

Abstract
Assessment of antigen-specific T-cell responses has been greatly facilitated by development of ELISPOT and intracellular cytokine flow cytometry (CFC) assays. The use of autologous antigen presenting cells transfected with in vitro transcribed RNA as stimulators allows in principle quantification of antigen-specific T-cells independent of the knowledge of the epitopes. We describe here a cytokine secretion assay that enables simultaneous assessment of both antigen-specific CD4+ as well as CD8+ T-cells directly from clinical samples without the need for generation of dendritic cells. To this aim, bulk PBMCs were electroporated with RNA encoding the antigen fused to trafficking signal sequences derived from a MHC class I molecule and used as stimulators. With human cytomegalovirus (HCMV) phosphoprotein 65 (pp65) as antigen we show that for measuring ex vivo T-cell responses in ELISPOT and CFC such stimulators are superior or at least equivalent to a pool of overlapping peptides representing the entire pp65 sequence as well as to untagged pp65 encoding RNA. This approach avoids the time consuming generation of dendritic cells as immune stimulators and, in particular when used in the context of the CFC, is robust, broadly applicable and fast.
Keywords: Immune monitoring, IVT-RNA, CFC
Introduction
Advancements in genomics and expression cloning technologies have boosted the discovery of potential target molecules for T-cell responses and thus have promoted a constantly growing number of immunotherapy trials in patients with cancer as well as infectious diseases. Using clinical endpoints for proving significant anti-cancer or anti-pathogen effects, such trials require enormous resources and only very few candidate antigens can be fully evaluated. Alternatively, studies based on surrogate parameters such as assessment of antigen-specific T-cell responses have been viewed as a way to prioritize agents for further clinical evaluation [1, 2].
Among the different methods for the enumeration of spontaneous as well as therapeutically induced antigen-specific T-cells, CFC, and ELISPOT assays have gained popularity as rapid, technically straightforward and highly sensitive stimulus-response methods for the detection of cytokine production [3–6].
For the detection of low-level responses, or definition of responses as positive or negative, the ELISPOT assay may be preferable [7, 8]. The CFC however, has a greater dynamic range and allows for phenotypic discrimination of responding cells using multiparameter flow cytometry [7–9]. Moreover, stimulation can be performed in whole blood, which facilitates application on clinical specimens [10, 11].
In both the CFC and ELISPOT assays a variety of approaches for stimulation have been evolving, including different types of cells such as dendritic cells, monocytes and PBMCs used as antigen presentors and a variety of antigen formulations including protein, single synthetic peptides or overlapping peptide pools, DNA or viral vectors [9, 12, 13].
Recently, it has been shown, that 5′ capped poly-adenylated RNA obtained by in vitro transcription (IVT) encoding an antigen of interest and introduced into autologous dendritic cells [14] or monocytes [15] can be successfully exploited for ex vivo detection and quantification of antigen-specific CD8+ T-cells [16, 17]. We were attracted by the possibility to measure T-cell responses against whole antigens using RNA and asked the question how this format can be exploited for development of a blood test, which is sufficiently versatile and convenient for clinical use, sensitive enough for ex vivo measurements, and allows quantification of both CD4+ and CD8+ T-cells. We assumed that overcoming two major technical challenges would be critical for meeting this objective.
First, the time of culture required for in vitro generation of dendritic cells as antigen presenters (APC) limits their utility for a fast blood test in a clinical setting. Peripheral mononuclear cells are much easier and faster to obtain but have a poorer immunostimulatory capacity. Recently we described structural modifications of the 3′ untranslated region and polyA-tail, which by increasing the stability and translational efficacy of IVT RNA augment the density of epitope/MHC complexes substantially [18] and may thus empower non-professional APCs.
Second, reports were controversial with regard to detection of CD4+ T-cell responses with IVT RNA in particular when using non-professional APCs [16, 17]. This has motivated linking of the antigens to trafficking sequences of molecules, which reside in MHC class II pathway, such as invariant chain protein and lysosome-associated membrane protein LAMP-1 [19–21]. We have recently observed that a N-terminal leader peptide together with an MHC class I trafficking signal attached to the C-terminus of an antigen dramatically improves the presentation of MHC class I and class II epitopes and leads to an almost 100-fold increase in stimulatory capacity (S. Kreiter et al., manuscript in preparation).
In the present study we report the suitability of IVT RNA combining both types of genetic modifications for efficient, versatile and simultaneous ex vivo quantification of CD4+ and CD8+ T-cells by CFC as well as ELISPOT assays. As model antigen for our studies we chose HCMV pp65, the immunodominant antigen of human cytomegalovirus, which bears multiple MHC class I and class II restricted epitopes and is frequently used for the validation of the sensitivity and specificity of T-cell immunoassays [9, 22, 23].
Materials and methods
Samples
Samples were collected from healthy HCMV seropositive or seronegative blood donors. PBMCs were isolated from whole blood by Ficoll-Hypaque (Amersham Biosciences, Uppsala, Sweden) density gradient centrifugation.
Peptides
A peptide pool (138 different peptides) consisting of 15 amino acid long peptides with 11 amino acid overlaps corresponding to HCMV pp65 sequence was used. A pool of overlapping peptides derived from the Wilms-Tumor antigen-1 (WT1) served as negative control. The peptides were synthesized by standard solid phase chemistry with free N and C termini by a commercial provider (Jerini, Berlin, Germany). Peptides were dissolved in DMSO to a final concentration of 1.0 mg/ml and kept at −70°C. Final concentrations of 1.75 μg/ml of each peptide were used in the assay. The HCMV pp65 peptide 495-503 (NLVPMVATV) [24] and a control peptide (KASEKIFYV) with no reported immunogenicity and derived from the human tumor-associated antigen SSX1 [25] were reconstituted in PBS 10% DMSO.
Vectors for in vitro transcription
All vectors used were variants of the pST1-A120 plasmid, which we engineered from the pCMV-Script vector (Stratagene, LaJolla, USA) by introducing a T7 promoter, a 120 bp poly(A) tail and the neomycin-resistance gene [see also Holtkamp et al. (2006) for a detailed description]. Starting from pST1-A120, we first inserted a HinDIII–PstI MHC class I signal peptide fragment amplified from activated PBMCs (primers: Sec-sense 5′-aag ctt agc ggc cgc acc atg cgg gtc acg gcg ccc cga acc-3′, Sec-antisense 5′-ctg cag gga gcc ggc cca ggt ctc ggt cag-3′) into the pST1-A120 vector. Moreover, a 3′ fragment of the MHC B transcript encoding the transmembrane and cytosolic domain together with the stop-codon flanked by BamHI and EcoRI sites was amplified from activated PBMCs via consensus primers (primers: MHCI-sense 5′-gga tcc atc gtg ggc att gtt gct ggc ctg gct-3′, MHCI-antisense 5′-gaa ttc agt ctc gag tca agc tgt gag aga cac atc aga gcc-3′) and inserted in frame behind the signal peptide. These modifications resulted in the MHCI vector backbone.
The genomic sequence of HCMV UL83 (pp65) was amplified from a lysate of HCMV-infected fibroblasts (BioWhitaker, Walkersville, USA) with and without stop-codon. BamHI sites were introduced by primers (primers: pp65-sense 5′-gga tcc acc atg gag tcg cgc ggt cgc cgt tgt ccc gaa atg-3′, pp65-No-Stop-antisense 5′-gga tcc acc tcg gtg ctt ttt ggg cgt cga ggc gat gc-3′, pp65-Stop-antisense 5′-gga tcc tca acc tcg gtg ctt ggg cgt cga ggc-3′). The BamHI-pp65-Stop fragment was cloned into the pST1-A120 to generate the pp65 vector. The BamHI-pp65-No-Stop fragment was inserted into the MHCI-vector backbone to generate pp65-MHCI.
As negative control NY-ESO-I cDNA flanked by BamHI was amplified from testis (primers NY-Eso-sense 5′-gga tcc gcc acc atg cag gcc gaa ggc cgg ggc aca-3′; NY-Eso-antisense 5′-gga tcc gcg cct ctg ccc tga ggg agg-3′) and cloned into the MHCI vector backbone. An IVT enhanced green fluorescence protein (eGFP) vector was generated by cloning BamHI-site-flanked eGFP-fragment amplified from peGFP-C1 vector (BD Biosciences, Heidelberg, Germany) into the pST1-A120 plasmid.
Generation of IVT RNA
pST1-A120 based plasmids were linearized behind the poly(A) tail, purified by phenol chloroform extraction and sodium acetate precipitation and used as templates for in vitro transcription (IVT) with Message-Machine Kit (Ambion, Austin, USA). The RNA concentration and quality were assessed by spectrophotometry and agarose/formaldehyde gel electrophoresis.
Generation of PBMCs, human monocytes and monocyte derived DCs as stimulator cells
Buffy coats were obtained from healthy blood bank donors. Monocytes were enriched from PBMCs with anti-CD14 microbeads (Miltenyi Biotec, Bergisch-Gladbach, Germany) according to the manufacturer’s instructions.
To obtain immature DCs, monocytes were differentiated in complete culture medium [RPMI 1640 with 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 1 mM sodium pyruvate, non-essential amino acids, 10% heat inactivated human AB-serum (all Invitrogen, Karlsruhe, Germany)] supplemented with 1,000 U/ml GM-CSF (Essex, Luzern, Switzerland) and 1,000 U/ml IL-4 (Strathmann Biotech, Hamburg, Germany). For generation of mature DCs, cells were harvested on day 5 and transferred into complete culture medium containing 500 U/ml IL-4, 800 U/ml GM-CSF, 10 ng/ml IL-1 β (Pharmingen, Hamburg, Germany), 10 ng/ml TNF-α (Sigma, Taufkirchen, Germany), 1,000 U/ml IL-6 (Strathmann Biotech) and 1 μg/ml PG-E2 (Sigma) for 1 day.
DCs were frozen in cryotubes at 1–5 × 106 DCs/tube in 1.8 ml of RPMI 1640 supplemented with 20% human AB-Serum and 10% DMSO and stored in liquid nitrogen until use. Prior to use, DCs were thawed in a 37°C water bath. Cold PBS was added, cells were pelleted in a precooled centrifuge (4°C) and resuspended in 5 ml of prewarmed culture medium.
Preparation of antigen loaded stimulator cells
Depending on the assay, PBMCs, purified monocytes or mature DCs were used as stimulators. Cells were washed twice, suspended in 250 μl X-VIVO-15 medium (Cambrex, Verviers, Belgium) and transferred into a 4 mm gap sterile electroporation cuvette (BioRad, Hercules, USA). After addition of 10 or 20 μg of IVT mRNA, cells were electroporated using a Gene-Pulser-II apparatus (Biorad) and electroporation conditions as specified in the text. Viability after electroporation was 89–96% for PBMCs, 84–95% for monocytes and 80–95% for DCs. For peptide pulsing, cells were diluted to a final concentration of 2–4 × 106 cells/ml in culture medium containing the peptide pool (each peptide 1.75 μg/ml) and were incubated for 3 h at 37°C. Cells were washed before being used as stimulators.
Enzyme-linked immunospot assay (ELISPOT)
Ninety-six-well microtiter plates (Millipore, Bedford, MA, USA) were coated overnight at 4°C with 0.1 μg/ml anti-IFN-γ antibody 1-D1k (Mabtech, Stockholm, Sweden). The antibody-coated plates were washed four times with PBS and blocked with 2% human albumin. Antigen presenting monocytes used as stimulator cells were plated in triplicates at a concentration of 4 × 104 target cells/well. CD4+ or CD8+ responder cells purified with magnetic beads (Miltenyi) were added at a concentration of 3–10 × 104 cells/well. The plates were incubated overnight (37°C, 5% CO2), washed four times with PBS 0.05% Tween 20, and incubated for 2 h with the anti IFN-γ biotinylated mAB 7-B6-1 (Mabtech) at a final concentration of 1 μg/ml at 37°C. After washing in PBS containing 0.05% Tween 20, avidin-bound horseradish peroxidase H (Vectastain Elite Kit; Vector Laboratories, Burlingame, USA) was added to the wells and incubated for 1 h at room temperature. The plates were washed once more with PBS containing 0.05% Tween 20 and developed with 3-amino-9-ethyl carbazole (Sigma). Spots were counted using a computer-based evaluation system and the KS-ELISPOT software version 4.4.35 (Carl Zeiss Vision, Eching, Germany).
Cytokine flow cytometry
For CFC PBMCs were either isolated by Ficoll-Hypaque density gradient centrifugation or obtained from whole blood by removal of red blood cells with hypotonic ammonium chloride (8.26 g NH4Cl, 0.86 g NaHCO3 and 0.037 g sodium EDTA in 1l water, pH 7.4). After peptide loading or RNA transfection 400 μl of cells diluted to a concentration of 1 × 107 cells/ml were incubated in a polypropylene tube for 6 h with anti-CD28 and anti-CD49d (BD Biosciences) at a final concentration of 1 μg/ml each for co-stimulation [9, 26]. Negative controls included cells incubated with a control peptide or cells transfected with control RNA.
Brefeldin A (Sigma) was added at a concentration of 10 μg/ml and the cells were incubated for an additional 5 h. Cells were washed, fixed for 5 min with 4% paraformaldehyde in PBS and thereafter permeabilized with FACS permeabilizing Solution (BD Biosciences). After an additional washing step, the cells were stained with fluorescein isothiocyanate (FITC)-conjugated anti-IFNγ, allophycocyanin (APC)-conjugated anti-CD4, R-phycoerythrin (PE)-conjugated anti-CD69 and peridinin chlorophyll protein (PerCP) conjugated anti-CD8 (all BD Biosciences) in a total volume of 50 μl for 30 min at room temperature. Following staining, the cells were washed with FACS buffer and resuspended in 400 μl 1% paraformaldehyde in PBS. Analysis was performed on a FACSCalibur (BD Biosciences) using CellQuest Pro software (BD Biosciences).
Results
Preparation of stimulators electroporated with IVT RNA
Whereas PBMCs transfected with antigen-encoding RNA induce proliferation and expansion of memory T-cells in vitro [15], they seem not to qualify as antigen presenters for quantitative ex vivo cytokine secretion assays [27]. Apparently, this low immunostimulatory capacity and inferior performance of bulk PBMCs or monocytes in ELISPOT and CFC can be easily overcome by higher densities of peptide/MHC complexes on the stimulators as achieved by pulsing with saturating amounts of synthetic peptides [28]. Accordingly, it appeared important to us to ensure efficient antigen delivery into these non-professional antigen presenters. Recently, we described structural modifications of the 3′-UTR and polyA-tail improving both surface presentation of epitopes and induction of T-cell responses by increasing the stability and translational efficiency of IVT RNA molecules in cells [18]. We used such genetically modified IVT RNA encoding eGFP and measured eGFP reporter protein by flow cytometry. In a series of experiments we tested different electroporation settings for PBMCs to be used as stimulators in CFC. We found 375 V and 150 μF to be optimal for both PBMCs obtained by Ficoll Hypaque gradient as well as leukocytes from whole blood after removal of erythrocytes by lysis. In typical experiments 70–80% of CD14+ monocytes within these bulk population were transfected (Fig. 1a). Other cell populations such as B- and T-cells were transfected as well (data not shown). For purified monocytes previously described electroporation conditions of 300 V and 150 μF [27] appeared to be optimal.
Fig. 1.

Efficient RNA transfer of IVT RNA into antigen presenting cells. a PBMCs obtained by Ficoll density centrifugation, whole blood leukocytes generated by erythrocyte lysis, and purified monocytes were electroporated with 20 μg eGFP RNA. Cells were cultured for 7 h and stained with anti CD14 antibody. Transfection efficiency was measured by flow cytometry. The numbers given refer to the transfection efficacy of the CD14+ population. This experiment is representative for six independent experiments. b PBMCs generated via Ficoll density centrifugation were electroporated with 20 μg eGFP RNA. The eGFP expression in the eGFP+ and CD14+ monocyte population was followed (black histogram) from 30 min to 24 h. PBMCs electroporated with control RNA were used as negative control (white histogram)
To assess the time kinetics of transgene expression, we quantified eGFP protein expression in monocytes at different time points after RNA transfer into PBMCs. Expression of protein was detectable as early as half an hour after electroporation. The mean fluorescence intensity of monocytes continuously increased until reaching a plateau 7 h after electroporation on which it remained during the observation period of 24 h (Fig. 1b). We concluded that measuring INF-γ secretion 6 h after initiation of stimulation as performed in standard CFC protocol may not ensure maximal density of surface MHC/peptide complexes. Since it has been reported that 12 h after antigen-transfer proliferative effects may start to confound IFN-γ secretion [8], we used monocytes for CFC 11 h after RNA transfer in all experiments described below.
Monocytes or bulk PBMCs pulsed with HCMV pp65 RNA allow ex vivo detection of specific CD8+ but not CD4+ T-cells
To assess whether IVT RNA can be used for antigen delivery to detect IFN-γ producing T-cells ex vivo, we analyzed immune responses of several HCMV-seropositive healthy donors. Purified CD8+ and CD4+ T lymphocytes were tested directly after isolation in ELISPOT assays for reactivity against autologous monocytes electroporated with IVT RNA encoding full length HCMV pp65 protein. IVT RNA encoding NY-ESO-I, for which detectable T-cell activity is absent in healthy donors, served as negative control. Stimulators pulsed with a pool of overlapping peptides spanning HCMV pp65 protein were used as a reference positive control to mimic the magnitude of IFN-γ production upon assumed availability of all potential MHC class I and II epitopes. For CFC analysis, bulk PBMCs were either electroporated with pp65 RNA or control RNA or they were pulsed with the peptide pool and reactive subsets of T-cells were visualized by co-staining with labeled anti-CD4, anti-CD8, anti-CD69 and anti-IFNγ-antibodies. We observed that for quantification of reactive CD8+ T-cells, RNA transfection was inferior to pulsing with the peptide pool in ELISPOT, whereas both formulations were comparably efficient in CFC for the majority of donors (Fig. 2a). The superior performance of the CFC is most likely attributable to its higher sensitivity for the detection of T-cells producing low amounts of interferon-γ as well as to the fact, that not only monocytes but also the lymphocyte subsets in the bulk PBMC population may serve as additional antigen presenters. In neither of these assays, however, robust detection of CD4+ T-cell responses with pp65 RNA electroporated APC was feasible (Fig. 2b). Reactive CD4+ T-cells were clearly measurable using stimulators pulsed with the pp65 peptide pool, indicating a lack of presentation of suitable amounts of pp65 HLA-class II epitopes in RNA transfected APC.
Fig. 2.

Inability of pp65 RNA to stimulate antigen-specific CD4+ lymphocytes in ex-vivo assays. PBMCs from healthy HCMV+ donors were tested in CFC for CD8+ (a) and CD4+ (b) reactivity against pp65 peptide pool (1.75 μg/ml; black bars), and pp65 RNA (10 μg each; gray bars). Each bar represents the percentage of IFN(secreting CD8+ or CD4+ lymphocytes after subtraction of background IFN(secretion against irrelevant control RNA encoding NY-ESO-I. CD8+ (a) and CD4+ (b) lymphocytes (1 × 105/well) from a healthy HCMV+ donor were stimulated in overnight IFNγ-ELISPOT with purified autologous monocytes (4 × 104/well) either transfected with 20 μg IVT mRNA or loaded with the HCMV pp65 peptide pool. Each bar represents the mean spot number of triplicates + SD. This result is representative for two independent experiments
Stimulators pulsed with HCMV pp65 fused to MHC class I transmembrane domain and cytoplasmic tail allow efficient ex vivo detection of specific T-cells of both subsets
As HCMV pp65 is a nuclear protein [29], we assumed that the inability of the APC transfected with pp65 RNA to stimulate CD4+ T-cells is most likely due to poor presentation of peptides on MHC class II molecules. We have recently shown that the combination of an N-terminal signal peptide with the cytoplasmic tail of MHC class I molecules as a C-terminal tag route antigens to compartments for peptide loading of MHC class II molecules. Moreover, we have observed that such fusion proteins exhibit improved antigen presentation efficiency also for MHC class I epitopes giving rise to increased densities of antigen derived peptide/MHC complexes and thus resulting in higher T-cell stimulatory capacity (S. Kreiter et al., manuscript in preparation). Based on these prior data, a plasmid construct for in vitro transcription was generated, encoding pp65 flanked N-terminally by a secretion signal and fused at the C-terminus to the transmembrane domain and cytoplasmic tail of a MHC class I molecule (pp65-MHCI; Fig. 3a). RNA encoding this pp65 fusion protein (pp65-MHCI) was electroporated into monocytes and compared to pp65 IVT RNA in an ELISPOT assay for its ability to induce IFN-γ secretion of sorted CD4+ or CD8+ T-cells obtained from a CMV seropositive donor. Background T-cell reactivity towards stimulators transfected with NY-ESO-I-MHCI RNA as irrelevant control was in the range of 2–6 spot-forming cells per 105 T lymphocytes regardless of which effector population was used. Remarkably, with pp65-MHCI as antigen formulation detection of antigen-specific CD4+ T-cells succeeded under the same ex vivo ELISPOT conditions as described above. Moreover, CD8+ T-cell reactivity measured with pp65-MHCI was more than threefold higher as compared to spot-induction by unmodified pp65 (Fig. 3b).
Fig. 3.
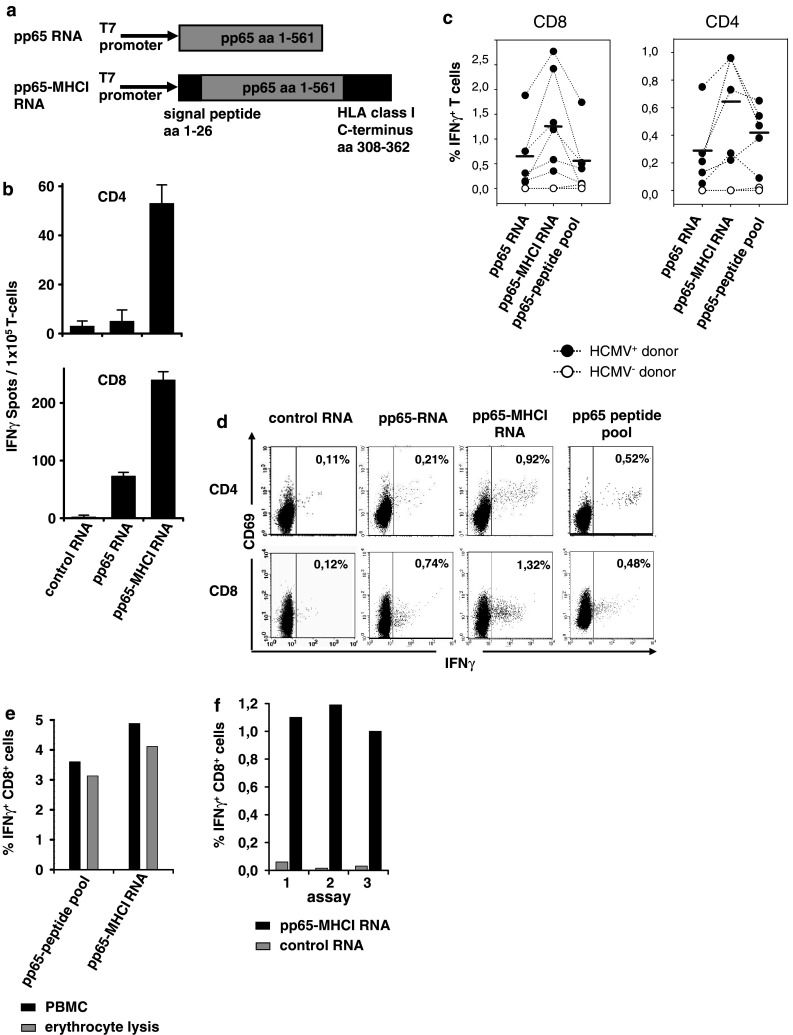
Simultaneous stimulation of CD4+ and CD8+ T-cells using IVT RNA of MHC fusion constructs. a Schematic diagram of the plasmid templates for in vitro transcription of RNA. b CD8+ and CD4+ T lymphocytes (1 × 105/well) from a healthy HCMV+ donor were stimulated in overnight IFNγ-ELISPOT with purified autologous monocytes (4 × 104/well) transfected with 20 μg IVT RNA. Each bar represents the mean spot number of triplicates + SD. This result is representative for three independent experiments. c The frequency of antigen-specific IFNγ secreting CD4+ and CD8+ T lymphocytes was measured in six healthy HCMV sero-positive (filled circles) and three HCMV sero-negative (open circles) donors by CFC. PBMCs were either loaded with pp65 peptide pool (1.75 μg/ml) or transfected with 10 μg pp65 or pp65-MHCI RNA. The background IFNγ reactivity against NY-ESO-I-MHCI RNA as irrelevant control was subtracted. Data points obtained with the PBMCs of the same donor are connected by a dotted line. The bar represents the mean frequency of antigen-specific T-cells. d Dot plots of a CFC from a representative donor are shown. The numbers represent the percentage of IFNγ secreting CD8+ or CD4+ T lymphocytes. e Frequencies of CD8+ IFNγ secreting lymphocytes were compared using either bulk populations obtained by Ficoll gradient purification or cells after erythrocyte lysis of whole blood. Cells were tested after electroporation with 10 μg of IVT RNA or loading with pp65 peptide pool (1.75 μg/ml). This result is representative for two independent experiments. f The frequency of CD8+ lymphocytes secreting IFNγ in PBMCs obtained from a healthy HCMV+ donor was measured in three independent runs
Analogously, we assessed induction of INF-γ secretion in CFC using PBMCs as stimulators. Six seropositive donors and three donors seronegative for HCMV infection were analyzed. Background IFN-γ secretion as measured for PBMCs electroporated with irrelevant control RNA was in the range of 0 to 0.1% of CD4+ and 0.01 to 0.16% of CD8+ T-cells.
For the majority of seropositive donors pp65-MHCI RNA was clearly superior to both pp65 RNA as well as peptide pool with regard to detection of CD4+ as well as CD8+ T-cells (Fig. 3c, with one representative example shown in Fig. 3d). Interestingly, the dynamic range of immune responses was highest when pp65-MHCI RNA was used for stimulation. T-cells from HCMV seronegative donors were clearly different in that only negligible numbers secreted IFN-γ.
CFC based protocols are attractive for clinical settings since they provide the option of fast processing directly from whole blood samples. As shown in Fig. 1a electroporation of IVT RNA into PBMCs obtained from whole blood by lysis of erythrocytes is efficient. Testing the utility of such stimulators for T-cell detection, we found that pp65-MHCI allows quantification of T-cells also from whole blood (Fig. 3e). It has been reported, that responses as measured by CFC are consistently higher in PBMCs than in whole blood [22], which is also supported by our data for antigens formulated as IVT RNA.
For the clinical setting, robustness and reproducibility of an assay are relevant. As exemplified by the CD8+ T-cell response of a defined donor measured in three independent runs by CFC, the use of IVT RNA does not impair reproducibility of this cytokine-based stimulus response assay (Fig. 3f).
Simultaneous detection of multiple T-cell epitopes using HCMV pp65-MHCI as antigen formulation
IVT RNA as antigen formulation encodes in principle all potential epitopes. To investigate whether the T-cell response measured by pp65-MHCI RNA electroporated stimulators is directed to multiple epitopes, we compared it to the reactivity of HLA A*0201 positive donors against the immunodominant HLA A*0201 restricted peptide pp65495–503.
Frequencies of IFN-γ positive cells as detected by pp65-MHCI RNA were found to be significantly higher compared to those detected by the single immunodominant peptide (Fig. 4a), suggesting that reactivity was only partially attributable to this epitope.
Fig. 4.

Simultaneous detection of multiple T-cell epitopes using HCMV pp65-MHCI RNA. a PBMCs from a healthy HCMV sero-positive HLA-A*0201+ donor were tested in CFC for CD8+ reactivity against the control peptide, pp65 peptide 495–503, NY-ESO-I-MHCI RNA as an irrelevant control, and pp65-MHCI RNA (10 μg each). Numbers represent the percentage of IFNγ secreting cells. Data are representative for three independent experiments. b CD8+ and CD4+ lymphocytes of a HCMV+ healthy donor were co-cultivated with autologous DCs loaded with four different sub pools of overlapping pp65 peptides (each peptide 1.75 μg/ml; see schematic drawing). c After expansion for 7 days each effector cell population (4 × 104/well) was tested in IFN(ELISPOT on autologous DCs (3 × 104/well) either loaded with the whole pp65 peptide pool, a pool of irrelevant peptides spanning the WT1 protein sequence (1.75 μg/ml), or transfected with pp65-MHCI RNA (20 μg). Each bar represents the mean spot number of duplicates. Due to the lack of adequate discrimination spot numbers were only quantified to a maximum of 800 per well
The capability of detecting polyepitopic responses was further confirmed using in vitro generated T-cell populations responding to different sets of epitopes within the pp65 antigen. To this aim, purified CD8+ as well as CD4+ T-cells were stimulated with autologous mature dendritic cells pulsed with four subpools of pp65 spanning peptides (Fig. 4b). At day 7 these in vitro generated effector cell populations were tested in ELISPOT against stimulators electroporated with pp65-MHCI or loaded with either the pp65 or an irrelevant control peptide pool. Autologous dendritic cells were used as stimulators as monocytes of sufficient viability could not be recovered after cryopreservation. Spot formation of individual effectors upon stimulation with pp65-MHCI was comparable to that with the peptide pool, confirming that different specificities within an oligoclonal population of responder T-cells can be comprehensively detected (Fig. 4c).
Discussion
The use of RNA based immunogens and their combination with freshly isolated PBMCs or monocytes as APCs provide a number of advantages as compared to alternative methods for quantification of antigen specific immune responses.
Pure RNA preparations can be easily obtained, preventing background reactivities against e.g., bacterial contaminants. Moreover, RNA does not need to enter the nucleus, but is translated in the cytosol immediately after electroporation enabling the use of non-dividing cells as stimulators.
Protocols were developed in which dendritic cells or purified monocytes transfected with antigen-encoding RNA were tested in stimulus-response assays for the identification of IFN-γ production by antigen-specific CD8+ T-cells [14, 15]. Most of these protocols use prior in vitro expansion of T-cell populations for quantification of antigen specific T-cells in particular CD4+ lymphocytes [30].
Here we extended the utility of antigen-encoding RNA as tool for the enumeration of antigen-specific T-cells by developing an assay system, which (1) utilizes PBMCs or monocytes as antigen presenters, (2) is sufficiently sensitive, and (3) allows to simultaneously detect antigen specific CD8+ and CD4+ T-cells.
Our comparative studies with blood samples obtained from CMV seropositive and CMV seronegative donors using a pp65 overlapping peptide pool as reference immunogen demonstrate, that antigen presentation by pp65-MHCI RNA electroporated stimulators is sufficiently potent to allow direct analysis of peripheral blood samples in a short time assay, without the need for prolonged in vitro stimulation or T-cell enrichment. Thus, in particular by using CFC, both T-cell subsets can be simultaneously quantified in a single test run within less than 24 h after blood samples have been collected from the patient. In case of retrospective analysis of collected samples, the typical drawbacks of using cryopreserved cells (for example, poor viability of monocytes) may apply.
Several strategies have been explored over the past few years to optimize MHC class II presentation of DNA or RNA encoded antigens. Antigens have been linked to the trafficking sequences of endosomal or lysosomal proteins known to reside in MHC class II antigen processing compartments, such as invariant chain [19, 31–33], lysosome-associated membrane proteins (LAMP1, LAMP2) and DC-LAMP [34–38]. The common molecular principle behind these approaches is that these proteins mediate translocation into defined subcellular compartments. Whereas invariant chain fusion proteins are mainly targeted to early endosomes, LAMP proteins are predominantly located in acidified late endosomes and lysosomes [39]. In each of these different compartments within the MHC class II processing route peculiar cleavage activities are prevalent and consecutively different types of epitopes are generated. This may explain, that the kind of MHC class II epitopes, which preferentially profit from this maneuvers, depends on the trafficking signal the antigen is fused to [38].
Our rationale to use MHC class I trafficking domains for efficient presentation of a comprehensive range of MHC class II epitopes was based on the observation that MHC class I molecules reside in or travel through all cellular compartments involved in MHC class II antigen processing and presentation [40].
We cannot answer so far, which “fraction” of CD8+ and CD4+ T-cell clones is detected by this assay and how “immunogenic” an antigen has to be to be suitable for this method. However, our data demonstrate that the use of the genetically modified pp65 results in the activation of larger CD4+ and CD8+ T cell subpopulations as compared with the hitherto best control (overlapping peptide pool). This indicates that indeed a large repertoire of T cells is adequately stimulated. Moreover, we have observed that not only viral proteins as presented in this paper but also less immunogenic tumor associated antigens (e.g., NY-ESO-I-MHCI) do profit from being linked to such MHC class I derived trafficking signals (S. Kreiter et al., manuscript in preparation), which make the potential of RNA to provide epitopes for any MHC class I and II allelotype exploitable.
To compensate for the poorer immunostimulatory capacity of monocytes and PBMCs we have resorted to optimized conditions for transfer and expression of IVT RNA and genetic modifications stabilizing this molecule, which we described previously [18].
Interestingly, with pp65-MHCI RNA for antigen delivery as compared to the pp65 peptide pool, frequencies of IFN-γ positive T-cells, in particular of the CD8+ subset are higher as is the dynamic range. One would think that a pool of overlapping peptides spanning the entire protein features in principle the complete spectrum of epitopes that can be processed from this particular protein. Two mechanisms of loading synthetic peptides on MHC molecules have been reported. On the one hand, exogenous peptides replace peptides in preloaded MHC molecules at the cell surface. Alternatively, they enter intracellular compartments and bind to de novo synthesized MHC molecules [41]. The relative contribution of these two mechanisms is not yet understood. However, it is clear that efficiency of each of these pathways will depend on the affinity and the stability of the exogenous peptides, which vary within a given peptide pool. Half-life of peptides in culture medium depends on several factors including hydrophobicity of the peptide and presence of peptidases and may be short [42]. Moreover, the synthetic peptides supplied in saturating concentrations within the pool have to compete against each other as well as against a background of endogenous peptides already occupying the MHC molecules [43]. Thereby, the efficiency with which exogenous peptides within the pool end up on MHC class I as well as class II molecules and the representativeness of the measured IFN-γ secretion for the authentic total immune response may be compromised.
The formulation of the antigen as IVT RNA and its improved processing and presentation by fusion to the MHC class I trafficking signal provide both a physiological composition of naturally processed peptide/MHC complexes as well as sufficient amounts of complexes for the induction of robust secretion of cytokines by responsive T-cells.
In summary, the approach we describe herein is ideal for rapid and broad analysis of the integrated cellular immune response in large cohorts of individuals e.g., during the course of clinical trials. Besides its application as an assay to enumerate antigen-specific, experienced T-cells, this system may be of particular interest to identify immunodominant antigens in screening approaches. Since DNA vector cassettes are used as templates for IVT RNA, the antigen coding sequence can be easily exchanged by standard molecular cloning techniques. This feature makes this short time blood test an attractive downstream tool for development of subunit vaccines against known as well as newly emerging infectious diseases. The application of advanced technologies such as high throughput sequencing, microarrays, proteomics and bioinformatics has provided new impulses to this research field facilitating the systematic mapping of genes and proteins of a pathogen [44–46]. Once a pathogen has been molecularly characterized on the sequence level, the subsequent determination of immunodominant antigens recognized by T-cells is a second major and underestimated challenge [47, 48]. Such reverse global approaches for the development of subunit vaccines may profit from this versatile assay allowing to determine within the pathogen proteome the comprehensive set of antigens recognized by immune effectors of infected hosts with protective immunity.
Acknowledgments
This study was supported by the Combined Project Grant SFB432, by the Heisenberg scholarship TU 115/3-1 of the Deutsche Forschungsgemeinschaft and by the Immunology Cluster of Excellence Mainz.
Footnotes
Sebastian Kreiter and Thorsten Konrad have contributed equally to this work. Özlem Türeci and Ugur Sahin have contributed equally to this work.
References
- 1.Clay TM, Hobeika AC, Mosca PJ, Lyerly HK, Morse MA. Assays for monitoring cellular immune responses to active immunotherapy of cancer. Clin Cancer Res. 2001;7:1127–1135. [PubMed] [Google Scholar]
- 2.Morse MA, Clay TM, Hobeika AC, Mosca PJ, Lyerly HK. Monitoring cellular immune responses to cancer immunotherapy. Curr Opin Mol Ther. 2001;3:45–52. [PubMed] [Google Scholar]
- 3.Czerkinsky C, Andersson G, Ekre HP, Nilsson LA, Klareskog L, Ouchterlony O. Reverse ELISPOT assay for clonal analysis of cytokine production. I. Enumeration of gamma-interferon-secreting cells. J Immunol Methods. 1988;110:29–36. doi: 10.1016/0022-1759(88)90079-8. [DOI] [PubMed] [Google Scholar]
- 4.Brosterhus H, Brings S, Leyendeckers H, Manz RA, Miltenyi S, Radbruch A, Assenmacher M, Schmitz J. Enrichment and detection of live antigen-specific CD4(+) and CD8(+) T cells based on cytokine secretion. Eur J Immunol. 1999;29:4053–4059. doi: 10.1002/(SICI)1521-4141(199912)29:12<4053::AID-IMMU4053>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 5.Kern F, Surel IP, Brock C, Freistedt B, Radtke H, Scheffold A, Blasczyk R, Reinke P, Schneider-Mergener J, Radbruch A, Walden P, Volk HD. T-cell epitope mapping by flow cytometry. Nat Med. 1998;4:975–978. doi: 10.1038/nm0898-975. [DOI] [PubMed] [Google Scholar]
- 6.Altman JD, Moss PA, Goulder PJ, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–96. doi: 10.1126/science.274.5284.94. [DOI] [PubMed] [Google Scholar]
- 7.Hobeika AC, Morse MA, Osada T, Ghanayem M, Niedzwiecki D, Barrier R, Lyerly HK, Clay TM. Enumerating antigen-specific T-cell responses in peripheral blood: a comparison of peptide MHC Tetramer, ELISpot, and intracellular cytokine analysis. J Immunother. 2005;28:63–72. doi: 10.1097/00002371-200501000-00008. [DOI] [PubMed] [Google Scholar]
- 8.Maino VC, Maecker HT. Cytokine flow cytometry: a multiparametric approach for assessing cellular immune responses to viral antigens. Clin Immunol. 2004;110:222–231. doi: 10.1016/j.clim.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 9.Karlsson AC, Martin JN, Younger SR, Bredt BM, Epling L, Ronquillo R, Varma A, Deeks SG, McCune JM, Nixon DF, Sinclair E. Comparison of the ELISPOT and cytokine flow cytometry assays for the enumeration of antigen-specific T cells. J Immunol Methods. 2003;283:141–153. doi: 10.1016/j.jim.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 10.Suni MA, Dunn HS, Orr PL, de Laat R, Sinclair E, Ghanekar SA, Bredt BM, Dunne JF, Maino VC, Maecker HT. Performance of plate-based cytokine flow cytometry with automated data analysis. BMC Immunol. 2003;4:9. doi: 10.1186/1471-2172-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maecker HT, Auffermann-Gretzinger S, Nomura LE, Liso A, Czerwinski DK, Levy R. Detection of CD4 T-cell responses to a tumor vaccine by cytokine flow cytometry. Clin Cancer Res. 2001;7:902s–908s. [PubMed] [Google Scholar]
- 12.Suni MA, Maino VC, Maecker HT. Ex vivo analysis of T-cell function. Curr Opin Immunol. 2005;17:434–440. doi: 10.1016/j.coi.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 13.Kern F, Faulhaber N, Frommel C, Khatamzas E, Prosch S, Schonemann C, Kretzschmar I, Volkmer-Engert R, Volk HD, Reinke P. Analysis of CD8 T cell reactivity to cytomegalovirus using protein-spanning pools of overlapping pentadecapeptides. Eur J Immunol. 2000;30:1676–1682. doi: 10.1002/1521-4141(200006)30:6<1676::AID-IMMU1676>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 14.Van Tendeloo VF, Ponsaerts P, Lardon F, Nijs G, Lenjou M, Van Broeckhoven C, Van Bockstaele DR, Berneman ZN. Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood. 2001;98:49–56. doi: 10.1182/blood.V98.1.49. [DOI] [PubMed] [Google Scholar]
- 15.Teufel R, Carralot JP, Scheel B, Probst J, Walter S, Jung G, Hoerr I, Rammensee HG, Pascolo S. Human peripheral blood monuclear cells transfected with messenger RNA stimulate antigen-specific cytotoxic T-lymphocytes in vitro. Cell Mol Life Sci. 2005;62(15):1755–1762. doi: 10.1007/s00018-005-5067-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Britten CM, Meyer RG, Frankenberg N, Huber C, Wolfel T. The use of clonal mRNA as an antigenic format for the detection of antigen-specific T lymphocytes in IFN-gamma ELISPOT assays. J Immunol Methods. 2004;287:125–136. doi: 10.1016/j.jim.2004.01.026. [DOI] [PubMed] [Google Scholar]
- 17.Britten CM, Meyer RG, Graf C, Huber C, Wolfel T. Identification of T cell epitopes by the use of rapidly generated mRNA fragments. J Immunol Methods. 2005;299:165–175. doi: 10.1016/j.jim.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 18.Holtkamp S, Kreiter S, Selmi A, Simon P, Koslowski M, Huber C, Tureci O, Sahin U. Modification of antigen encoding RNA increases stability, translational efficacy and T-cell stimulatory capacity of dendritic cells. Blood. 2006;108(13):4009–4017. doi: 10.1182/blood-2006-04-015024. [DOI] [PubMed] [Google Scholar]
- 19.Bonehill A, Heirman C, Tuyaerts S, Michiels A, Zhang Y, Van der Bruggen BP, Thielemans K. Efficient presentation of known HLA class II-restricted MAGE-A3 epitopes by dendritic cells electroporated with messenger RNA encoding an invariant chain with genetic exchange of class II-associated invariant chain peptide. Cancer Res. 2003;63:5587–5594. [PubMed] [Google Scholar]
- 20.Kavanagh DG, Kaufmann DE, Sunderji S, Frahm N, Le Gall S, Boczkowski D, Rosenberg ES, Stone DR, Johnston MN, Wagner BS, Zaman MT, Brander C, Gilboa E, Walker BD, Bhardwaj N. Expansion of HIV-specific CD4+ and CD8+ T cells by dendritic cells transfected with mRNA encoding cytoplasm- or lysosome-targeted Nef. Blood. 2006;107:1963–1969. doi: 10.1182/blood-2005-04-1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su Z, Vieweg J, Weizer AZ, Dahm P, Yancey D, Turaga V, Higgins J, Boczkowski D, Gilboa E, Dannull J. Enhanced induction of telomerase-specific CD4(+) T cells using dendritic cells transfected with RNA encoding a chimeric gene product. Cancer Res. 2002;62:5041–5048. [PubMed] [Google Scholar]
- 22.Hoffmeister B, Bunde T, Rudawsky IM, Volk HD, Kern F. Detection of antigen-specific T cells by cytokine flow cytometry: the use of whole blood may underestimate frequencies. Eur J Immunol. 2003;33:3484–3492. doi: 10.1002/eji.200324223. [DOI] [PubMed] [Google Scholar]
- 23.Nomura LE, Walker JM, Maecker HT. Optimization of whole blood antigen-specific cytokine assays for CD4(+) T cells. Cytometry. 2000;40:60–68. doi: 10.1002/(SICI)1097-0320(20000501)40:1<60::AID-CYTO8>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 24.Diamond DJ, York J, Sun JY, Wright CL, Forman SJ. Development of a candidate HLA A*0201 restricted peptide-based vaccine against human cytomegalovirus infection. Blood. 1997;90:1751–1767. [PubMed] [Google Scholar]
- 25.Gure AO, Tureci O, Sahin U, Tsang S, Scanlan MJ, Jager E, Knuth A, Pfreundschuh M, Old LJ, Chen YT. SSX: a multigene family with several members transcribed in normal testis and human cancer. Int J Cancer. 1997;72:965–971. doi: 10.1002/(SICI)1097-0215(19970917)72:6<965::AID-IJC8>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 26.Maecker HT, Dunn HS, Suni MA, Khatamzas E, Pitcher CJ, Bunde T, Persaud N, Trigona W, Fu TM, Sinclair E, Bredt BM, McCune JM, Maino VC, Kern F, Picker LJ. Use of overlapping peptide mixtures as antigens for cytokine flow cytometry. J Immunol Methods. 2001;255:27–40. doi: 10.1016/S0022-1759(01)00416-1. [DOI] [PubMed] [Google Scholar]
- 27.Ponsaerts P, Van den Bosch G, Cools N, Van Driessche A, Nijs G, Lenjou M, Lardon F, Van Broeckhoven C, Van Bockstaele DR, Berneman ZN, Van Tendeloo VF. Messenger RNA electroporation of human monocytes, followed by rapid in vitro differentiation, leads to highly stimulatory antigen-loaded mature dendritic cells. J Immunol. 2002;169:1669–1675. doi: 10.4049/jimmunol.169.4.1669. [DOI] [PubMed] [Google Scholar]
- 28.Schmittel A, Keilholz U, Bauer S, Kuhne U, Stevanovic S, Thiel E, Scheibenbogen C. Application of the IFN-gamma ELISPOT assay to quantify T cell responses against proteins. J Immunol Methods. 2001;247:17–24. doi: 10.1016/S0022-1759(00)00305-7. [DOI] [PubMed] [Google Scholar]
- 29.Schmolke S, Drescher P, Jahn G, Plachter B. Nuclear targeting of the tegument protein pp65 (UL83) of human cytomegalovirus: an unusual bipartite nuclear localization signal functions with other portions of the protein to mediate its efficient nuclear transport. J Virol. 1995;69:1071–1078. doi: 10.1128/jvi.69.2.1071-1078.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heine A, Grunebach F, Holderried T, Appel S, Weck MM, Dorfel D, Sinzger C, Brossart P. Transfection of dendritic cells with in vitro-transcribed CMV RNA induces polyclonal CD8+- and CD4+-mediated CMV-specific T cell responses. Mol Ther. 2006;13:280–288. doi: 10.1016/j.ymthe.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 31.Sanderson S, Frauwirth K, Shastri N. Expression of endogenous peptide-major histocompatibility complex class II complexes derived from invariant chain-antigen fusion proteins. Proc Natl Acad Sci USA. 1995;92:7217–7221. doi: 10.1073/pnas.92.16.7217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomson SA, Burrows SR, Misko IS, Moss DJ, Coupar BE, Khanna R. Targeting a polyepitope protein incorporating multiple class II-restricted viral epitopes to the secretory/endocytic pathway facilitates immune recognition by CD4+ cytotoxic T lymphocytes: a novel approach to vaccine design. J Virol. 1998;72:2246–2252. doi: 10.1128/jvi.72.3.2246-2252.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Diebold SS, Cotten M, Koch N, Zenke M. MHC class II presentation of endogenously expressed antigens by transfected dendritic cells. Gene Ther. 2001;8:487–493. doi: 10.1038/sj.gt.3301433. [DOI] [PubMed] [Google Scholar]
- 34.Bonehill A, Heirman C, Tuyaerts S, Michiels A, Breckpot K, Brasseur F, Zhang Y, Van Der Bosch P, Thielemans K. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J Immunol. 2004;172:6649–6657. doi: 10.4049/jimmunol.172.11.6649. [DOI] [PubMed] [Google Scholar]
- 35.Rowell JF, Ruff AL, Guarnieri FG, Staveley-O’Carroll K, Lin X, Tang J, August JT, Siliciano RF. Lysosome-associated membrane protein-1-mediated targeting of the HIV-1 envelope protein to an endosomal/lysosomal compartment enhances its presentation to MHC class II-restricted T cells. J Immunol. 1995;155:1818–1828. [PubMed] [Google Scholar]
- 36.Wu TC, Guarnieri FG, Staveley-O’Carroll KF, Viscidi RP, Levitsky HI, Hedrick L, Cho KR, August JT, Pardoll DM. Engineering an intracellular pathway for major histocompatibility complex class II presentation of antigens. Proc Natl Acad Sci USA. 1995;92:11671–11675. doi: 10.1073/pnas.92.25.11671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bonini C, Lee SP, Riddell SR, Greenberg PD. Targeting antigen in mature dendritic cells for simultaneous stimulation of CD4+ and CD8+ T cells. J Immunol. 2001;166:5250–5257. doi: 10.4049/jimmunol.166.8.5250. [DOI] [PubMed] [Google Scholar]
- 38.Bonehill A, Heirman C, Thielemans K. Genetic approaches for the induction of a CD4+ T cell response in cancer immunotherapy. J Gene Med. 2005;7:686–695. doi: 10.1002/jgm.713. [DOI] [PubMed] [Google Scholar]
- 39.Lizee G, Basha G, Jefferies WA. Tails of wonder: endocytic-sorting motifs key for exogenous antigen presentation. Trends Immunol. 2005;26:141–149. doi: 10.1016/j.it.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 40.Lizee G, Basha G, Tiong J, Julien JP, Tian M, Biron KE, Jefferies WA. Control of dendritic cell cross-presentation by the major histocompatibility complex class I cytoplasmic domain. Nat Immunol. 2003;4:1065–1073. doi: 10.1038/ni989. [DOI] [PubMed] [Google Scholar]
- 41.Day PM, Yewdell JW, Porgador A, Germain RN, Bennink JR. Direct delivery of exogenous MHC class I molecule-binding oligopeptides to the endoplasmic reticulum of viable cells. Proc Natl Acad Sci USA. 1997;94:8064–8069. doi: 10.1073/pnas.94.15.8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amoscato AA, Prenovitz DA, Lotze MT. Rapid extracellular degradation of synthetic class I peptides by human dendritic cells. J Immunol. 1998;161:4023–4032. [PubMed] [Google Scholar]
- 43.Luft T, Rizkalla M, Tai TY, Chen Q, MacFarlan RI, Davis ID, Maraskovsky E, Cebon J. Exogenous peptides presented by transporter associated with antigen processing (TAP)-deficient and TAP-competent cells: intracellular loading and kinetics of presentation. J Immunol. 2001;167:2529–2537. doi: 10.4049/jimmunol.167.5.2529. [DOI] [PubMed] [Google Scholar]
- 44.Holmes KV, Enjuanes L. The SARS coronavirus: a postgenomic era. Science. 2003;300:1377–1378. doi: 10.1126/science.1086418. [DOI] [PubMed] [Google Scholar]
- 45.Scarselli M, Giuliani MM, Adu-Bobie J, Pizza M, Rappuoli R. The impact of genomics on vaccine design. Trends Biotechnol. 2005;23:84–91. doi: 10.1016/j.tibtech.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 46.Pizza M, Scarlato V, Masignani V, Giuliani MM, Arico B, Comanducci M, Jennings GT, Baldi L, Bartolini E, Capecchi B, Galeotti CL, Luzzi E, Manetti R, Marchetti E, Mora M, Nuti S, Ratti G, Santini L, Savino S, Scarselli M, Storni E, Zuo PJ, Broeker M, Hundt E, Knapp B, Blair E, Mason T, Tettelin H, Hood DW, Jeffries AC, Saunders NJ, Granoff DM, Venter JC, Moxon ER, Grandi G, Rappuoli R. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science. 2000;287:1816–1820. doi: 10.1126/science.287.5459.1816. [DOI] [PubMed] [Google Scholar]
- 47.Milstien J, Lambert S. Emergency response vaccines—a challenge for the public sector and the vaccine industry. Vaccine. 2002;21:146–154. doi: 10.1016/S0264-410X(02)00436-X. [DOI] [PubMed] [Google Scholar]
- 48.Sturniolo T, Bono E, Ding JY, Raddrizzani L, Tuereci O, Sahin U, Braxenthaler M, Gallazzi F, Protti MP, Sinigaglia F, Hammer J. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol. 1999;17:555–561. doi: 10.1038/9858. [DOI] [PubMed] [Google Scholar]