

Abstract
Background
Recurrent glioblastoma is associated with a poor overall survival. Antiangiogenic therapy results in a high tumor response rate but has limited impact on survival. Immunotherapy has emerged as an efficient treatment modality for some cancers, and preclinical evidence indicates that anti-VEGF(R) therapy can counterbalance the immunosuppressive tumor microenvironment.
Methods
We collected peripheral blood mononuclear cells (PBMC) of patients with recurrent glioblastoma treated in a randomized phase II clinical trial comparing the effect of axitinib with axitinib plus lomustine and analyzed the immunophenotype of PBMC, the production of cytokines and expression of inhibitory molecules by circulating T cells.
Results
PBMC of 18 patients were collected at baseline and at 6 weeks after initiation of study treatment. Axitinib increased the number of naïve CD8+ T cells and central memory CD4+ and CD8+ T cells and reduced the TIM3 expression on CD4+ and CD8+ T cells. Patients diagnosed with progressive disease on axitinib had a significantly increased number of regulatory T cells and an increased level of PD-1 expression on CD4+ and CD8+ T cells. In addition, reduced numbers of cytokine-producing T cells were found in progressive patients as compared to patients responding to treatment.
Conclusion
Our results suggest that axitinib treatment in patients with recurrent glioblastoma has a favorable impact on immune function. At the time of acquired resistance to axitinib, we documented further enhancement of a preexisting immunosuppression. Further investigations on the role of axitinib as potential combination partner with immunotherapy are necessary.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-016-1836-3) contains supplementary material, which is available to authorized users.
Keywords: Glioblastoma, Antiangiogenesis, Axitinib, Regulatory T cells, Inhibitory molecules
Introduction
Glioblastoma (GBM) is the most common primary malignant brain tumor in adults and is associated with a poor prognosis [1]. For recurrent GBM, none of the currently available treatments has shown to improve overall survival (OS). In this patient population, treatment with cytotoxic drugs leads to a poor response rate of 5–10 % and a median OS of 25–30 weeks [2]. Neo-angiogenesis is a key feature of GBM, and antiangiogenic therapies have been investigated extensively for the treatment of GBM. Treatment with bevacizumab, a monoclonal antibody against vascular endothelial growth factor (VEGF), led to objective response rates (ORR) of 28.2 % [3]. Several VEGF-receptor (VEGFR)-targeting small molecules have been explored in GBM [4–6]. Despite an ORR of, respectively, 15.3 and 17.2 %, cediranib failed to improve survival of recurrent GBM patients when compared to lomustine (alkylating nitrosourea) alone [7].
We previously conducted a randomized, phase II clinical trial comparing axitinib, a tyrosine kinase inhibitor (TKI) against VEGFR-1, 2 and 3, with best standard of care in patients with recurrent GBM [8]. Treatment with axitinib resulted in an ORR of 28 % and an estimated 6-month progression-free-survival (6mPFS) rate of 34 % [9]. In a recently reported randomized phase II trial, the 9-month OS rate of recurrent GBM patients treated with a combination of bevacizumab and lomustine compared favorably to the outcome of patients treated with bevacizumab or lomustine as single agents (respectively, 59, 43 and 38 % 9-month OS was reported) [10]. We initiated a randomized phase II clinical trial with axitinib and axitinib plus lomustine combination therapy.
It has been shown in preclinical models that antiangiogenic treatment targeting the VEGF-VEGFR pathway can convert the immunosuppressive microenvironment of tumors into an immunostimulatory one [11]. We have previously shown in subcutaneous and intracranial melanoma mouse models that treatment with axitinib reduces the suppressive function of intratumoral monocytic myeloid-derived suppressor cells (moMDSCs), suggesting that treatment with axitinib can reprogram the immunosuppressive tumor microenvironment toward an immunostimulatory environment [12].
To date, different immunotherapies have been proposed as treatment for GBM. So far results of clinical trials have generated an indication for biologic activity but translated only into a limited impact on OS [13]. Nevertheless, it was recently reported that addition of rindopepimut, a peptide vaccine against the mutant endothelial growth factor receptor variant vIII (EGFRvIII), to bevacizumab in patients with recurrent EGFRvIII-positive GBM leads to a 6mPFS rate of 27 % (control vaccine + bevacizumab 6mPFS 11 %) [14]. These results suggest that combination of tumor antigen-specific immunotherapy combined with antiangiogenic treatment can improve the outcome of recurrent GBM patients.
In this study, we investigated whether the selective VEGFR inhibitor axitinib would be a good candidate for combination with immunotherapy by investigating the immunophenotype of PBMC from recurrent GBM patients treated in the ongoing axitinib plus lomustine trial.
Materials and methods
Patient specimens and study protocol
Fifty-six patients with histologically confirmed glioblastoma (WHO grade IV glioma) were included in a phase II, non-comparative, randomized, two-arm, open-label clinical trial with axitinib alone or axitinib plus lomustine conducted at four medical centers (NCT01562197). At progression, patients in the axitinib treatment arm were allowed to continue axitinib and initiate lomustine at the condition that the treating physician believed that patients could benefit from continued axitinib treatment. The ethical committees of each medical center and Belgian competent authorities approved the protocol, and all patients provided written informed consent before study participation. Peripheral blood samples were collected at baseline and at 6 weeks post-treatment. Radiological assessments to evaluate tumor response were performed at baseline and every 6 weeks after treatment initiation. Tumor response assessments were determined according to the RANO criteria. If patients had a partial response (PR) or stable disease (SD) at the first assessment at 6 weeks, it had to be confirmed at the next assessment at 12 weeks to meet the criteria for objective tumor response. If this was not the case, patients were considered as having unconfirmed tumor response (uCR or uPR) at 6 weeks and a progressive disease (PD) at 12 weeks.
Sample preparation
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood by Ficoll-Paque (Lymphoprep™, Axis-shield) centrifugation. PBMCs were frozen until needed, and upon use, cells were rested overnight in an incubator at 37°C with 5 % CO2.
Flow cytometry
For characterization of the T cell populations, PBMCs were stained with eF605NC-conjugated anti-human CD3 (eBioscience), PECy7-conjugated anti-human CD4 (BD Biosciences), APC-H7-conjugated anti-human CD8 (BD Biosciences), PE-conjugated anti-human CD25 (Miltenyi Biotec), FITC-conjugated CD127 (BD Biosciences) and an intranuclear staining using the FoxP3 Staining Buffer Set, according to the manufacturer’s instructions with APC-conjugated anti-human FoxP3 (eBioscience).
For characterization of the dendritic cells (DCs) and moMDSCs PBMCs were stained with PacificBlue-conjugated anti-human CD11b (BD Biosciences), APC-conjugated anti-human CD11c (BD Biosciences), PECy7-conjugated anti-human CD33 (BD Biosciences), APC-H7-conjugated anti-human CD14 (BD Biosciences), PE-conjugated anti-human HLA-DR (BD Biosciences) and FITC-conjugated anti-human CD15 (BD Biosciences).
For the determinations of T cell subsets and inhibitory molecules, PBMCs were cultured at a density of 5 × 105 cells per well in a 96-well plate in RPMI-1640 medium (Sigma) supplemented with 1 % human AB-serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, 1 mM sodium pyruvate and non-essential amino acids and were either left unstimulated or were stimulated with anti-CD3/CD28 microbeads (Dynabeads®, Invitrogen) for 24 h. Afterward, PBMCs were harvested and part of the cells was stained with eF605NC-conjugated anti-human CD3, PECy7-conjugated anti-human CD4, APC-H7-conjugated anti-human CD8, PE-conjugated anti-human CD45RO (BD Biosciences) and APC-conjugated anti-human CD62L (BD Biosciences). The remaining PBMCs were stained with PE-CF594-conjugated anti-human CD3 (BD Biosciences), PECy7-conjugated anti-human CD4, FITC-conjugated anti-human CD8 (Biolegend), eF710-conjugated anti-human LAG3 (eBioscience), eF450-conjugated anti-human TIM3 (eBioscience) and PE-conjugated anti-human PD-1 (BD Biosciences), for investigation of the expression of inhibitory molecules.
ICS staining
PBMCs were cultured at a density of 2 × 106 cells per well in a 48-well plate in RPMI-supplemented medium and stimulated with anti-CD3/CD28 beads for 24 h in the presence of brefeldin A (GolgiPlug, BD Biosciences). After 24 h, PBMCs were harvested and stained using the following antibodies: PECy7-conjugated anti-human CD4, APC-H7-conjugated anti-human CD8, PerCP-eF710-conjugated anti-human LAG3, eF450-conjugated anti-human TIM3 and PE-conjugated anti-human PD-1. Fixation and permeabilization of the cells were performed using the Cytofix/Cytoperm kit (BD Biosciences), according to the manufacturer’s instructions. Intracellular stainings were performed using the following antibodies: FITC-conjugated anti-human IFN-γ (BD Biosciences), BV605-conjugated TNF-α (BD Biosciences) and APC-conjugated IL-2 (eBioscience). Aspecific T cell responses, measured upon culture in the absence of T cell stimulus, were considered as background.
Luminex analysis
After polyclonal stimulation, supernatants were collected for detection of cytokine secretion. IFN-γ, TNF-α, IL-2, IL-5, IL-10, IL-13 and IL-17 were quantified by cytokine magnetic bead array (Bio-Rad Laboratories Inc, Hercules, CA, cat. nr. 171-304070) with a Bio-Plex 200 System Luminex reader (Bio-Rad Laboratories Inc, Hercules, CA) using Bio-Plex Manager 4.1.1 software.
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 5.0 software (GraphPad Software). A Wilcoxon matched-pairs signed-rank test was used to compare data obtained at baseline and at 6 weeks. For comparison between treatment groups, a Mann-Whitney U test was performed. Data are represented as median with interquartile range (*p < 0.05; **p < 0.01).
Results
Patient characteristics and clinical response to axitinib or axitinib plus lomustine
Of the 56 patients randomized in the two treatment arms, we obtained peripheral blood from 21 patients (supplementary Table 1). Three patients died before the assessment at 6 weeks and were excluded from the analysis. Of the 18 remaining patients (7F/11 M), 10 patients were randomized in the axitinib arm and 8 patients in the axitinib plus lomustine arm. The median age of the total population was 52 years (range 24–69) and 57 years (range 24–69) and 48 years (range 28–63) for the axitinib and the combination arm, respectively. Axitinib treatment was always initiated at 5 mg bid, and depending on the occurrence of side effects or increased blood pressure, a respective dose reduction or dose escalation was implied. Lomustine was given at a dose of 90 mg/m2 simultaneously with the initiation of axitinib treatment. At 6 weeks the majority of patients were treated with 5 mg axitinib bid. For only 4 patients, the dose was escalated to 7 mg axitinib bid and 2 patients needed to reduce their dose of axitinib (2 mg bid and 3 mg bid, respectively).
Among patients treated with axitinib alone, 3/10 had a PR (30 %), 1/10 had a SD (10 %), and 6/10 had a PD (60 %); among those treated in the combination arm, 2/8 patients had a PR (25 %), 3/8 had a SD (37.5 %), and 3 had a PD (37.5 %).
Axitinib does not affect the numbers of different immune cell populations
The effect of axitinib or axitinib plus lomustine on several immune cell populations was documented and is summarized in Fig. 1. Axitinib treatment did not significantly influence the number of CD4+ T cells, CD8+ T cells, CD11b+ cells or moMDSCs (CD11b+CD33+CD14+HLADRlow cells). There was a trend toward increased dendritic cell numbers (CD11c+ cells, 2.1 vs. 3.6 %) (Fig. 1c). When lomustine was added to axitinib treatment, a reduction in the number of T cells, mainly of CD8+ T cells (24 vs. 12.65 %), DCs and moMDSCs, was observed. There was also a nonsignificant increase in the number of Treg (1.2 vs. 2.1 %) (Fig. 1d).
Fig. 1.
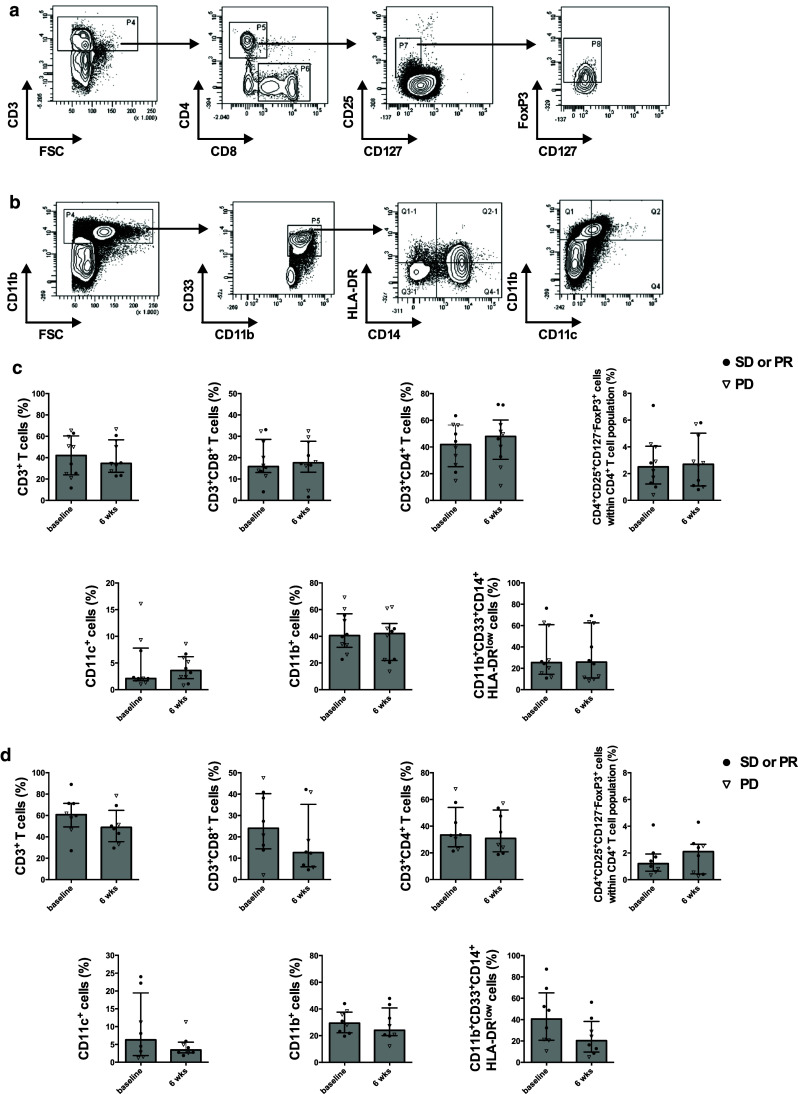
Axitinib treatment does not affect the immune cell populations. Gating strategy for the determination of the percentages of CD4+ T cells, CD8+ T cells, Treg (a), CD11c+ cells, CD11b+ cells and moMDSCs (b). Percentages of specific immune cell populations of patients treated with axitinib (n = 10) (c) and patients treated with axitinib plus lomustine (n = 8) (d)
Axitinib increases the number of central memory CD4+ and CD8+ T cells and the number of naïve CD8+ T cells
Since we did not observe a difference in the number of CD4+ T cells or CD8+ T cells upon axitinib treatment, we further investigated its effect on different subsets of T cells. We specifically studied the naïve T cell population (CD45RO−CD62L+CD4+ or CD45RO−CD62L+CD8+ T cells), the central memory T cells (TCM; CD45RO+CD62L+CD4+ T cells or CD45RO+CD62L+CD8+ T cells) and the effector memory T cells (TEM; CD45RO+CD62L−CD4+ T cells or CD45RO+CD62L−CD8+ T cells).
We first investigated the influence of the treatments on the naïve CD4+ and CD8+ T cells. For the CD4+ T cells we did not find significant differences at baseline and at 6 weeks (Fig. 2a). However, within the CD8+ T cell population, we found that axitinib increased the number of naïve CD8+ T cells (8 vs. 14.5 %). This effect was not found in the combination arm (Fig. 2b). After polyclonal stimulation we observed a comparable reduction in the number of naïve CD4+ T cells at baseline (14.15 vs. 4 %) and at 6 weeks (15.35 vs. 5.35 %) in both treatment arms (Fig. 2a). A similar reduction in the number of naive CD8+ T cells at baseline (14.5 vs. 8 %, p = 0.055) and 6 weeks (14.5 vs. 4.5 %, p = 0.023) was found after axitinib treatment. However, in the combination arm the reduction of the naïve T cell population was less pronounced at 6 weeks as compared to baseline (resp., 3.95 vs. 1.6 % and 2.1 vs. 0.95 %) (Fig. 2b).
Fig. 2.
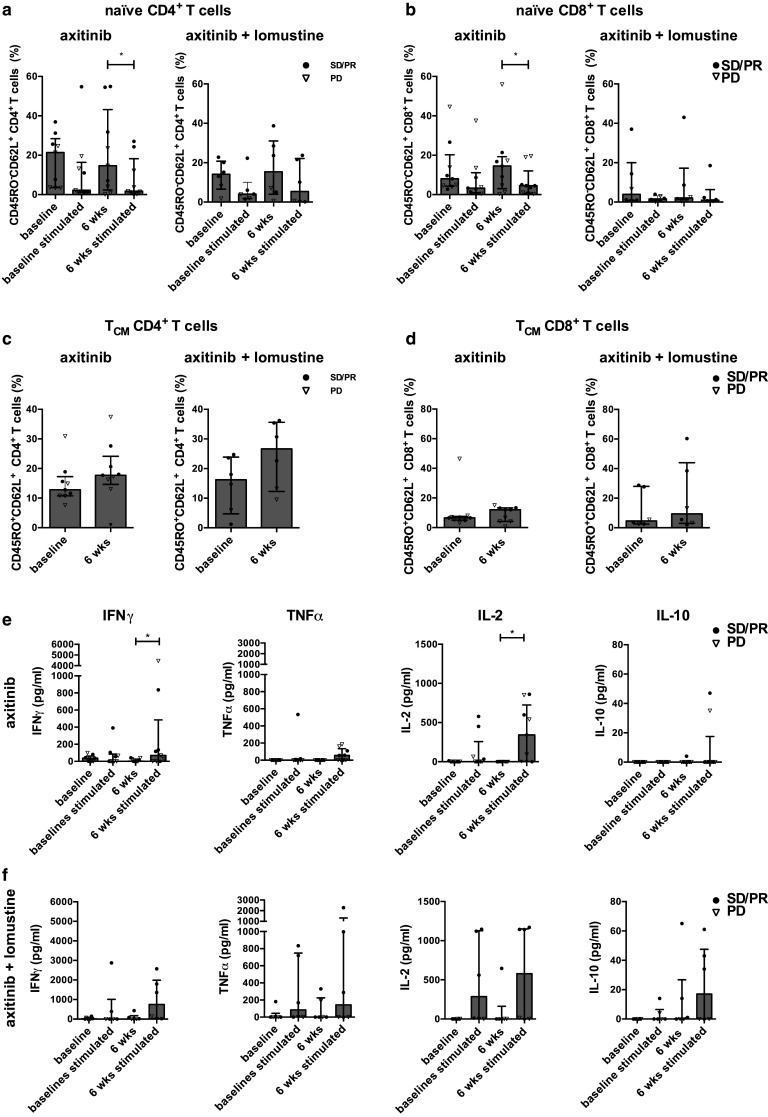
Axitinib treatment increases the number of naïve CD8+ T cells and central memory T cells. Percentages of the naïve CD4+ T cell subsets (a) and naïve CD8+ T cell subsets (b) determined in the absence or presence of anti-CD3/CD28 beads for 24 h at baseline and at 6 weeks after axitinib (n = 9) or axitinib plus lomustine treatment (n = 6). Percentages of TCM CD4+ T cells (c) and TCM CD8+ T cell subsets (d) determined at baseline and at 6 weeks after treatment. In the supernatants of unstimulated and stimulated PBMCs, IFN-γ, TNF-α, IL-2 and IL-10 were quantified by cytokine magnetic bead array after axitinib (n = 9) (e) or axitinib plus lomustine treatment (n = 6) (f)
Next we investigated the effects on the central memory T cells (TCM). After 6 weeks of treatment we observed increased numbers of TCM CD4+ T cells and TCM CD8+ T cells in both treatment arms. However, this did not reach statistical significance (Fig. 2c, d). After polyclonal stimulation no significant changes in the numbers of TCM CD4+ and CD8+ T cells were observed after stimulation at baseline and after 6 weeks in both treatment arms (supplementary Fig. 1b and 1c). No specific changes were found in the number of TEM CD4+ and CD8+ T cells (supplementary Fig. 1d and 1e).
Axitinib significantly increases the expression of LAG3 on CD4+ T cells and reduces the expression of TIM3 on CD4+ and CD8+ T cells
Next, we investigated the expression of PD-1, LAG3 and TIM3 on CD4+ and CD8+ T cells before and after in vitro polyclonal stimulation. Axitinib significantly increased the LAG3 expression on CD4+ T cells (5.02 vs. 6.82 %, p = 0.039) and nonsignificantly on CD8+ T cells (1.4 vs. 2.2 %). This increased LAG3 expression was not observed in the combination arm. In both treatment arms, a LAG3 upregulation on CD4+ and CD8+ T cells was observed after stimulation. The LAG3 upregulation on CD8+ T cells was significantly higher after 6 weeks of axitinib treatment as compared to baseline (resp., 2.2 vs. 9.9 %, p = 0.023) (Fig. 3a, b, left panel).
Fig. 3.

Axitinib treatment is associated with increased LAG3 expression on CD4+ T cells and reduced TIM3 expression on CD4+ and CD8+ T cells. Percentages of LAG3+ CD4+ T cells (left panel), TIM3+ CD4+ T cells (middle panel) and PD-1+ CD4+ T cells (right panel) as determined in the absence or presence of anti-CD3/CD28 beads at baseline and at 6 weeks after axitinib (n = 9) (upper panel) or axitinib plus lomustine treatment (n = 6) (lower panel). a Percentages of LAG3+ CD8+ T cells (left panel), TIM3+ CD8+ T cells (middle panel) and PD-1+ CD8+ T cells (right panel) as determined in the absence or presence of anti-CD3/CD28 beads at baseline and at 6 weeks after axitinib (n = 9) (upper panel) or axitinib plus lomustine treatment (n = 6) (lower panel) (b)
Next we found that axitinib reduced TIM3 expression on CD4+ and CD8+ T cells (resp., 1.2 vs. 0.7 % and 2.5 vs. 1.8). In the combination arm, a nonsignificant increase of TIM3 expression was observed on CD8+ T cells after 6 weeks of treatment (0.9 vs. 1.5 %). In both treatment arms, TIM3 upregulation was apparent on CD4+ and CD8+ T cells after stimulation. In the combination arm, a significantly higher TIM3 expression was observed on CD4+ T cells at 6 weeks as compared to baseline (resp., 2.4 vs. 4.55 %, p = 0.031) (Fig. 3a, b, middle panel).
In the absence of polyclonal stimulation, no specific changes were observed for the PD-1 expression. However, after stimulation, we found that the PD-1 upregulation on CD8+ T cells was lower after stimulation at 6 weeks as compared to baseline after axitinib treatment (resp., 3.3 vs. 1.5 %, p = 0.055). The PD-1 upregulation on CD4+ T cells was absent (Fig. 3a, b, right panel). No significant differences were observed in the co-expression of these inhibitory molecules (supplementary Fig. 2).
Axitinib treatment leads to enhanced production of IFN-γ, TNF-α and IL-2
Since we found some differences in the T cell subsets and in the expression of inhibitory molecules, we further analyzed the supernatants for the presence of different cytokines before and after 24 h of stimulation. At baseline, almost no production of cytokines was observed after stimulation. However, in both treatment arms we observed an increased IFN-γ, IL-2 and TNF-α secretion after 6 weeks of treatment. In the axitinib arm this increase was significant for IFN-γ (p = 0.031) and IL-2 (p = 0.031) (Fig. 2e, f).
Progressive disease is associated with a significant increase of Treg numbers
We further investigated whether we could find differences between patient groups with a different disease status independent of the treatment arm. We analyzed the different immune cell populations at baseline and at 6 weeks of treatment. A significant increase in the number of Treg in patients with PD was observed after 6 weeks of treatment (2.2 vs. 28 %, p = 0.0041) (Fig. 4a). In both patient populations a nonsignificant reduction of moMDSC was found after 6 weeks of treatment. However, it was more pronounced in patients with SD/PR (48.8 vs. 27.30 % and 20.50 vs. 12.50 %) (Fig. 4b). We did not find specific differences in the other immune cell populations (data not shown).
Fig. 4.
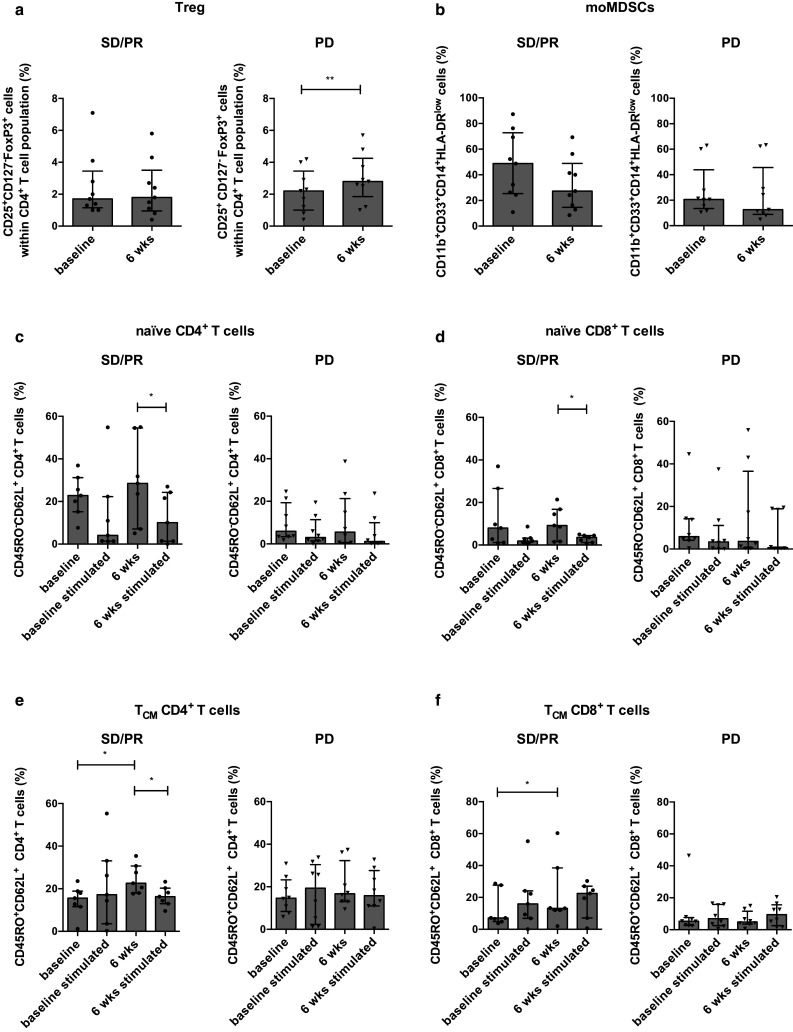
Different T cell subsets according to antitumor response independent of treatment. Percentages of Treg (CD4+ CD25+ CD127− FoxP3+ T cells) (a) and moMDSCs (CD11b+ CD33+ CD14+ HLA-DRlow cells) (b) determined at baseline and at 6 weeks after treatment for patients with SD/PR (filled circle; n = 9) or PD (inverted triangle; n = 9). Percentages of naïve CD4+ T cells (c) and naïve CD8+ T cells (d) subsets determined in the absence and presence of anti-CD3/CD28 beads at baseline and at 6 weeks for patients with SD/PR (filled circle; n = 7) or PD (inverted triangle; n = 8). Percentages of TCM CD4+ T cells (e) and TCM CD8+ T cells (f) determined in the absence and presence of anti-CD3/CD28 beads at baseline and at 6 weeks for patients with SD/PR (filled circle; n = 7) or PD (inverted triangle; n = 8)
Partial response or stable disease is associated with a significant increase of central memory CD4+ and CD8+ T cells
Next we investigated whether there were alterations in the different T cell subsets at baseline and after 6 weeks of treatment in both patient populations. We first studied the naïve T cell numbers and found no significant differences in the naïve T cell numbers at baseline and at 6 weeks. However, in the patients with SD/PR the number of naïve CD4+ and CD8+ T cells was significantly reduced after stimulation at 6 weeks of treatment (resp., 28.50 vs. 10.10 %, p = 0.016 and 9.2 vs. 3.6 %, p = 0.047). In contrast, no changes in the numbers of naïve CD4+ and CD8+ T cells were observed after stimulation for the patients with PD (Fig. 4c, d). Next we investigated the numbers of TCM. We found that patients with SD/PR had a significant increase in the number of TCM CD4+ and CD8+ T cells after 6 weeks of treatment (resp., 15.60 vs. 22.60 %, p = 0.016 and 7.10 vs. 13.20 %, p = 0.031). After stimulation, patients with SD/PR had a significant reduction of the TCM CD4+ T cells (22.60 vs. 16.30 %, p = 0.047) and a further increase of the TCM CD8+ T cells was observed after stimulation at 6 weeks (13.20 vs. 22.50 % %) (Fig. 4e, f). No specific differences were found in the TEM CD4+ or CD8+ T cells.
Progressive disease is associated with PD-1 upregulation on CD4+ and CD8+ T cells
We further investigated the expression of different inhibitory molecules at baseline and at 6 weeks in both patient populations. A nonsignificant PD-1 upregulation on CD4+ and CD8+ T cells was observed in patients with PD (resp., 1.45 vs. 3.2 % and 0.9 vs. 1.6 %), and in patients with SD/PR, a nonsignificant PD-1 downregulation on CD8+ T cells was observed (1.8 vs. 0.4 %) (Fig. 5a).
Fig. 5.

Progressive disease is associated with an upregulation of PD-1 and with dysfunctional T cells. Percentages of PD1+ CD4+ T cells and PD1+ CD8+ T cells (a), LAG3+ CD4+ and LAG3+ CD8+ T cells (b) and TIM3+ CD4+ and TIM3+ CD8+ T cells (c) determined in the absence or presence of anti-CD3/CD28 beads at baseline and at 6 weeks for patients with SD/PR (filled circle; n = 7) or PD (inverted triangle; n = 8). IFN-γ, TNF-α, IL-2 and IL-10 were quantified in the supernatants in the absence or presence of anti-CD3/CD28 beads at baseline and at 6 weeks for patients with SD/PR (filled circle; n = 7) or PD (inverted triangle; n = 8) (d)
No significant differences in LAG3, TIM3, or co-expression of these molecules at baseline and at 6 weeks were observed (resp., Fig. 5b, c and data not shown). However, in patients with SD/PR a significant LAG3 upregulation on CD4+ and CD8+ T cells after stimulation at baseline (resp., 2.7 vs. 4.7 %, p = 0.0469 and 0.9 vs. 11.5 %, p = 0.0156) and at 6 weeks was found (resp., 2.8 vs. 9.8 % p = 0.0156 and 1.9 vs. 15.4 %, p = 0.0156). In the patients with PD the LAG upregulation was almost absent after stimulation at 6 weeks (Fig. 5b).
Although the differences were more pronounced in the patients with SD/PR, TIM3 expression changed to a comparable extent in both patient populations (Fig. 5c).
Stable disease or partial response is associated with higher cytokine secretion
Since no reduction in the number of naïve CD4+ and CD8+ T cells was observed upon stimulation in patients with PD, we hypothesized that higher numbers of less functional, more exhausted T cells were present in these patients. Therefore, the cytokine secretion after stimulation was investigated. In the supernatants of patients with SD/PR we found a significant increase in IFN-γ (p = 0.0156), TNF-α (p = 0.0313) and IL-2 (p = 0.0313) secretion after stimulation at 6 weeks. Moreover, significantly increased TNF-α (p = 0.0313) and IL-2 (p = 0.0156) levels were also present at baseline. We also found a nonsignificant increase in IL-10 production after stimulation at 6 weeks. The cytokine production was below the limit of detection in the majority patients with PD (Fig. 5d).
Expression of inhibitory molecules LAG3, TIM3 and PD-1 on CD4+ and CD8+ T cells is associated with reduction of cytokine production
Since LAG3 and TIM3 are upregulated after stimulation and since these molecules are associated with T cell exhaustion, we investigated whether the LAG3- or TIM3-expressing CD4+ and CD8+ T cells are still capable of cytokine production. Through simultaneous intracellular cytokine staining, we found that CD4+ T cells and CD8+ T cells expressing LAG3, TIM3 or PD-1 produced almost no IFN-γ, TNF-α or IL-2 (Fig. 6a, b).
Fig. 6.
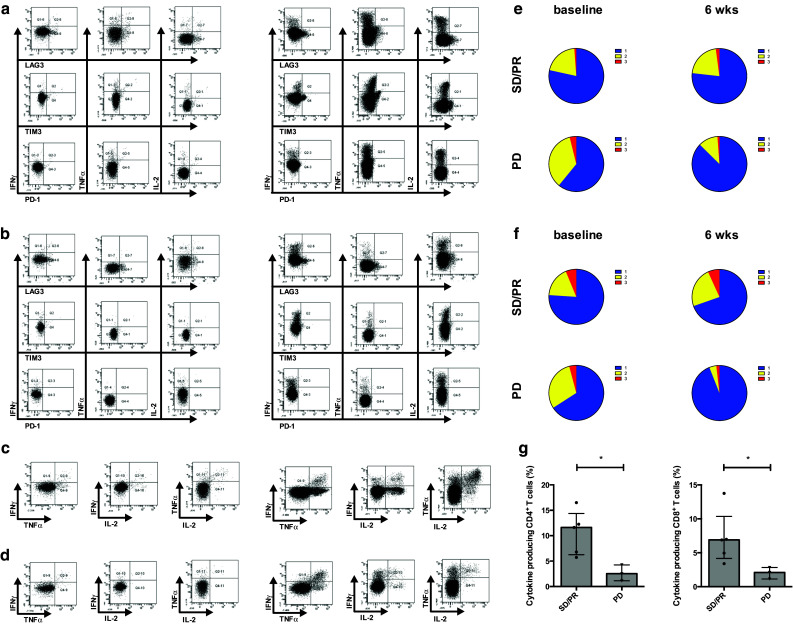
Expression of inhibitory molecules on CD4+ and CD8+ T cells and progressive disease is associated with reduced production of cytokines. Representative flow cytometry profiles showing LAG3, TIM3 or PD-1 expression profiles on CD4+ T cells (a) and CD8+ T cells (b) simultaneous with ICS for IFN-γ, TNF-α and IL-2 and representative flow cytometry profiles of the production of IFN-γ, TNF-α and IL-2 on CD4+ T cells (c) and CD8+ T cells (d) of patient 16 (axitinib, SD) after stimulation with anti-CD3/CD28 beads at baseline (left panel) and at 6 weeks (right panel) post-treatment. Pie charts showing the proportion of CD4+ T cells (e) and CD8+ T cells (f) displaying 1–3 functions determined after polyclonal stimulation at baseline and at 6 weeks after treatment for patients with SD/PR (upper panel; n = 5) or PD (lower panel; n = 3). Percentages of total cytokine-producing CD4+ and CD8+ T cells as determined at 6 weeks comparing patients with SD/PR (filled circle; n = 5) and patients with PD (inverted triangle; n = 3) (g)
We further analyzed the T cell functionality by determining the proportion of cells producing multiple cytokines (Fig. 6c, d). An increased number of bi- and polyfunctional CD4+ and CD8+ T cells [resp., 1.22 vs. 1.90 % and 0.99 vs. 1.52 % (2 cytokines) and 0.07 vs. 0.19 % and 0.35 vs. 0.44 % (3 cytokines)] and a higher number of cytokine-producing CD4+ and CD8+ T cells (resp., 5.98 vs. 8.99 % and 5.59 vs. 6.49 %) were observed in patients with SD/PR after 6 weeks of treatment (Fig. 6e, f upper panel). In contrast, patients with PD had a strong reduction of the number of bi- and polyfunctional CD4+ and CD8+ T cells [resp., 2.17 vs. 0.29 % and 1.65 vs. 0.09 % (2 cytokines) and 0.24 vs. 0.03 % and 0.23 vs. 0.04 % (3 cytokines)] and a lower number of cytokine-producing CD4+ and CD8+ T cells (resp., 6.17 vs. 2.53 % and 5.52 vs. 2.17 %) after 6 weeks of treatment (Fig. 6e, f, lower panel). Furthermore, a significantly lower number of cytokine-producing CD4+ and CD8+ T cells was observed at 6 weeks after treatment in patients with PD as compared to patients with SD/PR (Fig. 6g).
Discussion
In this study we show that PD in patients with recurrent GBM treated with axitinib or axitinib plus lomustine is associated with increased Treg numbers, PD-1 upregulation on CD4+ and CD8+ T cells and reduced T cell functionality. In addition, we found that treatment with axitinib leads to increased numbers of naïve and TCM CD8+ T cells and TCM CD4+ T cells.
Whereas the effects of different small molecules on immune cells have been studied extensively in preclinical studies, to our knowledge this is the first time that the effect of axitinib on the immune cells of patients was investigated [12, 15–17].
In our patient population axitinib did not influence the different immune cell populations in peripheral blood. However, within the T cell population we observed an increased number of naïve CD8+ T cells and TCM CD4+ and CD8+ T cells. In addition, we found an increased production of IFN-γ, TNF-α and IL-2 by T cells after 6 weeks of treatment. Previously, it was shown that following adoptive cell transfer, TCM CD8+ T cells have a better antitumor efficacy compared to TEM [18, 19]. Furthermore, in patients with SD/PR the number of TCM CD4+ and CD8+ T cells was significantly increased. Moreover, when we investigated the expression of the inhibitory molecules we observed that axitinib reduced the TIM3 expression on CD4+ and CD8+ T cells. In melanoma patients it has been shown that upregulation of TIM3 and PD-1 is associated with dysfunction of antigen-specific CD8+ T cells and that the expression of these molecules plays a role in regulating the expansion of vaccine-induced CD8+ T cells [20, 21]. Thus, the reduced TIM3 expression combined with the increased numbers of naïve and TCM CD8+ T cells and TCM CD4+ T cells suggests that axitinib could lead to better antitumor immune responses in patients with recurrent glioblastoma. The addition of lomustine to the axitinib treatment reduced the number of CD8+ T cells and diminished the effect of axitinib to increase the number of naïve CD8+ T cells. Lomustine is known to cause lymphopenia [22]; consequently, these results were not unexpected and highlight its probable interference with an antitumor immune response.
However, we also observed a higher LAG3 expression on CD4+ T cells after axitinib treatment. LAG3 is upregulated on exhausted T cells, and blockade of LAG3 can enhance antitumor T cell responses [23, 24]. This is in contrast with our previously suggested hypothesis that axitinib has beneficial effects on the immune system. An explanation of this finding could be that the majority of the patients treated with axitinib were progressive. Although we did not find differences in LAG3 expression between patients with SD/PR and those with PD after 6 weeks of treatment, we found that patients with PD lacked LAG3 upregulation after stimulation. In normal conditions, upon T cell activation LAG3 expression is detectable approximately 24 h post-activation and peaks on day 3 or day 4 after stimulation [24]. This suggests that patients with PD possess T cells that are less responsive to polyclonal stimulation.
In patients with PD we also found a PD-1 upregulation. It has already been shown that PD-1 expression on CD4+ and CD8+ T cells in the peripheral blood is higher in patients with high-grade glioma as compared to patients with low-grade glioma [25]. Our results show that PD-1 expression is further increased on CD4+ and CD8+ T cells of patients who are progressive during treatment with axitinib or axitinib plus lomustine. Furthermore, we observed that CD4+ and CD8+ T cells expressing PD-1, LAG3 or TIM3 had a reduced cytokine production. Additionally, we found that patients with PD had a strong reduction of T cell functionality and of the total number of cytokine-producing CD4+ and CD8+ T cells. Moreover, they had a significantly lower proportion of cytokine-producing T cells as compared to patients with SD/PR. Together with the increased numbers of naïve CD4+ and CD8+ T cells, the lack of LAG3 upregulation after stimulation and the increased PD-1 expression, these results suggest that PD is associated with T cell exhaustion. Treatment with axitinib did not affect the PD-1 expression on CD4+ T cells and non-stimulated CD8+ T cells. After stimulation we found a higher PD-1 expression on CD8+ T cells at baseline and at 6 weeks of axitinib treatment. This effect was completely abrogated when axitinib was combined with lomustine. Since PD-1 is upregulated after T cell activation, this suggests that lomustine negatively affects the CD8+ T cell activation. The effect of TKIs on the expression of inhibitory molecules has not been studied extensively. In preclinical models it has been shown that sunitinib reduces the expression of PD-1 and CTLA-4 on Treg and CD8+ T cells, respectively. However, sunitinib did not affect the PD-1 expression on CD4+ and CD8+ T cells. Sorafenib also reduced the expression of CTLA-4 on Treg in a preclinical model for HCC [26, 27]. In patients with renal cell carcinoma, Guislain et al. found that sunitinib improves the expansion of TILs and found that sunitinib-pretreated patients had a higher PD-1 expression on expanded CD8+ TILs [28]. To our knowledge, this is the first time that the effect of TKI treatment on LAG3 and TIM3 expression has been examined.
In this study, we also observed an increased number of Treg in patients with PD. It has been shown that increased proportions of Treg are present in the tumor and peripheral blood of glioblastoma patients. Moreover, removal of Treg from the peripheral blood of patients leads to restoration of the T cell function. However, it was shown that in contrast to other cancers [29], Treg are not predictors for patient survival in glioma [30–32]. Thus, in this study we show that patients with recurrent glioblastoma that are resistant to antiangiogenic treatment display a further enhancement of a preexisting immunosuppression.
Recently, tumor regression and long-term survival in a syngeneic intracranial mouse glioma model were obtained when inhibiting CTLA-4, PD-L1 and indoleamine 2,3-dioxygenase [33]. These results suggest that modulation of the immunosuppression to obtain an antitumor immune response in glioblastoma is optimal when several pathways are simultaneously inhibited. Several research groups have shown that antiangiogenic treatments have beneficial effects on the immune system [11, 34]. Moreover, antiangiogenic treatments are known to reduce peritumoral edema, reducing the use of immunosuppressive corticosteroids in glioblastoma patients [3, 35, 36].
Conclusion
In this study we investigated the effects of axitinib on the circulating immune cells. Although our results did not always reach statistical significance, probably due to the limited number of patients, they suggest that combining axitinib with immunotherapy in glioblastoma patients could have synergistic effects.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We would like to acknowledge the patients who consented to participate in this study, their families, and Pfizer Belgium for the provision of axitinib and a research grant for conducting the clinical trial. We would also like to thank the data manager Katrien Van den Bossche and Kathleen Mooren from the University Hospital Brussels (Universitair Ziekenhuis Brussel, UZ Brussel) for their help with the data collection, and Ludwig Van den Hove, PhD, Pfizer Belgium, for his support and critical review of the manuscript. Sarah K Maenhout and Stephanie Du Four are funded by a PhD grant from the Agency for Innovation by Science and Technology in Flanders (IWT). Brenda De Keersmaecker is funded by a research grant Emmanuel van der Schueren from the Flemish League against Cancer (Vlaamse Liga Tegen Kanker, VLK). This work is supported by a grant from the Research Foundation Flanders [Fonds voor Wetenschappelijk Onderzoek (FWO, G023411 N)] to Kris Thielemans and Joeri L Aerts. The FACSAria III cell sorter and the LSR Fortessa were purchased with support from the Hercules Foundation to Kris Thielemans and Joeri L Aerts (Grant UABR/09/002) and the Foundation against Cancer (Stichting Tegen Kanker), respectively. The clinical study was supported by a research grant from Pfizer.
Abbreviations
- 6mPFS
6-Month progression-free survival
- CR
Complete response
- EGFRvIII
Endothelial growth factor receptor variant III
- GBM
Glioblastoma
- LAG3
Lymphocyte-activation gene 3
- ORR
Objective response rate
- PD
Progressive disease
- PDGFR
Platelet-derived growth factor receptor
- PR
Partial response
- SD
Stable disease
- TCM
Central memory T cells
- TEM
Effector memory T cells
- TIM3
T cell immunoglobulin domain and mucin domain 3
- TKI
Tyrosine kinase inhibitor
- Treg
Regulatory T cells
- VEGF
Vascular endothelial growth factor
- VEGFR
Vascular endothelial growth factor receptor
Compliance with ethical standards
Conflict of interest
None.
References
- 1.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Lamborn KR, Chang SM, Prados MD. Prognostic factors for survival of patients with glioblastoma: recursive partitioning analysis. Neuro Oncol. 2004;6:227–235. doi: 10.1215/S1152851703000620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 4.Stupp R, Hegi ME, Gorlia T, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:1100–1108. doi: 10.1016/S1470-2045(14)70379-1. [DOI] [PubMed] [Google Scholar]
- 5.Hutterer M, Nowosielski M, Haybaeck J, et al. A single-arm phase II Austrian/German multicenter trial on continuous daily sunitinib in primary glioblastoma at first recurrence (SURGE 01-07) Neuro Oncol. 2014;16:92–102. doi: 10.1093/neuonc/not161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee EQ, Kuhn J, Lamborn KR, et al. Phase I/II study of sorafenib in combination with temsirolimus for recurrent glioblastoma or gliosarcoma: North American Brain Tumor Consortium study 05-02. Neuro Oncol. 2012;14:1511–1518. doi: 10.1093/neuonc/nos264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Batchelor TT, Mulholland P, Neyns B, et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol. 2013;31:3212–3218. doi: 10.1200/JCO.2012.47.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neyns B, Duerinck J, Du Four S, et al. Randomized phase II study of axitinib versus standard of care in patients with recurrent glioblastoma. ASCO Meet Abstr. 2014;32:2018. [Google Scholar]
- 9.Duerinck J, Du Four S, Bouttens F, Neyns B (2016) Randomized phase II study of axitinib versus physicians best alternative choice of therapy in patients with recurrent glioblastoma. J Neurooncol. doi:10.1007/s11060-016-2092-2 [DOI] [PubMed]
- 10.Taal W, Oosterkamp HM, Walenkamp AME, et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a randomised controlled phase 2 trial. Lancet Oncol. 2014;15:943–953. doi: 10.1016/S1470-2045(14)70314-6. [DOI] [PubMed] [Google Scholar]
- 11.Huang Y, Goel S, Duda DG, et al. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013;73:2943–2948. doi: 10.1158/0008-5472.CAN-12-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du Four S, Maenhout SK, De Pierre K, et al. Axitinib increases the infiltration of immune cells and reduces the suppressive capacity of monocytic MDSCs in an intracranial mouse melanoma model. Oncoimmunology. 2015;4:e998107. doi: 10.1080/2162402X.2014.998107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson CM, Lim M, Drake CG. Immunotherapy for brain cancer: recent progress and future promise. Clin Cancer Res. 2014;20:3651–3659. doi: 10.1158/1078-0432.CCR-13-2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reardon DA, Schuster J, Tran DD, et al. ReACT: overall survival from a randomized phase II study of rindopepimut (CDX-110) plus bevacizumab in relapsed glioblastoma. ASCO Meet Abstr. 2015;33:2009. [Google Scholar]
- 15.Chung AS, Wu X, Zhuang G, et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat Med. 2013;19:1114–1123. doi: 10.1038/nm.3291. [DOI] [PubMed] [Google Scholar]
- 16.Doloff JC, Waxman DJ. VEGF receptor inhibitors block the ability of metronomically dosed cyclophosphamide to activate innate immunity-induced tumor regression. Cancer Res. 2012;72:1103–1115. doi: 10.1158/0008-5472.CAN-11-3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stehle F, Schulz K, Fahldieck C, et al. Reduced immunosuppressive properties of axitinib in comparison with other tyrosine kinase inhibitors. J Biol Chem. 2013;288:16334–16347. doi: 10.1074/jbc.M112.437962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gattinoni L, Klebanoff CA, Palmer DC, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8 + T cells. J Clin Invest. 2005;115:1616–1626. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klebanoff Christopher, Gattoni Luca, Restifo N. Sorting through subsets: which T cell populations mediate highly effective adoptive immunotherapy? J Immunother. 2012;35:651–660. doi: 10.1097/CJI.0b013e31827806e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fourcade J, Sun Z, Pagliano O, et al. CD8 + T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res. 2012;72:887–896. doi: 10.1158/0008-5472.CAN-11-2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fourcade J, Sun Z, Pagliano O, et al. PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8 + T cells induced by melanoma vaccines. Cancer Res. 2014;74:1045–1055. doi: 10.1158/0008-5472.CAN-13-2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clary S, Nagarkatti PS, Nagarkatti M. Immunomodulatory effects of nitrosoureas on the phenotype and functions of T cells in the thymus and periphery. Immunopharmacology. 1990;20:153–164. doi: 10.1016/0162-3109(90)90029-E. [DOI] [PubMed] [Google Scholar]
- 23.Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3—potential mechanisms of action. Nat Rev Immunol. 2015;15:45–56. doi: 10.1038/nri3790. [DOI] [PubMed] [Google Scholar]
- 24.Goldberg MV, Drake CG. LAG-3 in cancer immunotherapy. Curr Top Microbiol Immunol. 2011;344:269–278. doi: 10.1007/82_2010_114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei B, Wang L, Zhao X, et al. The upregulation of programmed death 1 on peripheral blood T cells of glioma is correlated with disease progression. Tumor Biol. 2014;35:2923–2929. doi: 10.1007/s13277-013-1376-9. [DOI] [PubMed] [Google Scholar]
- 26.Ozao-Choy J, Ma G, Kao J, et al. The novel role of Tyrosine Kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69:2514–2522. doi: 10.1158/0008-5472.CAN-08-4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen ML, Yan BS, Lu WC, et al. Sorafenib relieves cell-intrinsic and cell-extrinsic inhibitions of effector T cells in tumor microenvironment to augment antitumor immunity. Int J Cancer. 2014;134:319–331. doi: 10.1002/ijc.28362. [DOI] [PubMed] [Google Scholar]
- 28.Guislain A, Gadiot J, Kaiser A, et al. Sunitinib pretreatment improves tumor—infiltrating lymphocyte expansion by reduction in intratumoral content of myeloid—derived suppressor cells in human renal cell carcinoma. Cancer Immunol Immunother. 2015;64:1241–1250. doi: 10.1007/s00262-015-1735-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 30.Choi BD, Fecci PE, Sampson JH. Regulatory T cells move in when gliomas say “I DO”. Clin Cancer Res. 2012;18:6086–6088. doi: 10.1158/1078-0432.CCR-12-2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fecci PE, Mitchell DA, Whitesides JF, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66:3294–3302. doi: 10.1158/0008-5472.CAN-05-3773. [DOI] [PubMed] [Google Scholar]
- 32.Thomas AA, Fisher JL, Rahme GJ, et al. Regulatory T cells are not a strong predictor of survival for patients with glioblastoma. Neuro Oncol. 2015;17:801–809. doi: 10.1093/neuonc/nou363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wainwright DA, Chang AL, Dey M, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. 2014;20:5290–5301. doi: 10.1158/1078-0432.CCR-14-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Motz GT, Santoro SP, Wang L-P, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014;20:607–615. doi: 10.1038/nm.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Batchelor TT, Reardon DA, de Groot JF, et al. Antiangiogenic therapy for glioblastoma: current status and future prospects. Clin Cancer Res. 2014;20:5612–5619. doi: 10.1158/1078-0432.CCR-14-0834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Franchimont D. Overview of the actions of glucocorticoids on the immune response: a good model to characterize new pathways of immunosuppression for new treatment strategies. Ann N Y Acad Sci. 2004;1024:124–137. doi: 10.1196/annals.1321.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.