

Abstract
Purpose
CD4+CD25+ regulatory T (Treg) cells are present in increased numbers in patients with advanced cancer and CD25+ T cell depletion potentiates tumour immunity in animal models. The aim of this study was to assess the feasibility and safety of adoptive transfer of CD25+ depleted autologous T cells in patients with advanced renal cell carcinoma and to examine resulting changes in lymphocyte subsets.
Patients and methods
Six patients with advanced renal cell carcinoma underwent leukapheresis followed by conditioning chemotherapy with cyclophosphamide and fludarabine. The autologous leukapheresis product was depleted of CD25+ cells using CliniMACS® System then re-infused into the patient.
Results
Efficient CD25+ depletion from all leukapheresis products was achieved and 0.55–5.87 × 107/kg CD3+ cells were re-infused. Chemotherapy related haematological toxicity was observed, but blood counts recovered in all patients allowing discharge after a mean inpatient stay of 21 days. One patient subsequently developed a rapidly progressive neurological syndrome. A transient reduction in CD25+ subset was noted in the peripheral blood of 5 out of 6 patients with evidence of increased T cell responses to PHA in 4 out of 6 patients. One patient showed increased specific proliferative responses to the tumour associated antigen h5T4 coinciding with the nadir of Treg cells.
Conclusions
Given the transient nature of the reduction in CD25+ subset and the observed toxicity there is a need to explore further strategies to improve the safety and efficacy of this approach. Nevertheless, the results provide proof of concept in potentiation of tumour antigen T cell responses when Treg cell levels are depleted.
Keywords: Regulatory T cell, CD25, FOXP3, Adoptive cell therapy, Conditioning chemotherapy
Introduction
The adaptive immune system has evolved several systems to handle the problem of randomly generated receptors that recognise self-antigens. Whilst key cell-intrinsic mechanisms include clonal deletion and anergy of autoreactive cells, the role of regulatory T (Treg) cells in exerting a cell-extrinsic, dominant form of self-tolerance is becoming increasingly apparent [1]. CD4+CD25+ Treg cells are characterised by expression of high levels of CD25 (the IL-2Rα chain), the transcription factor forkhead box P3 (FOXP3), glucocorticoid-induced TNFR (GITR) and T lymphocyte-associated antigen 4 (CTLA-4) [2]. They constitute 5–10% of the peripheral CD4+ T cell compartment in humans and mice [3, 4] and data exist to support the concept that they develop in the thymus and represent a distinct cell lineage [5]. The fundamental role that Treg cells play as mediators of self-tolerance is reflected in the severe immunopathologies seen in both humans and mice with mutations in FOXP3 (IPEX syndrome and scurfy respectively) [6–9].
The T cell receptor (TCR) repertoire of Treg cells is diverse, but appears to be skewed towards recognizing MHC class II bound self-peptides from the periphery [10]. Given that many tumour-associated antigens (TAA) are self-antigens, Treg cells engaged in the maintenance of self-tolerance may concurrently inhibit the antitumour potential of effector T cells that recognise these TAAs. Indeed, evidence is now emerging to support the hypothesis that Treg cells play an important role in the context of cancer. For example several studies have found increased numbers of Treg cells in the peripheral blood of patients with advanced cancer compared to normal controls [11–13]. Antigen-specific Treg cells have also been demonstrated in tumour infiltrating lymphocytes (TILs) derived from fresh tumour samples which suppress the proliferation of naïve CD4+ T cells and inhibit IL-2 secretion by effector T cells upon activation by tumour specific ligands [14].
Adoptive cell therapy (ACT) is an approach that may enable effective manipulation of Treg cells to enhance antitumour activity. Increasingly ACT is being used in conjunction with prior host lymphodepletion since various preclinical studies have shown that it can significantly improve the antitumour efficacy of transferred T cells [15, 16]. Under conditions of lymphodepletion, adoptively transferred T cells undergo homeostatic proliferation. The resultant T cells acquire characteristics of memory and effector cells as measured by phenotype, hypersensitivity to antigen stimulation and increased production of IFNγ [17]. The mechanisms by which lymphodepletion enhances T cell responses are not fully elucidated, but are likely to include the elimination of cells that compete for activating cytokines (minimizing cytokine sinks), improved antigen presentation and the elimination of Treg cells [18].
Lymphodepletion has been shown to be effective in clinical trials involving the adoptive transfer of TILs in patients with metastatic melanoma. In one series, 35 patients were treated with lymphodepleting (but non-myeloablative) chemotherapy then adoptive transfer of TILs followed by systemic IL-2 therapy [19]. Approximately 50% of the patients had objective tumour responses. Unfortunately therapies requiring isolation and expansion of TILs from tumours other than melanoma have proven difficult, thus there is a need for alternative approaches to be developed.
In an ACT strategy in nude mice, it has been shown that transfer of splenic-derived T cells depleted of CD25+ cells can elicit potent immune responses to syngeneic tumours in vivo resulting in tumour eradication [20]. Transfer of non-depleted cells or a mixture of CD25+ and non-depleted cells did not result in tumour eradication indicating that in this model removal of CD25+ T cells (including Treg cells) can break immunological unresponsiveness to autologous tumours.
In this study, the role of CD25+ T cell depletion in breaking immunological tolerance in six patients with metastatic renal cell carcinoma was examined. This tumour type was selected because it is well recognised that renal cell carcinoma can show susceptibility to immune-based therapies [21]. We have used the same lymphodepleting chemotherapy regime previously used in the TIL ACT studies [19] and also in non-myeloablative allogeneic stem-cell transplantation in renal cell carcinoma patients [21]. In our study, prior to administration of chemotherapy, a leukapheresis product was subject to CD25+ depletion using magnetic beads (CliniMACS® CD25 Reagent System) to eliminate the CD4+CD25+ Treg cell population. The aim of the lymphodepleting chemotherapy was to profoundly lymphodeplete the patients of remaining endogenous Treg cells and to provide a lymphopenic environment to promote homeostatic proliferation of the adoptively transferred CD25+ depleted cells. The safety, toxicity and clinical outcomes as well as the effect on the phenotype of peripheral blood lymphocytes were examined. In addition, changes in the functional immune repertoire were assessed using PHA (phytohemagglutinin) to measure polyclonal T cell activation whilst h5T4 was selected as a suitable TAA to look for an increase in specific immune responses. h5T4 is an oncofoetal protein not expressed at significant levels on normal adult human tissues, but overexpressed in many tumours including renal cell carcinoma [22]. It appears to be a good target for immunotherapy approaches since previous vaccine studies have shown that it is possible to break tolerance to h5T4 [23].
Methods
Patient selection
All patients had histologically confirmed renal cell carcinoma that was metastatic or unresectable, were 18 years of age or over and provided written, informed consent to participate. Eligibility criteria also included WHO performance status of 0 or 1, life expectancy of greater than 3 months and measurable disease on CT or MRI scan according to RECIST criteria. Patients were required to have adequate haematological function (haemoglobin ≥ 10.0 g/dL, neutrophils ≥ 2.0 × 109 L−1, lymphocytes ≥ 1.0 × 109 L−1, platelets ≥ 100 × 109 L−1) hepatocellular function (bilirubin ≤ 1.5× upper limit of normal (ULN), ALT, AST and ALP all ≤5× ULN), serum creatinine ≤ 0.14 mmol/L and adequate cardiac function. All patients had failed at least one line of standard therapy and had completed this at least 1 month prior to entry into the trial with toxicities from previous therapies having resolved. Patients were excluded if they were at high medical risk because of non-malignant systemic disease including autoimmune disease, had clinical evidence of cerebral metastases or were felt likely to require systemic steroid therapy during the course of the trial. This study was conducted in accordance with the declaration of Helsinki and the EU directive on good clinical practice and ethical approval was obtained from South Manchester Research Ethics Committee. Written informed consent was obtained from each patient.
Study objectives, endpoints and sample size
The primary clinical objective was to assess the feasibility and safety of this approach. The outcome measures were to show that efficient (>2 log) depletion of CD25+ could be achieved and the depleted product re-infused into patients as laid out in the clinical protocol. Safety was measured by descriptive analysis of toxicities experienced by patients graded according to NCI common toxicity criteria adverse events version 3 (CTCAEv3) (http://ctep.cancer.gov/reporting/ctc.html). The primary laboratory objective was to look for phenotypic changes in the peripheral blood as a result of therapy. The endpoint was achieved by measurement of change in the level of Treg cells (identified by the expression of CD4, CD25 and FOXP3) in peripheral blood by flow cytometric analysis following treatment. Secondary endpoints were to look for early evidence of clinical responses by CT scan and to explore changes in the functional repertoire of cellular immunity by changes in proliferative responses of PBMC to the tumour associated antigen h5T4, which is over-expressed in renal cell carcinoma.
The initial planned sample size was up to a total of 14 patients with a protocol specified interim analysis after the recruitment of 6 patients. The interim analysis highlighted the transient nature of depletion of Treg cells and recruitment was thus discontinued at this stage.
Preparation of CD25 depleted T cells
Peripheral blood mononuclear cells (PBMCs) were collected from patients using standard leukapheresis protocols and the cells were depleted of CD25+ cells using CliniMACS® CD25 Reagent following the manufacturer’s instructions (Miltenyi Biotec, Germany). Briefly, the leukapheresis product was first washed with CliniMACS PBS/EDTA buffer containing 0.5% HSA, then centrifuged (300g 15 min) and resuspended in 95 mL of buffer. To the cell suspension, 7.5 mL CliniMACS CD25 Reagent was added and incubated for 30 min between 19 and 25°C. After a further wash, the cells were resuspended in buffer to a final volume of 100 mL and a sample taken for flow cytometry analysis. The labelled cells were then separated on the CliniMACS Instrument using the Miltenyi Biotec DEPLETION 2.1 protocol. The collected CD25 depleted and non-depleted cell populations were then cryopreserved in HSA containing 10% DMSO and stored in the vapour phase of liquid nitrogen until required for re-infusion into the patient.
Flow Cytometry analysis of leukapheresis product
Samples from the leukapheresis product were analysed by flow cytometry both pre- and post-CD25 depletion to determine CD3, CD4, CD8, CD19, CD25 and CD45 absolute counts. All staining was direct using a standard lyse/no wash procedure with the exception of the CD25 staining. For an optimal staining result, an indirect assay using an antihuman CD25 antibody conjugated to biotin together with a phycoerythrin conjugated antibiotin antibody (Miltenyi Biotec, Germany) was used following the manufacturers instructions. CD3-FITC, CD4-PE, CD8-PE, CD19-PE and CD45-FITC antibodies were all obtained from Immunotech (Beckman Coulter, CA).
Treatment regimen
Patients received intravenous cyclophosphamide (60 mg/kg) per day for 2 days followed by intravenous fludarabine (25 mg/m2) per day for 5 days, as inpatients at Christie Hospital NHS trust Clinical Trials Inpatient Unit (Fig. 1). Autologous CD25+ depleted cells were thawed and re-infused into the patients 1 day after completion of chemotherapy. Haematological indices and toxicities were monitored closely. Prophylactic granulocyte-colony stimulating factor (GCSF; Granocyte® 263 μg subcutaneous injection once daily) was administered until neutrophil count recovered to greater than 1.0 × 109 L−1. Blood and platelet support was given as required. Patients were discharged when they were no longer GCSF, blood or platelet transfusion dependants. They received prophylactic antimicrobials for a total of 6 weeks and were followed up as outpatients every 2 weeks for the first 3 months then on a monthly basis. Blood samples for trial assays were taken when the patients attended for follow-up. PBMC were isolated by Ficoll-Hypaque medium and then cryopreserved until required for analysis. Re-assessment CT scans were carried out at 6 weeks, 3 months and 6 months.
Fig. 1.

Treatment regimen. Leukapheresis product was CD25+ depleted then cryopreserved. Patients received cyclophosphamide 60 mg/kg on day 7 and on day 6, followed by fludarabine 25 mg/m2 day 5–1. On day 0 the cryopreserved CD25+ depleted cells were thawed and reinfused. The patients were given prophylactic GCSF and antimicrobials and monitored as inpatients until their blood counts had recovered to appropriate levels for discharge
Phenotypic identification of Treg cells
Treg cells were identified by expression of CD4, CD25 and FOXP3 transcription factor using the human regulatory T cell staining kit (eBioscience, San Diego, CA, USA), according to the manufacturer’s instruction. Briefly, frozen PBMC were thawed and resuspended in phosphate-buffered saline (PBS). Cells were first stained for extracellular CD4 and CD25 markers using CD4/25 cocktail (a cocktail of antihuman CD4-FITC and antihuman CD25-APC). Following fixation and permeabilization, the cells were washed and blocked for non-specific binding sites using normal rat serum. Antihuman FOXP3-PE or rat IgG2a-PE isotype negative control was then added for 30 min before washing twice and flow cytometric analysis using the FACSCalibur (Becton Dickinson, CA, USA).
Proliferation assays
One day prior to setting up the proliferation assays, PBMC from pre-treatment time point were thawed and monocytes isolated by plastic adherence. Then they were either infected with 2 pfu/cell of MVA-LacZ as a control or MVA-h5T4 [24] for 90 min at 37°C. The virus was then removed and the monocytes cultured overnight in Iscoves modified D MEM (Invitrogen) supplemented with 10% heat inactivated human AB serum (IMDM + AB) to allow h5T4 expression. The following day, frozen PBMC from various time points were thawed into IMDM + AB. MVA-Infected monocytes were collected and cultured with 1.5 × 105 PBMC at monocyte:PBMC ratio of 1:10 into 96 well U bottom tissue culture plates (Falcon). In addition, 1.5 × 105 PBMC/well were cultured with pools of three 32 mer peptides spanning the whole sequence of h5T4 protein (10 μg/mL of each peptide), or mock-stimulated with equivalent concentration of DMSO as a negative control. PHA-M (Sigma, MO) at 1, 2 and 5 μg/mL was also added as a polyclonal stimulant.
Plates were incubated for 5 days at 37°C before pulsing with 1 μCi/mL of [3H]TdR for the last 18 h then harvested using a Packard cell harvester and counted on a Packard Topcount. The stimulation indices (SI) were calculated by dividing the count per minute (cpm) of the test wells by the cpm of control wells (DMSO). For MVA-infected monocytes, the results are expressed as the mean cpm of the MVA-h5T4-infected monocytes compared to the mean cpm for the MVA-LacZ-infected monocytes.
Results
Patient characteristics
Six patients were recruited and underwent leukapheresis. All patients had undergone previous nephrectomies and had metastatic disease. The patient characteristics are described further in Table 1.
Table 1.
Patient characteristics
| Patient number | Age | Sex | Histology and Fuhrman grade of tumour | Prior therapy | Sites of metastatic disease | WHO performance status | Clinical features at screening | ||
|---|---|---|---|---|---|---|---|---|---|
| Hb (g/dL) | LDH (IU/L) | Ca (mmol/L) | |||||||
| 1 | 63 | M | Papillary cell Grade 3 | scIL-2 | Ly, Ad, Li | 0 | 13.0 | 376 | 2.36 |
| 2 | 38 | M | Clear cell grade 2 | IL-2/IFN/5FU | Ly, Lu, Ad | 0 | 14.9 | 417 | 2.55 |
| 3 | 55 | M | Clear cell grade 4 | IVIL-2 | Lu | 0 | 11.9 | 357 | 2.27 |
| 4 | 58 | M | Clear cell grade2 | IVIL-2 | Lu, Bo | 0 | 12.9 | 303 | 2.25 |
| 5 | 66 | M | Clear cell grade 2 (with small foci of grade 4) | IFN, SU | Lu | 1 | 10.5 | 400 | 2.18 |
| 6 | 58 | F | Clear cell grade 2-3 | IFN, antiIL-6 antibody | Li, Ad, Lu, Gb, Bo | 0 | 12.8 | 527 | 2.27 |
sc Subcutaneous, 5FU 5 Fluorouracil, SU sutent, Ly lymphnode, Ad adrenal, Li liver, Lu lung, Bo bone, Gb gallbladder, WHO World Health Organisation, Hb haemaglobin (normal range 13.0–18.0 g/dL), LDH lactate dehydrogenase (normal range 200–550 IU/L), Ca adjusted calcium (normal range 2.10–2.65 mmol/L)
Efficiency of CD25+ cell depletion
The leukapheresis product was depleted of CD25+ cells by magnetic selection using the CliniMACS® System which gave >2 log depletion of CD25+ lymphocytes in five out of six patients (Table 2). An example of the CD25 biotin flow cytometry staining for patient 1 at different stages of the depletion process is shown in Fig. 2.
Table 2.
Composition of leukapheresis product before and after CliniMACS® CD25 depletion
| Patient | 1 | 2a | 2a | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|---|---|
| Before CD25 depletion | Cell viability (%) | 98.3 | 98.3 | 99 | 97.6 | 97.2 | 89 | 99.3 |
| CD3+(×108) | 46.5 | 63 | 148 | 41.7 | 46 | 104 | 114 | |
| CD3+CD25+(×108) | 2.1 | 2.3 | 10 | 2.8 | 1.9 | 1.3 | 4.4 | |
| After CD25 depletion | Cell viability (%) | 86.7 | 95 | 97 | 91.2 | 94.5 | 77 | 97.9 |
| CD3+(×108) | 16.3 | 10.3 | 9.8 | 11 | 17 | 68 | 53 | |
| CD3+CD25+(×108) | 0.004 | 0.002 | 0 | 0.008 | 0.01 | 0.03 | 0.04 | |
| Fold depletion CD25+ cells | 5.25 × 102 | 1.15 × 103 | >104 | 3.55 × 102 | 1.9 × 102 | 0.4 × 102 | 1.15 × 102 | |
| Total dose of infused CD3+ cells × 107/kg body weight | 1.33 | 0.55a | 0.9 | 1.7 | 5.87 | 2.68 | ||
aDue to insufficient cell numbers following processing, this patient was leukapheresed twice and the cells from both depletions were pooled to make up the re-infused product
Fig. 2.

CD25 biotin flow cytometry staining during the depletion process for patient 1. Leukapheresis product analysed by flow cytometry at various stages during the depletion process. The dot plots a–d show CD3 and CD25 staining before depletion (a), post-bead attachment (b), after depletion (c) and the enriched product (d)
Clinical feasibility and toxicities
The conditioning chemotherapy was administered in all cases as per protocol over a 7-day period. Autologous CD25+ depleted leukapheresis product was re-infused into all patients, the day after completion of chemotherapy. There were no immediate toxicities from the infusion of CD25+ depleted cells. The chemotherapy resulted in expected haematological toxicities with all six patients experiencing grade IV neutropenia and grade IV lymphopenia. Four patients had episodes of fever associated with neutropenia, which was treated with intravenous antibiotics. In addition four patients developed grade IV thrombocytopenia requiring platelet support and the same four patients also required blood transfusions. Other expected toxicities associated with the chemotherapy including nausea, vomiting and diarrhoea were grades I–II. Figure 3 shows the recovery of the patients’ blood counts, which were in line with expectation. They remained as inpatients for a mean of 21 days (range 18–33) prior to discharge.
Fig. 3.
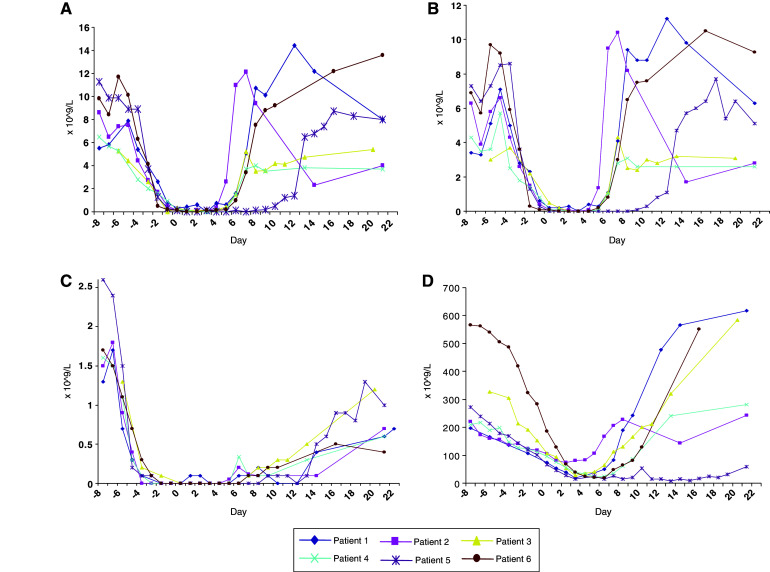
Peripheral blood counts for patients during therapy. a Total white blood cell (WBC) counts. b Neutrophil counts. c Lymphocyte counts. d Platelet counts
Patient 5 had delayed bone marrow recovery and developed aspergillous pneumonia, which was treated with appropriate antifungal agents. He also required local brachytherapy for an endobronchial metastasis that was threatening his right main bronchus, but made a gradual recovery and was discharged from hospital 33 days after admission.
Patient 6 was re-admitted 3 weeks after discharge complaining of fatigue, diarrhoea and was found to have deranged liver function tests. The diarrhoea initially settled with conservative treatment and stool cultures were negative. Prophylactic antifungals were discontinued resulting in an improvement in liver function tests and general condition. Over subsequent days, however, the patient complained of visual disturbuance and became profoundly depressed. During a 2-week period, she developed progressive neurological symptoms of upper motorneurone weakness and dementia. Compared to pre-treatment, MRI brain scans showed increasing white matter signal consistent with a non-specific leukoencephalopathy. CSF assessment showed glucose and protein levels within the normal range and two lymphocytes per mm3. The CSF was negative by PCR for the following viruses: HSV type 1 and 2, EBV, CMV, HHV6, HHV7, JCV, BK, enterovirus and parechovirus. She was treated with high dose intravenous steroids and immunoglobulin, but unfortunately failed to respond and died 1 month later. Post mortem examination revealed leukoencephalopathy with myelin loss and severe axonal damage affecting predominantly the occipital lobes. There was no inflammation within the cortex to suggest paraneoplastic or immune-mediated encephalitis
Patients underwent CT scans at 6 weeks, 3 months and 6 months. None of the patients had objective responses according to RECIST criteria. One patient had a liver metastasis that measured 1.6 × 1.4 cm on the pre-treatment scan, that reduced in size at 6 weeks and was not identifiable on the scan at 3 months, but disease elsewhere slowly increased in size and by 6 months there was clear overall progression by RECIST criteria. A second patient had an initial minor decrease in the size of a lung metastasis, from 1.4 cm long diameter on the pre-treatment scan to 0.9 cm at 6 weeks, but by 3 months there was evidence of progression. The surviving patients all had progressive disease by the 3 month scan and were taken off study.
Immunological phenotypic analysis
Patient 1 was the only patient who showed a level of Treg cells, which was higher than the normal range documented in normal volunteers [13] but these patients had all received some previous treatment. The proportion of lymphocytes, which are Treg cells (CD4+CD25+FOXP3+) is shown for patient 1 in Fig. 4a, b during the follow-up period. At week 2 there is a decrease in the percentage of CD4+CD25+FOXP3+ cells which rebounds, peaking at week 8 before returning to pre-treatment levels by week 16. Similar initial transient reduction in CD4+CD25+FOXP3+ cell levels were seen at 2 weeks (patient 2, 4 and 5) and at 4 weeks for patient 3 but patient 6 showed an increase in Treg cells up to week 4 (Fig. 4c).
Fig. 4.
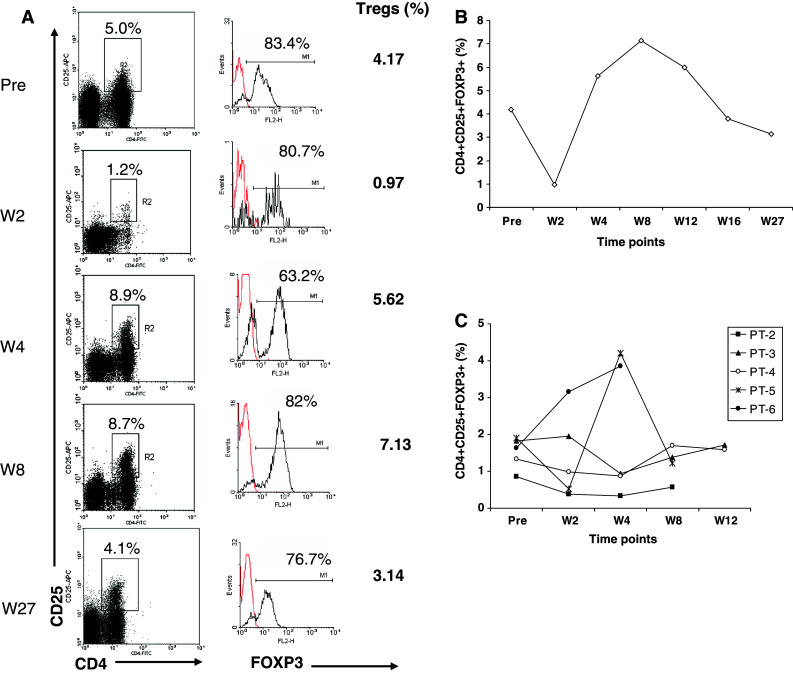
Frequency of T regulatory cells in the peripheral blood of patients prior to and after treatment. Peripheral blood mononuclear cells were stained for CD4, CD25 and FOXP3, and analysed by flow cytometry. Lymphocytes are gated according to forward and side scatter characteristics. The dot plots in a show examples of staining of CD4 and CD25 before treatment and at weeks 2, 4, 8 and 27 after treatment for patient 1. The R2 gate identifies the CD4+CD25high fraction within the lymphogate. The histograms to the right of each plot show the percentages of FOXP3+ cells within R2 gate. The proportion of CD4+CD25highFOXP3+ T regulatory cells at each time point is calculated and indicated to the right of histograms. The change in Treg frequency measured for patient 1 at all time points is shown in b and for all other patients in c
Assessing immune function
Figure 5 shows the PHA response of patient lymphocytes at pre-treatment and weeks 2, 4, 8 and 12. For patients 1, 2, 4 and 5 there is a potentiation of the SI at 2 weeks compared to pre-treatment and this level is higher than any subsequent measurement. Patient 6 showed no difference at week 2 compared to the pre-treatment and patient 3 had a very high pre-treatment response level to PHA and subsequent responses were successively reduced. h5T4 specific immune responses were measured using proliferation to monocytes infected with MVA-h5T4 or control MVA-LacZ or to pools of overlapping 32 mer peptides covering the 5T4 sequence. Only patient 1, who had higher levels of Treg cells pre-treatment, showed clear evidence of a potentiation of 5T4 proliferative responses (Fig. 6). In the MVA-h5T4 proliferation, this increased level was also seen at week 4. Using the peptide pools, the maximum proliferation was seen only at week 2 coincident with the nadir of Treg cell levels.
Fig. 5.

Proliferative response to 5 μg/mL PHA. Peripheral blood mononuclear cells isolated from all patients at different time points before and after treatment were stimulated with 5 μg/mL PHA. Following 5-day incubation, [3H]TdR was added for the last 18 h and proliferation was measured. Results are shown as the mean SI for triplicate measurements and bars refer to the standard error of the mean
Fig. 6.

Proliferative response to h5T4. Peripheral blood mononuclear cells isolated from patient 1 before and at weeks 2, 4, 8 and 12 after treatment were incubated with media (ie unstimulated cells), MVA-LacZ or MVA-h5T4 (a) or with pools of three 32 mer peptides spanning the whole sequence of h5T4 protein (b). Following 5-day incubation, [3H]TdR was added for the last 18 h and proliferation was measured. Results are shown as the mean CPM for MVA-LacZ and MVA-h5T4 or SI for the h5T4 peptide pools of triplicate measurements. Bars refer to the standard error of the mean
Discussion
This study demonstrates that CD25+ cell depletion from a leukapheresis product followed by autologous re-infusion into lymphopenic patients is feasible and results in reduction of host Treg cell population. There are a number of possible explanations for why the depletion of Treg cells in the patients was, however, short-lived. Firstly low levels of CD4+CD25+Treg cells in the leukapheresis product, on re-infusion into a lymphopenic environment, could have expanded rapidly. Secondly, the conditioning regimen that was used is lymphodepleting, but not myeloablative as demonstrated by the recovery in blood counts without the need for stem cell rescue. Compared to complete myeloablation, sub-total lymphodepletion is likely to result in residual host T cells including Treg cells and increased competition for activating cytokines limiting the activation and proliferation of the adoptively transferred cells. In this context, the recovery in Treg cells may reflect repopulation of residual host Treg cells. The same chemotherapy regime as used in this study has previously been effective as part of a non-myeloablative allogeneic stem-cell transplantation strategy in metastatic renal cell carcinoma in which sustained regression of disease was demonstrated [21]. There is, however, experimental evidence to suggest that complete myeloablation followed by autologous (or syngeneic) haematopoetic stem cell rescue might further enhance treatment efficacy in ACT [25]. Nevertheless, in the context of haematological malignancies stem-cell transplantation strategies using completely myeloablative conditioning regimes are associated with substantial morbidity and mortality and this must be balanced with the patient’s best interest in the development of new therapies.
A third possible explanation for the rapid regeneration of Treg cells could be the conversion of non-regulatory T cells into CD4+CD25+ Treg cells following adoptive transfer. It is an area of active debate within the field whether Treg cells are either a dedicated cell lineage that develops exclusively in the thymus or can be derived from mature non-regulatory T cells in the periphery [5, 26]. Several studies have suggested that, in the presence of cytokines such as transforming growth factor-β (TGF-β) during antigen stimulation, naïve CD4+CD25−FOXP3+ T cells can be converted to CD4+CD25+ FOXP3+ T cells [27, 28]. In the majority of cases minor contamination with natural Treg cell populations that were selectively expanded could not be ruled out. In a specific model system, however, the proof of concept of differentiation to natural Treg cells outside the thymus has been established [29].
A recently published study in patients with metastatic melanoma also found that there was rapid Treg cell repopulation after conditioning chemotherapy and re-infusion of autologous lymphocytes depleted of CD25+ cells [30]. They found that as high as 63% of peripheral CD4+ T cells expressed FOXP3 shortly after cell infusion. Crucially, however, patients in that study received high-dose intravenous IL2 following infusion of the CD25 depleted cell product. CD25 is the α-chain of the high affinity IL2 receptor (IL-2R) and evidence exists to show that IL2 plays an important role in the generation and maintenance of Treg cells [31]. Indeed, it has been shown that when IL2 is administered to lymphopenic patients, the CD4+CD25HI Treg compartment is markedly increased compared to patients who did not receive IL2 [32]. Therefore in our study we avoided the administration of IL2 following re-infusion of the CD25+ depleted leukapheresis product.
A number of other ACT strategies have been explored in renal cell carcinoma including the use of lymphokine-activated killer (LAK) cells [33], TILs [34] and autolymphocyte therapy [35]. In LAK and TIL therapy, cells were cultured ex vivo with IL2 and usually administered in combination with exogenous IL2. Thus the cells were exposed to conditions favourable for the expansion of Treg cells. Conversely, in autolymphocyte therapy, no IL2 was added to the ex vivo cell culture media [36]. Instead T cells were exposed to cytokines secreted by autologous PBMCs, which had been activated by antiCD3 monoclonal antibodies. In this case it was shown that ex vivo lysis of tumour targets was mediated by CD45RO+ memory T cells [37]. Although specific depletion of Treg cells was not described, the lack of exogenous IL2 may have restrained expansion of what are now known as Treg cells. However, despite some early clinical promise [35], the results of larger trials were not confirmatory [38] and autolymphcyte therapy has not been pursued in recent years.
The immune response against h5T4 seen in patient 1 in our study is intriguing, particularly since this patient also had resolution of a liver metastasis on CT scan (although there was documented progression elsewhere) and was also the only patient with papillary cell histological subtype. In addition, this patient had high Treg level at presentation; therefore, it might be important to target patients with high baseline Treg levels. Unfortunately the immune modulation in other patients was limited to polyclonal response to PHA. It remains unclear whether the activation noted was related to re-infusion of Treg depleted leukapheresis product or another effect of the therapy such as the lymphodepletion or a chemotherapy effect.
Although the conditioning chemotherapy was not myeloablative, it was still associated with significant toxicity. All six patients experienced grade IV haematological toxicity. Two patients had life threatening non-haematological toxicity. The first developed aspergillous pneumonia, which was treated with appropriate antifungal medication, and the patient made a gradual recovery. A second patient developed a delayed, but rapidly progressive neurological syndrome and died 2 months after receiving the cell therapy. Post mortem examination revealed demyelination and severe axonal damage affecting predominantly the occipital lobes. The clinical and pathological pictures were remarkably similar to those described in patients who received high doses of fludarabine during early dose finding studies for the drug [39]. The syndrome has also been reported in around 0.2–0.5% of patients treated with the standard-dose fludarabine used in this trial (25 mg/m2 daily for 5 days) [40]. The typical reported picture is of delayed neurotoxicity at around 21–43 days after starting treatment and consisting of optic neuritis, cortical blindness, altered mental status, generalised weakness and in some cases seizures. Post mortem examinations consistently show acute demyelination. In a number of cases, brain biopsies have been positive for JC virus suggesting this may be a causative agent. JC virus infects oligodendroglial cells during childhood and re-activation is associated with severe immunosuppression, whether from lymphoproliferative disorders or from therapeutic causes, resulting in multiple foci of demyelination [41]. However, there was no evidence of JC virus reactivation either in the CSF or at postmortem in the case reported here. In a series of 35 reported patients with metastatic melanoma who received the same conditioning treatment prior to TIL infusion, one patient developed cortical blindness attributed to fludarabine [19]. Interestingly there is emerging data, which suggest that leucoencephalopathy may also be associated with other immunotherapies such as rituximab [42]. The small number of patients in our trial precludes any conclusions as to whether these patients were at increased risk over and above their level of immunosuppression resulting from advanced cancer and receiving fludarabine. It is, however, clearly something that will require close monitoring in future ACT studies using fludarabine and other conditioning regimens. In a broader context, the toxicity of the currently used non-specific chemotherapy is not inconsiderable and the identification of less toxic, ideally more specific, conditioning therapy is an important goal.
Recent publications have demonstrated the power of ACT after host preconditioning by lymphodepletion as an anticancer strategy [19, 43]. There is increasing interest in the use of genetic modification of T cells as part of an ACT strategy, for example with tumour specific TCRs or with antibody based chimeric immune receptors (CIRs). Whilst preconditioning with lymphodepleting chemotherapy appears to play a key role, presumably at least in part by depletion of host Treg cells, their success may be affected by the presence of Treg cells within the adoptively transferred cellular product. This study examined the use of CD25+ cell depletion of the leukapheresis collection as one mechanism to reduce the impact of these transferred Treg cells.
The primary clinical endpoint of feasibility was met as the leukapheresis product was successfully depleted (>2 log) in five of six cases with no observed toxicities relating directly to the re-infusion of the CD25+ depleted cells. However, the documented side effects relating to the chemotherapy-conditioning regimen, including the death of one patient from a progressive neurological syndrome indicates that the regimen carries a risk of significant toxicity. The transient nature of the reduction in numbers of Treg cells and any changes in lymphocyte functionality resulting from the therapy appeared short-lived and alternative strategies for effective lymphodepletion therefore need to be considered. Possible approaches include the use of FOXP3 antisense molecules [44], or directed immunotoxins [45, 46]. Nevertheless, the results of this study provide proof of concept in potentiation of tumour antigen T cell responses when Treg cell levels are depleted. In addition to improving the lymphodepletion, future studies will look to augment this potentiation, for example through the incorporation of vaccination against tumour antigens into the regimen.
Acknowledgments
We thank all the bodies that funded this work including CRUK, The ATTACK Project (LSH-CT-2005-018914) an Integrated Project funded under the Sixth EU Framework Programme and The Kay Kendall Leukaeamia Fund. Thanks also go to Miltenyi Biotec for their support and provision of training and CliniMACS® CD25 Reagent for the study. We are also grateful to Maria Hulston and Lesley McDonald for their invaluable assistance with the CD25 depletion. EBA is supported by the National Blood Service, NHS Blood and Transplant and with funding from the NHS R&D Directorate.
References
- 1.Schwartz RH. Natural regulatory T cells and self-tolerance. Nat Immunol. 2005;6:327–330. doi: 10.1038/ni1184. [DOI] [PubMed] [Google Scholar]
- 2.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 3.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 4.Dieckmann D, Plottner H, Berchtold S, Berger T, Schuler G. Ex vivo isolation and characterization of CD4(+)CD25(+) T cells with regulatory properties from human blood. J Exp Med. 2001;193:1303–1310. doi: 10.1084/jem.193.11.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nat Immunol. 2005;6:331–337. doi: 10.1038/ni1179. [DOI] [PubMed] [Google Scholar]
- 6.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 7.Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, Bowcock AM. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest. 2000;106:R75–R81. doi: 10.1172/JCI11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, Bricarelli FD, Byrne G, McEuen M, Proll S, Appleby M, Brunkow ME. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27:18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- 9.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 10.Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY. Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity. 2004;21:267–277. doi: 10.1016/j.immuni.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 11.Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res. 2003;9:606–612. [PubMed] [Google Scholar]
- 12.Ichihara F, Kono K, Takahashi A, Kawaida H, Sugai H, Fujii H. Increased populations of regulatory T cells in peripheral blood and tumor-infiltrating lymphocytes in patients with gastric and esophageal cancers. Clin Cancer Res. 2003;9:4404–4408. [PubMed] [Google Scholar]
- 13.Griffiths RW, Elkord E, Gilham DE, Ramani V, Clarke N, Stern PL, Hawkins RE. Frequency of regulatory T cells in renal cell carcinoma patients and investigation of correlation with survival. Cancer Immunol Immunother. 2007;56:1743–1753. doi: 10.1007/s00262-007-0318-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang HY, Lee DA, Peng G, et al. Tumor-specific human CD4+ regulatory T cells and their ligands: implications for immunotherapy. Immunity. 2004;20:107–118. doi: 10.1016/S1074-7613(03)00359-5. [DOI] [PubMed] [Google Scholar]
- 15.Cheever MA, Greenberg PD, Fefer A. Specificity of adoptive chemoimmunotherapy of established syngeneic tumors. J Immunol. 1980;125:711–714. [PubMed] [Google Scholar]
- 16.North RJ. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J Exp Med. 1982;155:1063–10744. doi: 10.1084/jem.155.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baccala R, Gonzalez-Quintial R, Dummer W, Theofilopoulos AN. Tumor immunity via homeostatic T cell proliferation: mechanistic aspects and clinical perspectives. Springer Semin Immunopathol. 2005;27:75–85. doi: 10.1007/s00281-004-0196-9. [DOI] [PubMed] [Google Scholar]
- 18.Gattinoni L, Powell DJ, Jr, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. 2006;6:383–393. doi: 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163:5211–5218. [PubMed] [Google Scholar]
- 21.Childs R, Chernoff A, Contentin N, Bahceci E, Schrump D, Leitman S, Read EJ, Tisdale J, Dunbar C, Linehan WM, Young NS, Barrett AJ. Regression of metastatic renal-cell carcinoma after nonmyeloablative allogeneic peripheral-blood stem-cell transplantation. N Engl J Med. 2000;343:750–758. doi: 10.1056/NEJM200009143431101. [DOI] [PubMed] [Google Scholar]
- 22.Griffiths RW, Gilham DE, Dangoor A, Ramani V, Clarke NW, Stern PL, Hawkins RE. Expression of the 5T4 oncofoetal antigen in renal cell carcinoma: a potential target for T-cell-based immunotherapy. Br J Cancer. 2005;93:670–677. doi: 10.1038/sj.bjc.6602776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harrop R, Connolly N, Redchenko I, Valle J, Saunders M, Ryan MG, Myers KA, Drury N, Kingsman SM, Hawkins RE, Carroll MW. Vaccination of colorectal cancer patients with modified vaccinia Ankara delivering the tumor antigen 5T4 (TroVax) induces immune responses which correlate with disease control: a phase I/II trial. Clin Cancer Res. 2006;12:3416–24. doi: 10.1158/1078-0432.CCR-05-2732. [DOI] [PubMed] [Google Scholar]
- 24.Mulryan K, Ryan MG, Myers KA, et al. Attenuated recombinant vaccinia virus expressing oncofetal antigen (tumor-associated antigen) 5T4 induces active therapy of established tumors. Mol Cancer Ther. 2002;1:1129–1137. [PubMed] [Google Scholar]
- 25.Wrzesinski C, Paulos CM, Gattinoni L, et al. Hematopoietic stem cells promote the expansion and function of adoptively transferred antitumor CD8 T cells. J Clin Invest. 2007;117:492–501. doi: 10.1172/JCI30414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.von Boehmer H. Mechanisms of suppression by suppressor T cells. Nat Immunol. 2005;6:338–344. doi: 10.1038/ni1180. [DOI] [PubMed] [Google Scholar]
- 27.Chen W, Jin W, Hardegen N, et al. Conversion of peripheral CD4 + CD25- naive T cells to CD4 + CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng SG, Wang JH, Gray JD, Soucier H, Horwitz DA. Natural and induced CD4 + CD25+ cells educate CD4+CD25-cells to develop suppressive activity: the role of IL-2, TGF-beta, and IL-10. J Immunol. 2004;172:5213–5221. doi: 10.4049/jimmunol.172.9.5213. [DOI] [PubMed] [Google Scholar]
- 29.Apostolou I, von Boehmer H. In vivo instruction of suppressor commitment in naive T cells. J Exp Med. 2004;199:1401–1408. doi: 10.1084/jem.20040249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Powell DJ, Jr, de Vries CR, Allen T, Ahmadzadeh M, Rosenberg SA. Inability to mediate prolonged reduction of regulatory T Cells after transfer of autologous CD25-depleted PBMC and interleukin-2 after lymphodepleting chemotherapy. J Immunother. 2007;30:438–447. doi: 10.1097/CJI.0b013e3180600ff9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malek TR, Bayer AL. Tolerance, not immunity, crucially depends on IL-2. Nat Rev Immunol. 2004;4:665–674. doi: 10.1038/nri1435. [DOI] [PubMed] [Google Scholar]
- 32.Zhang H, Chua KS, Guimond M, Kapoor V, Brown MV, Fleisher TA, Long LM, Bernstein D, Hill BJ, Douek DC, Berzofsky JA, Carter CS, Read EJ, Helman LJ, Mackall CL. Lymphopenia and interleukin-2 therapy alter homeostasis of CD4 + CD25+ regulatory T cells. Nat Med. 2005;11:1238–1243. doi: 10.1038/nm1312. [DOI] [PubMed] [Google Scholar]
- 33.Parkinson DR, Fisher RI, Rayner AA, Paietta E, Margolin KA, Weiss GR, Mier JW, Sznol M, Gaynor ER, Bar MH, et al. Therapy of renal cell carcinoma with interleukin-2 and lymphokine-activated killer cells: phase II experience with a hybrid bolus and continuous infusion interleukin-2 regimen. J Clin Oncol. 1990;8:1630–1636. doi: 10.1200/JCO.1990.8.10.1630. [DOI] [PubMed] [Google Scholar]
- 34.Figlin RA, Thompson JA, Bukowski RM, Vogelzang NJ, Novick AC, Lange P, Steinberg GD, Belldegrun AS. Multicenter, randomized, phase III trial of CD8(+) tumor-infiltrating lymphocytes in combination with recombinant interleukin-2 in metastatic renal cell carcinoma. J Clin Oncol. 1999;17:2521–2529. doi: 10.1200/JCO.1999.17.8.2521. [DOI] [PubMed] [Google Scholar]
- 35.Osband ME, Lavin PT, Babayan RK, Graham S, Lamm DL, Parker B, Sawczuk I, Ross S, Krane RJ. Effect of autolymphocyte therapy on survival and quality of life in patients with metastatic renal-cell carcinoma. Lancet. 1990;335:994–998. doi: 10.1016/0140-6736(90)91064-H. [DOI] [PubMed] [Google Scholar]
- 36.Dillman RO. Lymphocyte therapy of renal cell carcinoma. Expert Rev Anticancer Ther. 2005;5:1041–1051. doi: 10.1586/14737140.5.6.1041. [DOI] [PubMed] [Google Scholar]
- 37.Gold JE, Ross SD, Krellenstein DJ, LaRosa F, Malamud SC, Osband ME. Adoptive transfer of ex vivo activated memory T-cells with or without cyclophosphamide for advanced metastatic melanoma: results in 36 patients. Eur J Cancer. 1995;31A:698–708. doi: 10.1016/0959-8049(94)00523-8. [DOI] [PubMed] [Google Scholar]
- 38.Lavin PT, Maar R, Franklin M, Ross S, Martin J, Osband ME. Autolymphocyte therapy for metastatic renal cell carcinoma: initial clinical results from 335 patients treated in a multisite clinical practice. Transplant Proc. 1992;24:3059–3064. [PubMed] [Google Scholar]
- 39.Warrell RP, Jr, Berman E. Phase I and II study of fludarabine phosphate in leukemia: therapeutic efficacy with delayed central nervous system toxicity. J Clin Oncol. 1986;4:74–79. doi: 10.1200/JCO.1986.4.1.74. [DOI] [PubMed] [Google Scholar]
- 40.Gonzalez H, Bolgert F, Camporo P, Leblond V. Progressive multifocal leukoencephalitis (PML) in three patients treated with standard-dose fludarabine (FAMP). Hematol Cell Ther. 1999;41:183–186. doi: 10.1007/s00282-999-0183-7. [DOI] [PubMed] [Google Scholar]
- 41.Sweet TM, Del Valle L, Khalili K. Molecular biology and immunoregulation of human neurotropic JC virus in CNS. J Cell Physiol. 2002;191:249–256. doi: 10.1002/jcp.10096. [DOI] [PubMed] [Google Scholar]
- 42.Freim Wahl SG, Folvik MR, Torp SH. Progressive multifocal leukoencephalopathy in a lymphoma patient with complete remission after treatment with cytostatics and rituximab: case report and review of the literature. Clin Neuropathol. 2007;26:68–73. doi: 10.5414/npp26068. [DOI] [PubMed] [Google Scholar]
- 43.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Veldman C, Pahl A, Beissert S, Hansen W, Buer J, Dieckmann D, Schuler G, Hertl M. Inhibition of the transcription factor Foxp3 converts desmoglein 3-specific type 1 regulatory T cells into Th2-like cells. J Immunol. 2006;176:3215–3222. doi: 10.4049/jimmunol.176.5.3215. [DOI] [PubMed] [Google Scholar]
- 45.Attia P, Powell DJ, Jr, Maker AV, Kreitman RJ, Pastan I, Rosenberg SA. Selective elimination of human regulatory T lymphocytes in vitro with the recombinant immunotoxin LMB-2. J Immunother. 2006;29:208–214. doi: 10.1097/01.cji.0000187959.45803.0c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dannull J, Su Z, Rizzieri D, et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest. 2005;115:3623–3633. doi: 10.1172/JCI25947. [DOI] [PMC free article] [PubMed] [Google Scholar]