: 10.1084/jem.178.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kennedy R, Celis E. Multiple roles for CD4+ t cells in anti-tumor immune responses. Immunol Rev. 2008;222:129–144. doi: 10.1111/j.1600-065X.2008.00616.x. [DOI] [PubMed] [Google Scholar]
- 26.Bioley G, Jandus C, Tuyaerts S, Rimoldi D, Kwok WW, Speiser DE, Tiercy JM, Thielemans K, Cerottini JC, Romero P. Melan-a/mart-1-specific cd4 T cells in melanoma patients: identification of new epitopes and ex vivo visualization of specific t cells by MHC class II tetramers. J Immunol. 2006;177:6769–6779. doi: 10.4049/jimmunol.177.10.6769. [DOI] [PubMed] [Google Scholar]
- 27.Manici S, Sturniolo T, Imro MA, Hammer J, Sinigaglia F, Noppen C, Spagnoli G, Mazzi B, Bellone M, Dellabona P, Protti MP. Melanoma cells present a MAGE-3 epitope to CD4(+) cytotoxic t cells in association with histocompatibility leukocyte antigen DR11. J Exp Med. 1999;189:871–876. doi: 10.1084/jem.189.5.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chaux P, Vantomme V, Stroobant V, Thielemans K, Corthals J, Luiten R, Eggermont AM, Boon T, van der Bruggen P. Identification of mage-3 epitopes presented by HLA-DR molecules to CD4(+) T lymphocytes. J Exp Med. 1999;189:767–778. doi: 10.1084/jem.189.5.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. Syfpeithi: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.