

Abstract
Immunotherapy for leukemia is a promising targeted strategy to eradicate residual leukemic cells after standard therapy, in order to prevent relapse and to prolong the survival of leukemia patients. However, effective anti-leukemia immune responses are hampered by the weak immunogenicity of leukemic cells. Therefore, much effort is made to identify agents that could increase the immunogenicity of leukemic cells and activate the immune system. Synthetic agonists of Toll-like receptor (TLR)7 and TLR8 are already in use as anticancer treatment, because of their ability to activate several immune pathways simultaneously, resulting in effective antitumor immunity. However, for leukemic cells little is known about the expression of TLR7/8 and the direct effects of their agonists. We hypothesized that TLR7/8 agonist treatment of human acute myeloid leukemia (AML) cells would lead to an increased immunogenicity of AML cells. We observed expression of TLR7 and TLR8 in primary human AML cells and AML cell lines. Passive pulsing of primary AML cells with the TLR7/8 agonist R-848 resulted in increased expression of MHC molecules, production of proinflammatory cytokines, and enhanced allogeneic naïve T cell-stimulatory capacity. These effects were absent or suboptimal if R-848 was administered intracellularly by electroporation. Furthermore, when AML cells were cocultured with allogeneic PBMC in the presence of R-848, interferon (IFN)-γ was produced by allogeneic NK and NKT cells and AML cells were killed. In conclusion, the immunostimulatory effect of the TLR7/8 agonist R-848 on human AML cells could prove useful for the design of TLR-based immunotherapy for leukemia.
Keywords: TLR7, TLR8, R-848, AML, Immunotherapy
Introduction
To prolong remission and survival of patients with acute myeloid leukemia (AML), there is a strong need for the development of novel adjuvant treatment options, such as immunotherapy, to eliminate residual leukemic cells after standard therapy. The aim of leukemia immunotherapy is to specifically eradicate leukemic cells by activation of the immune system. However, effective anti-leukemia immune responses are hampered by the weak immunogenicity of leukemic cells. Toll-like receptors (TLR) are a family of pattern-recognition receptors (PRR) that are considered to be very important in the induction of effective immune responses. Expression of TLR has been shown for several immune cells, whereby the amount of expression and the combination of TLR differs from one cell type to another [1, 2]. Signaling pathways activated by TLR ligands lead to NF-κB and MAPK activation, cytokine gene transcription (e.g., IL-6, IFN-α, and IL-12) and increased costimulatory molecule expression (e.g., CD40, CD80, and CD86) [3].
Before ssRNA was identified as the natural ligand for TLR7 and TLR8, imidazoquinolines (adenosine analogs) were already used as agonists of TLR7 and TLR8 [4]. In animal models and in clinical studies, it was shown that these synthetic ligands of TLR7/8 provoked a profound antitumoral immune response [5]. The imidazoquinoline imiquimod stimulates the secretion of proinflammatory cytokines by human blood cells through activation of transcription factors, like NF-κB, resulting in a T helper (Th)1-polarized immune response [6]. Administration of imiquimod to human myeloid or plasmacytoid dendritic cells (DC), the most professional antigen-presenting cells (APC), leads to both cytokine production and to the ability to kill tumor cells [7, 8]. The use of imiquimod as a cancer vaccine adjuvant also results in CTL and NK cell activation [5]. Resiquimod (R-848) is another imidazoquinoline that binds to both TLR7 and TLR8 [4], resulting in an enhanced activity profile as compared to imiquimod [9]. Several animal studies have shown that resiquimod can enhance specific T cell responses when administered topical or systemic as vaccine adjuvant [10–13]. Therefore, R-848 has become the subject of active research to determine its effects on human cells for immunotherapeutic purposes [14]. A clinical study has already demonstrated that topical application of resiquimod is safe and effective at activating the local immune response [15]. Progenitor cells [16] and cancer cells [17] show detectable levels of TLR7 and TLR8 expression [18]. For plasma cells and cell lines from multiple myeloma (MM) patients, it was shown that addition of TLR ligands like R-848 resulted in increased survival and proliferation of the plasma cells, providing a possible link between inflammation and progression of MM [17, 19]. For chronic lymphocytic leukemia (CLL) cells however, activation of TLR7 did not result in a strong proliferation, but in an increased susceptibility to chemotherapeutic agents [20]. It was also shown that TLR7 activation of CLL cells resulted in a heterogeneous increase in expression of costimulatory molecules and production of proinflammatory cytokines, further enhanced by the simultaneous addition of protein kinase C (PKC) agonists [21].
As there is a growing interest in imidazoquinolines as cancer vaccine adjuvants, we wanted to assess the effect of TLR7/8 ligands on primary AML cells and cell lines. Therefore, we determined the expression of TLR7 and TLR8 in AML cells and we measured the effects of R-848 on cell viability, costimulatory molecule expression, cytokine production, and immunogenicity of AML cells, as well as the ability of AML cells to stimulate activation of allogeneic T cells, NK cells, and NKT cells in the presence of R-848.
Materials and methods
Source of cell lines and primary cells
Peripheral blood from healthy donors was obtained from buffy coats provided by the Antwerp Blood Transfusion Center. Peripheral blood and bone marrow samples from 21 newly diagnosed or relapsed patients with AML (Table 1) were collected after obtaining informed consent. Only patients with >75% CD33+ or CD34+ AML cells in blood or bone marrow were included in this study. Experiments were performed on fresh or thawed AML cells. Peripheral blood mononuclear cells (PBMC) and bone marrow mononuclear cells (BMMC) were isolated by Ficoll-Paque Plus gradient separation (Amersham Biosciences, Uppsala, Sweden). The human myeloid leukemia cell lines NB-4 and U-937 were obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany) and the American Type Culture Collection (Rockville, MD, USA), respectively. Cells were cultured in complete medium consisting of Iscove’s modified Dulbecco’s medium (IMDM; Cambrex Bio Science, Verviers, Belgium) supplemented with L-glutamine (584 mg/L), 4-(2-hydroxy-ethyl)-1-piperazineethane-sulfonic acid (HEPES; 25 mM), gentamicin (10 mg/L; Invitrogen, Merelbeke, Belgium), amphotericin B (1 mg/L Fungizone; Invitrogen), and 10% fetal bovine serum (FCS; Perbio Science, Erembodegem, Belgium). Cells were maintained in logarithmic growth phase at 37°C in a humidified atmosphere supplemented with 5% CO2.
Table 1.
Demographic and clinical features of acute myeloid leukemia patients
| Patient | Gender | Age (years) | PB or BM | FAB | Karyotype |
|---|---|---|---|---|---|
| AML P1 | F | 79 | BM | M2 | Normal |
| AML P2 | M | 53 | BM | M1 | Normal |
| AML P3 | F | 77 | PB | M6 | 46, XX, t(5;17) |
| AML P4 | F | 78 | PB | M1 | Normal |
| AML P5 | M | 63 | PB | M5a | 46, XY/73–78, XXXY, 1q-(3:14) |
| AML P6 | M | 62 | PB | Sec AML | Normal |
| AML P7 | M | 62 | PB | M2 | 46, XY, der(17) t(8;17) |
| AML P8 | M | 77 | BM | Sec AML | 46, XY, t(9;22) |
| AML P9 | M | 75 | BM | M5b | 47, XY, +8, add(10)(p15) |
| AML P10 | M | 77 | BM | M2 | Normal |
| AML P11 | F | 73 | PB | M5b | Normal |
| AML P12 | M | 23 | BM | M2 | Normal |
| AML P13 | M | 69 | PB/BM | M5a | Normal |
| AML P14 | M | 36 | PB/BM | M2 | Normal |
| AML P15 | M | 71 | PB | M2 | Normal |
| AML P16 | M | 62 | PB | M2 | Normal |
| AML P17 | F | 71 | PB | Sec AML | Normal |
| AML P18 | F | 25 | PB | M5a | Normal |
| AML P19 | M | 30 | PB | M3 | Normal |
| AML P20 | F | 67 | BM | M2 | 46, XX, del(5q) |
| AML P21 | F | 61 | BM | M2 | 46, XX, inv(16) |
Abbreviations: M, male; F, female; PB, peripheral blood; BM, bone marrow; FAB, French–American–British classification system; Sec AML, secondary AML
Treatment of AML cells with R-848
The AML cells were treated with R-848 (PharmaTech, Shanghai, China) by means of passive pulsing or electroporation. Resiquimod (R-848; 4-amino-2-ethoxymethyl-a,a-demethyl-1H-imidazo-{4,5-c}quinoline-1-ethanol) is a member of the imidazoquinoline family (adenosine analogs). Passive pulsing of AML cells with R-848 was performed by adding 5 μg/mL R-848 during 24 h (unless otherwise stated). The cells were extensively washed to remove excess of R-848. Electroporation of R-848 was performed as described previously for poly(I:C) [22, 23] with minor modifications. After washing, 0.2 mL of cell suspension was mixed with 10 μg R-848 and electroporated for 7 ms in a 0.4 cm cuvette (Thermo Fisher Scientific, Zellik, Belgium) at 300 V using a GenePulser Xcell device (Bio-Rad Laboratories, Nazareth, Belgium). Immediately after electroporation, cells were washed twice to remove extracellular R-848. Mock-electroporation was performed similarly, but without the addition of R-848 in the electroporation protocol. About 24 h after R-848 treatment of AML cells, cytokines in the supernatant were measured using a multiplex fluorescent bead immunoassay (Human Th1/Th2 11plex Kit; Bender MedSystems, Vienna, Austria). Interferon (IFN)-α production by AML cells upon R-848 treatment was performed by IFN-α enzyme-linked immunosorbent assay (ELISA; PBL Biomedical Laboratories, New Jersey, USA).
Priming of naïve T cells
After 24 h of R-848 treatment, primary AML cells were extensively washed to remove all produced cytokines and the excess of R-848 when passively pulsed. Washed AML cells were plated with allogeneic naïve T cells in 48-well plates at ratio 1:1. Naïve CD4+CD45RA+ T cells were isolated from PBMC using the naïve CD4+ T cell enrichment kit, according to the protocol (Easysep, StemCellTechnologies, Grenoble, France). After 3 days, IL-2 (30 U/mL; Biosource) was added and at day 5, supernatant was taken to determine the level of IFN-γ by ELISA (Biosource).
Addition of R-848 to AML/PBMC cocultures
To determine the effect of R-848 on cytokine production in cocultures of AML cells and PBMC, thawed primary AML cells or NB-4 cells were cocultured with thawed PBMC at ratio 1:1 in 48-well plates in the presence or absence of 5 μg/mL R-848. In the transwell experiments, the AML cells were put in transwells (pore size 0.4 μm; BD) during coculture with the PBMC. After 3 days, supernatant was harvested for multiplex immunoassay.
For the intracellular cytokine staining, primary AML or NB-4 cells were cocultured with thawed PBMC at ratio 1:1 with or without R-848 (5 μg/mL). After 30 h, brefeldine A (Golgi Plug; BD, Erembodegem, Belgium) was added overnight to block cytokine secretion. Thereafter, membrane staining was performed for CD3 and CD56 with specific PerCP- and PE-labeled antibodies, respectively. Then, cells were lysed and permeabilized (BD) before staining with a specific FITC-labeled monoclonal antibody directed against IFN-γ (Pharmingen, Erembodgem, Belgium). To determine the effect of type I IFN on cocultures of AML cells and PBMC, IFN-α (PeproTech, London, UK) was added at 1 × 104 U/mL.
A flow cytometric cytotoxicity assay was performed as described previously with minor modifications [24]. Briefly, AML cells were labeled with PKH67 (Sigma) according to the manufacturer’s protocol. The PKH67-labeled AML cells were cocultured for 16 h with fresh PBMC or highly purified NK cells (isolation by negative selection; Miltenyi Biotec, Utrecht, The Netherlands) at effector:target ratio 5:1. Thereafter, cells were stained with annexin V and PI to determine viability of the PKH67-positive AML cells. The percentage of killing was determined by the fraction of non-viable (cells that stained positive for annexin V and/or PI) PKH67-positive cells. Neutralizing antibodies directed against human IL-6 and TNF-α were purchased from R&D (Oxon, UK) and were used at 5 μg/mL.
Flow cytometry
Apoptosis was measured by staining with annexin V (FITC-conjugated; BD) and propidium iodide (Sigma, Bornem, Belgium) to distinguish between apoptotic and necrotic cells. Staining of B7 and MHC molecules on AML cells was performed using FITC- or PE-labeled monoclonal antibodies for CD80, CD86, HLA-ABC, and HLA-DR (BD). Propidium iodide was added to include only viable cells in the analysis. Intracellular staining of TLR7 and TLR8 on AML cells was performed using monoclonal antibodies for TLR7 and TLR8 (FITC- and PE-labeled, respectively; Imgenex, San Diego, USA), costained with monoclonal antibodies directed against CD33 or CD34 (BD). Before staining the cells with TLR7 and TLR8 antibodies, cells were fixed for 10 min at room temperature using lyse/fix solution (BD) and permeabilized for 10 min at room temperature using Perm2 solution (BD). Three-color flow cytometry was performed on a FACScan (BD). Flow cytometry in the cytotoxicity assay was performed on a CyFlow (Partec, Münster, Germany). Relevant fluorochrome-labeled isotype-matched antibodies were used as negative controls. Routinely, 1 × 104 events per target population were analyzed using CellQuest software (BD) or FlowJo software version 7.2 (Tree Star, Ashland, USA).
Reverse transcriptase-polymerase chain reaction (RT-PCR)
Total RNA was isolated from 1 to 5 × 106 cells using an RNeasy kit (Qiagen, Antwerp, Belgium). DNase treatment was performed during the RNA isolation according to manufacturer’s protocol. cDNA was obtained by incubating 1–3 μg total RNA with 1 mM dNTP, 0.2 μg random hexamer primers, 20 units of RNasin, and 40 units of Moloney murine leukemia virus reverse transcriptase in a total volume of 20 μL for 10 min at 25°C, 1 h at 42°C, and 10 min at 70°C according to manufacturer’s protocol (Fermentas, St. Leon-Rot, Germany).
Quantitative PCR was performed as described previously [22]. Primer pairs were used specific for human TLR7 (forward 5′-AGT GTC TAA AGA ACC TGG-3′; reverse 5′-CTT GGC CTT ACA GAA ATG-3′; amplicon: 545 bp) [25], human TLR8 (forward 5′-CTT CGA TAC CTA AAC CTC TCT AGC AC-3′; reverse 5′-AAG ATC CAG CAC CTT CAG ATG A-3′; amplicon 90 bp) [26], and ABL1 (forward 5′-AGC ATC TGA CTT TGA GCC-3′; reverse 5′-CCC ATT GTG ATT ATA GCC TAA GAC-3′; amplicon: 194 bp). RT-PCR products were analyzed in a 2% agarose gel stained with 0.5 μg/mL ethidium bromide (Sigma) and visualized by UV light.
Statistical analysis
Quantitative experiments were analyzed using Student’s t-test. All P-values were obtained by two-tailed tests and differences were considered statistically significant when P < 0.05.
Results
Expression of TLR7 and TLR8 in AML cells
First, we checked the expression of TLR7 and TLR8 in unmanipulated AML cells by RT-PCR (purity ≥90%). Expression of both TLR7 and TLR8 was clearly detectable in all AML samples tested [11 primary AML patient samples (Fig. 1a) and 2 AML cell lines (NB-4, U-937; data not shown)]. We also checked protein expression by using specific antibodies for intracellular staining. Flow cytometric detection showed that 6 out of 8 primary AML patient samples tested had detectable expression of TLR7, whereas all 8 samples had TLR8 expression (Fig. 1b and data not shown). NB-4 and U-937 AML cells showed only little expression of TLR7, but had a high expression of TLR8 by intracellular staining and flow cytometric detection (Fig. 1b).
Fig. 1.

Expression of TLR7/8 by primary AML cells. a Expression of TLR7 and TLR8 mRNA in AML cells, determined by RT-PCR. Representative results are shown for 3 patients out of 11 tested. Right side of the gel: DNA ladder. ABL1, c-abl oncogene 1 (used as housekeeping gene to check cDNA quality); AML P, AML patient. b Expression of TLR7 and TLR8 protein, determined by intracellular flow cytometry. Histogram overlays show the level of TLR7 and TLR8 expression (filled gray histograms) compared to isotype control staining (open black histograms) (n = 2 per AML cell type tested)
Effect of R-848 treatment on AML cells: viability, phenotype, and cytokine production
To check if the synthetic TLR7/TLR8 ligand R-848 (Resiquimod) induces apoptosis of AML cells, we treated AML cells with 5 μg/mL R-848 and stained the cells with annexine V and PI 24 h after R-848 treatment. Viable cells were defined as both annexine V- and PI-negative cells. No statistically significant decrease in viability of primary AML cells could be detected (P = 0.15; n = 5), although there was a tendency to decreased viability following R-848 treatment (Fig. 2a). For the tested AML cell lines NB-4 and U-937, we could not detect any influence of R-848 treatment on viability of the AML cells (data not shown). As it was shown that TLR7/8 activation can enhance the proliferation of MM cells [17, 19], we monitored the number of AML cells from patients or AML cell lines 24 h after R-848 treatment, as an indicator of cell proliferation. Resiquimod treatment did not result in a statistically significant increase in the amount of AML cells (P = 0.66; n = 7). In conclusion, treatment of AML cells with R-848 does not significantly alter apoptosis, or the number of AML cells.
Fig. 2.

Effect of R-848 treatment on primary AML cells: viability, phenotype, and cytokine production. a Effect of R-848 treatment on cell viability of primary AML cells. The chart shows the mean percentage ± SD of viable cells (n = 5). Annexin V- and PI-negative cells were defined as viable. b Effect of R-848 treatment on the membrane expression of MHC molecules on primary AML cells. Flow cytometry results are expressed as mean fluorescence intensity (MFI; Y-axis) (n = 8). c Effect of R-848 treatment on cytokine production by primary AML cells. The concentration of the cytokines IL-6, IL-1β, and TNF-α was measured in the supernatant 24 h after addition of R-848 to AML cells (n = 10). * P < 0.05. Unmod, unmodified AML cells; R-848, AML cells pulsed with 5 μg/mL R-848
As it is known that TLR activation results in upregulation of costimulatory molecules and MHC molecules on myeloid antigen-presenting cells [27, 28], we hypothesized that R-848 could also upregulate the expression of CD80, CD86, and MHC molecules on the membrane of AML cells. Treatment of primary AML cells (n = 11) or the AML cell lines NB-4 and U-937 with R-848 did not result in de novo expression or upregulation of the costimulatory molecules CD80 or CD86 (data not shown). However, we observed an increase in expression of MHC class I and II molecules on the membrane of primary AML cells (n = 8), measured by flow cytometry 24 h after treatment with R-848. Administration of R-848 by passive pulsing resulted a statistically significant increase in mean fluorescence intensity (MFI) for HLA-ABC (P = 0.01), but not for HLA-DR (P = 0.06) (Fig. 2b). Interestingly, when we focused on the AML cells of 4 patients with intrinsic HLA-DR expression, we observed a statistically significant upregulation of HLA-DR following R-848 treatment in this subgroup (P = 0.04). The AML cell lines NB-4 and U-937, however, did not upregulate MHC molecules after treatment with R-848 (data not shown). In conclusion, primary AML cells but not AML cell lines, respond to treatment with R-848 by increased expression of MHC molecules.
Next, we checked the production of cytokines by AML cells after R-848 treatment. We detected a statistically significant increase in IL-6 production after passive pulsing of primary AML cells with R-848, compared to untreated AML cells (Fig. 2c; n = 10). Similar trends were observed for IL-1β and TNF-α (Fig. 2c). No cytokine production was observed after R-848-treatment of NB-4 and U-937 AML cell lines (data not shown). For IFN-α, 5 out of 12 AML patients secreted detectable amounts of IFN-α (22.1 ± 5.1 pg/1 × 106 AML cells/24 h; n = 5) after R-848 pulsing. In conclusion, passive pulsing of primary AML cells with R-848 results in production of the proinflammatory cytokines IL-6, TNF-α, IL-1β, and/or IFN-α.
Increased immunogenicity of R-848-treated AML cells
To investigate whether R-848 treatment would increase the ability of AML cells to stimulate allogeneic T cells toward a Th1 response, primary AML cells (unmodified or R-848-exposed) were put into coculture with purified allogeneic naïve CD4+CD45RA+ T cells. At day 5, we performed IFN-γ ELISA on the supernatant of the coculture. Primary AML cells exposed to R-848 led to a 6- to 285-fold increase (P < 0.05) in IFN-γ production as compared to naïve T cells cocultured with untreated AML cells for 2 out of 3 patients tested (Fig. 3). In contrast, no increase in IFN-γ was detected in supernatant of cocultures of PBMC when R-848 was introduced into AML cells by means of electroporation (Fig. 3). In conclusion, passive pulsing of primary AML cells with R-848 can increase their immunogenicity, as determined by IFN-γ production by allogeneic naïve T cells.
Fig. 3.

Effect of R-848 treatment on the immunogenicity of primary AML cells. The concentration of IFN-γ detected in the supernatant taken after 5 days of coculturing allogeneic naïve T cells and primary AML cells. The AML cells were treated with or without R-848 prior to coculture with allogeneic T cells. The results shown are representative of 2 AML patients. AML Unmod, unmodified AML cells; AML EP Mock, mock-electroporated AML cells; AML EP R-848, AML cells electroporated with R-848; AML PP R-848, AML cells passively pulsed with R-848; CD4+CD45RA+ T cells, naïve CD4+ T cells. * P < 0.05
Synergistic cytokine effects of R-848 in cocultures of PBMC and AML cells
Based on the initial observation that passive pulsing of primary AML cells with R-848 increased their immunogenicity, we investigated the effect of R-848 administration to cocultures of AML cells and allogeneic PBMC. In this setting, PBMC could also benefit from the immunostimulatory effects of R-848 administration. After 3 days of coculture, cytokine concentrations were measured by multiplex immunoassay. Both primary AML cells and NB-4 cells were used as AML cells. While only low levels of IFN-γ were induced upon exposure of AML cells or PBMC alone to R-848, coculture of primary AML cells and PBMC resulted in marked levels of IFN-γ. This IFN-γ production could synergistically be boosted by adding R-848 to the coculture (Fig. 4a). Transwell experiments pointed out that this boosted IFN-γ production was strictly dependent on cell contact between primary AML cells and allogeneic PBMC (Fig. 4a). The same trend was seen when NB-4 cells were used as AML cell source (Fig. 4b). In conclusion, the presence of R-848 in cocultures of PBMC and AML cells has a synergistic effect on the production of IFN-γ.
Fig. 4.

Effect of R-848 addition to cocultures of PBMC and AML cells. The concentration of IFN-γ detected in supernatant after 3 days of coculturing AML cells (AML), allogeneic PBMC, and/or R-848. The mean ± SD shown is of 3 independent experiments with AML cells from AML P21 (a) or NB-4 cells (b). −TW, without transwell; +TW, with transwell. * P < 0.05
To find the nature of the cells responsible for the observed IFN-γ production in the coculture of PBMC, AML cells, and R-848, we performed intracellular cytokine staining (ICS) by multiparametric flow cytometry as described in the section “Materials and methods”. As shown in Fig. 5, the ICS data confirmed that IFN-γ was predominantly present in PBMC cocultured with AML cells (primary AML cells or NB-4 cells) in the presence of R-848. Interferon-γ was detected in the CD3−CD56+ NK cell, the CD3+CD56+ NKT cell, and CD3+CD56− T cell population, with the highest percentage of IFN-γ-positive cells in the NK and NKT cell population (Fig. 5). In conclusion, simultaneous treatment of PBMC/AML cell cocultures with R-848 results in potent activation of NK and NKT cells.
Fig. 5.
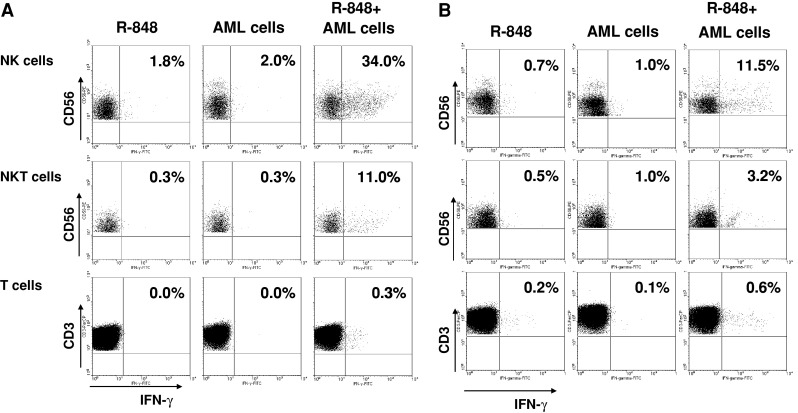
Determination of IFN-γ-producing cells after coculturing PBMC, AML cells, and R-848. Intracellular staining of IFN-γ production in NK cells, NKT cells, and T cells after coculturing PBMC, AML cells, and/or R-848. Dot plots shows the percentage of IFN-γ-positive cells within the NK cell population (above), NKT cell population (middle), and T cell population (below). a Representative result of cocultures of allogeneic PBMC with primary AML cells from AML P3 (n = 3). The results are representative for experiments done with AML cells from 4 different patients. b Representative result of cocultures of allogeneic PBMC with NB-4 cells (n = 4)
Killing of AML cells by addition of R-848 to PBMC and AML cocultures
Subsequently, we investigated if the addition of R-848 to PBMC and AML cocultures resulted in killing of AML cells. Therefore, AML cells were stained with the lipophilic fluorescent probe PKH67 and the percentage of annexin V- and/or PI-positive AML cells was determined 16 h after coculture with allogeneic PBMC in the presence of R-848. Interestingly, for 2 out of 4 patients tested, addition of R-848 to a coculture of AML cells and allogeneic PBMC resulted in decreased viability of AML cells (decrease of 20 ± 2%; Fig. 6). Highly purified NK cells were used to see if this cytotoxic effect could be obtained by NK cells only. As shown in Fig. 6, the decrease in cell viability was less pronounced (6 ± 0%) when using purified NK cells as compared to PBMC. In order to further explore the mechanism of action, neutralizing antibodies directed against the proinflammatory cytokines IL-6 and TNF-α were added to the cocultures. These antibodies were used to block cytokine production by primary AML cells in response to R-848 treatment and to verify if these cytokines contributed to the decreased AML cell viability. However, neutralization of IL-6 and TNF-α did not abrogate the effect of R-848 on decreased AML cell viability in cocultures of PBMC and primary AML cells. If NB-4 cells were cocultured with allogeneic PBMC, cell viability of NB-4 cells also decreased by adding R-848 with 12 ± 7% (n = 3). In conclusion, NB-4 cells and primary AML cells from 2 out of 4 patients tested were killed in the presence of allogeneic PBMC and R-848. Production of IL-6 and TNF-α did not contribute to the decrease in cell viability.
Fig. 6.

Effect of R-848 on killing of primary AML cells by allogeneic PBMC or NK cells. Flow cytometric detection of AML cell viability following coculture of primary AML cells and allogeneic PBMC (black bars) or NK cells (striped bars) in the presence of R-848. PKH67-positive cells that were positive for annexin V and/or PI were defined as non-viable AML cells. (a) Coculture of effector cells with primary AML cells from AML P17. (b) Coculture of effector cells with primary AML cells from AML P16. Results are representative of experiments done with two different effector cell donors.+ R-848, addition of R-848; + Ab, addition of neutralizing antibodies directed against IL-6 and TNF-α; + R-848 + Ab, addition of both R-848 and neutralizing antibodies against IL-6 and TNF-α
Discussion
In this study, we demonstrate that the immunogenicity of AML cells can be increased by treating them with the TLR7/8 agonist R-848 and that addition of R-848 to cocultures of AML cells and allogeneic PBMC results in marked IFN-γ production and killing of AML cells.
To our knowledge, this is the first report on the expression of TLR7 and TLR8 in AML cells. Previously, we already demonstrated the expression of TLR3 in AML cells and the responsiveness of AML cells to the TLR3 ligand poly(I:C) [22]. Expression of TLR7 in leukemic cells was previously found in ALL [29] and CLL cells [21], whereas MM cell lines express both TLR7 and TLR8 [17, 19]. Expression of other TLR in leukemic cells was reported by Maratheftis et al. showing the expression of TLR4 in THP-1, a human monocytic leukemia cell line [30] and of TLR1-4 in bone marrow-derived mononuclear cells (BMMC) and CD34+ hematopoietic progenitor cells from myelodysplastic syndrome (MDS) patients [31].
To check the effect of TLR7/8 agonists on AML cells, we used R-848, because it is a synthetic agonist of both TLR7 and TLR8 [4]. Furthermore, R-848 has been shown to stimulate cytokine secretion, macrophage activation, and cellular immunity to a greater extent than the TLR7 agonist imiquimod [6, 9, 32, 33]. In accordance with these results, we showed that primary AML cells responded to R-848 treatment with increased expression of MHC molecules and secretion of proinflammatory cytokines.
Although it was previously reported that TLR agonists induce apoptosis [22, 34], we could not detect a statistically significant increase in apoptosis or necrosis by treatment of primary AML cells with resiquimod. These results correspond to a report of Schön et al. [34], wherein resiquimod did not induce apoptosis of skin cancer cells either, in contrast to imiquimod. Furthermore, we could not detect a significant change in the number of AML cells by R-848 treatment. The viability and cell count results are in favor of the hypothesis that resiquimod treatment does not lead to AML cell proliferation. It was previously reported that passive pulsing of MM cells with R-848 or loxoribine (as a TLR7 agonist) induced proliferation and increased survival of the tumor cells [17, 19]. In addition, this effect on proliferation of MM cells was partially due to autocrine IL-6 signaling. We also observed increased secretion of IL-6 by passive pulsing of AML cells, but no increase in number of cells. From these data, we deduce that the effects of resiquimod on apoptosis and cell proliferation are dependent on tumor type and therefore need to be carefully examined per cell type. Furthermore, the reported effects of IL-6 on immune responses are contradictory. Although IL-6 is a proinflammatory cytokine, suppressive effects on the activation of immune cells like T cells, NK cells, and NKT cells are also attributed to this cytokine [35–37]. However, we observed high activation of T cells in allo-MLR reactions and activation of NK and NKT cells despite the presence of IL-6.
As shown in the allo-MLR experiments, the immunogenicity of primary AML cells was enhanced after pulsing with R-848 for 2 out of 3 patients tested. It was shown by Caron et al. that R-848 can act directly on CD4+ T cells, leading to the induction of proliferation and cytokine production [25]. Therefore, primary AML cells were thoroughly washed after R-848 treatment to ensure that no remnants of R-848 could be responsible for the observed CD4+ T cell activation. Interestingly, we observed donor-derived heterogeneity in the increase of expression of MHC molecules and the amount of secreted cytokines after treatment of AML cells with R-848, in concordance with the results obtained by Spaner et al. for CLL cells treated with the TLR7 agonist S28690 [21] and with PBMC from healthy donors treated with different TLR7 agonists [38]. This heterogeneity might explain the lack of increased immunogenicity of R-848-pulsed AML cells from 1 patient, but we were not able to confirm this hypothesis, due to the absence of experimental data about increase in MHC molecules or cytokine production for this particular patient. However, the heterogeneity of the AML cell response to R-848 can be considered to be rather limited, because for all 10 AML patients tested (AML P1–10), AML cells from only 1 patient showed a decrease in production of 1 of the 3 cytokines tested (IL-6, IL-1β, and TNF-α) after R-848 treatment (i.e., AML P3 for IL-6). We could not find any link between karyotype or FAB class and degree of responsiveness of the primary AML cells to R-848. Remarkably, only primary AML cells responded to R-848 treatment, in contrast to the AML cell lines NB-4 and U-937, despite expression of TLR7 and TLR8. This non-responsiveness of the AML cell lines NB-4 and U-937 to R-848 treatment warrants further investigation.
We found a striking superiority of R-848 pulsing above electroporation of primary AML cells in terms of upregulation of MHC molecules, cytokine production (data not shown), and immunogenicity (Fig. 3). In previous reports, only passive pulsing or lipofection of cells with TLR7/8 agonists were evaluated, showing that these are suitable administration methods, resulting in TLR7 and/or TLR8 activation [4, 39, 40]. From our observations, electroporation proves less successful in directing the TLR7/8 agonist to its cognate receptors. Since we have previously shown that electroporation of AML cells with the TLR3 ligand poly(I:C) was superior over pulsing [22], we conclude that it is useful to test different transfer techniques for each TLR ligand.
After the initial observations that only passive pulsing and not electroporation with R-848 increased the immunogenicity of primary AML cells, we decided to add R-848 to AML cell/PBMC cocultures. In this way, we could test the potency of R-848 as immune response enhancer in vitro, acting both on AML cells and immune cells. Strikingly, we observed a synergistic effect on IFN-γ production when adding R-848 to AML/PBMC cocultures for AML cells from all 3 patients tested. The amount of secreted IFN-γ exceeded the sum of IFN-γ produced by R-848-pulsed PBMC, by R-848-pulsed AML cells, and by AML/PBMC cocultures, thereby revealing an interaction between AML cells and PBMC after addition of R-848. In order to identify the nature of the cells responsible for the production of IFN-γ, we performed intracellular staining experiments. In these experiments we observed that mainly NK and NKT cells became highly activated by addition of R-848 to AML/PBMC cocultures. Both cell types are important for the antitumor response against AML cells [41–45]. The production of IFN-γ by NK and NKT cells is thought to be important to further polarize the adaptive immune response to a Th1 response [45–47]. Moreover, alloreactive NK cells play a crucial role in the eradication of leukemic cells in allogeneic transplantation models, as part of the graft-versus-leukemia effect [42, 47–49].
It was previously reported that NK cell activation by TLR7/8 agonists is dependent on contact with or factors (e.g., IL-12 or type I IFN) produced by accessory cells in PBMC [50–54]. However, in our experiments, R-848 did not induce comparable NK cell activation (measured by IFN-γ production) when added to PBMC as when added to PBMC/AML cells cocultures. These results show that an NK cell activating factor was provided by the AML cells themselves. This factor could be either a secreted cytokine or a membrane-bound molecule. It is known that NK cells can be activated by IL-2, IL-12, or type I IFN [55]. In our experiments, we showed that R-848-pulsing of AML cells induced only low levels of IFN-α for 5 out of 12 patients and not IL-2 or IL-12 (data not shown). Moreover, addition of IFN-α to PBMC/AML cocultures could not replace R-848 for the induction of high NK-cell activation (data not shown), showing that the effect of R-848 cannot be explained by the presence of IFN-α. Conversely, by performing transwell experiments, we were able to show that a membrane-bound molecule is involved, because NK cell activation in the presence of R-848 was highly dependent on cell-to-cell contact between PBMC and AML cells. NK cells contain a large number of inhibiting and activating receptors [56]. MHC class I molecules are able to bind to inhibitory NK cell receptors, thereby preventing NK cell-mediated killing of normal cells. If tumor cells have low or no expression of HLA class I molecules, they might activate the killing function of NK cells [57]. However, in our experiments, R-848 treatment resulted in an upregulation of MHC class I molecules on AML cells, making it unlikely that NK cells would become activated by the lack of these inhibitory ligands. Conversely, activating ligands might be upregulated on the membrane of R-848-treated AML cells. Therefore, we checked the expression of the ligands for activating NK-cell receptor NKG2D (MICA, MICB, and ULBPs) [58] by using specific antibodies and flow cytometric detection. We were not able to show upregulation of the NKG2D ligands MICA, MICB, or ULBP3 on primary AML cells following R-848 treatment (data not shown). However, other activating NK cell ligands may still be involved [57, 59].
Next to activation of NK cells, we also showed that addition of R-848 to AML/PBMC cocultures resulted in apoptosis and necrosis of AML cells. Cytokines produced by AML cells after R-848 treatment (IL-6 and TNF-α) did not contribute to the decreased AML cell viability in the AML/PBMC cocultures. Furthermore, we showed that cell viability of AML cells was more decreased if AML cells were cocultured with PBMC than when cocultured with isolated NK cells. This suggests that additional factors are provided by accessory cells in PBMC to boost the cytotoxic potential of the killer cells.
In conclusion, we observed that primary AML cells respond to the TLR7/8 agonist R-848 by increased expression of MHC molecules, production of proinflammatory cytokines, and enhanced T cell stimulatory capacity. Furthermore, in the presence of R-848 and AML cells, allogeneic NK and NKT cells were activated and the AML cells were killed. These results support the use of R-848 in TLR-based immunotherapy of AML.
Acknowledgments
This work was supported by Grant no. G.0370.08 and no. G.0082.08 of the Fund for Scientific Research, Flanders, Belgium (FWO-Vlaanderen), by research grants of the Foundation Against Cancer (Stichting tegen Kanker), by Grant no. 802 of the Antwerp University Concerted Research Action (BOF-GOA), by a Methusalem grant of the Antwerp University and by a part of the Interuniversity Attraction Poles (IAP) programme #P6/41 financed by the Belgian Government. Part of this research was kindly supported by the Antwerp University Hospital (UZA). E. L. J. M. Smits holds a fellowship of the Stichting Emmanuel van der Schueren of the Vlaamse Liga tegen Kanker (VLK). E. Lion was funded by a PhD grant of the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT-Vlaanderen).
References
- 1.Hornung V, Rothenfusser S, Britsch S, et al. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168:4531–4537. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- 2.Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. 2002;168:554–561. doi: 10.4049/jimmunol.168.2.554. [DOI] [PubMed] [Google Scholar]
- 3.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 4.Jurk M, Heil F, Vollmer J, et al. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat Immunol. 2002;3:499. doi: 10.1038/ni0602-499. [DOI] [PubMed] [Google Scholar]
- 5.Schon MP, Schon M. TLR7 and TLR8 as targets in cancer therapy. Oncogene. 2008;27:190–199. doi: 10.1038/sj.onc.1210913. [DOI] [PubMed] [Google Scholar]
- 6.Wagner TL, Ahonen CL, Couture AM, et al. Modulation of TH1 and TH2 cytokine production with the immune response modifiers, R-848 and imiquimod. Cell Immunol. 1999;191:10–19. doi: 10.1006/cimm.1998.1406. [DOI] [PubMed] [Google Scholar]
- 7.Gibson SJ, Lindh JM, Riter TR, et al. Plasmacytoid dendritic cells produce cytokines and mature in response to the TLR7 agonists, imiquimod and resiquimod. Cell Immunol. 2002;218:74–86. doi: 10.1016/S0008-8749(02)00517-8. [DOI] [PubMed] [Google Scholar]
- 8.Stary G, Bangert C, Tauber M, et al. Tumoricidal activity of TLR7/8-activated inflammatory dendritic cells. J Exp Med. 2007;204:1441–1451. doi: 10.1084/jem.20070021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wagner TL, Horton VL, Carlson GL, et al. Induction of cytokines in cynomolgus monkeys by the immune response modifiers, imiquimod, S-27609 and S-28463. Cytokine. 1997;9:837–845. doi: 10.1006/cyto.1997.0239. [DOI] [PubMed] [Google Scholar]
- 10.Vasilakos JP, Smith RM, Gibson SJ, et al. Adjuvant activities of immune response modifier R-848: comparison with CpG ODN. Cell Immunol. 2000;204:64–74. doi: 10.1006/cimm.2000.1689. [DOI] [PubMed] [Google Scholar]
- 11.Tomai MA, Imbertson LM, Stanczak TL, et al. The immune response modifiers imiquimod and R-848 are potent activators of B lymphocytes. Cell Immunol. 2000;203:55–65. doi: 10.1006/cimm.2000.1673. [DOI] [PubMed] [Google Scholar]
- 12.Otero M, Calarota SA, Felber B, et al. Resiquimod is a modest adjuvant for HIV-1 gag-based genetic immunization in a mouse model. Vaccine. 2004;22:1782–1790. doi: 10.1016/j.vaccine.2004.01.037. [DOI] [PubMed] [Google Scholar]
- 13.Thomsen LL, Topley P, Daly MG, et al. Imiquimod and resiquimod in a mouse model: adjuvants for DNA vaccination by particle-mediated immunotherapeutic delivery. Vaccine. 2004;22:1799–1809. doi: 10.1016/j.vaccine.2003.09.052. [DOI] [PubMed] [Google Scholar]
- 14.Smits EL, Ponsaerts P, Berneman ZN, et al. The use of TLR7 and TLR8 ligands for the enhancement of cancer immunotherapy. Oncologist. 2008;13:859–875. doi: 10.1634/theoncologist.2008-0097. [DOI] [PubMed] [Google Scholar]
- 15.Sauder DN, Smith MH, Senta-McMillian T, et al. Randomized, single-blind, placebo-controlled study of topical application of the immune response modulator resiquimod in healthy adults. Antimicrob Agents Chemother. 2003;47:3846–3852. doi: 10.1128/AAC.47.12.3846-3852.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sioud M, Floisand Y. TLR agonists induce the differentiation of human bone marrow CD34+ progenitors into CD11c+ CD80/86+ DC capable of inducing a Th1-type response. Eur J Immunol. 2007;37:2834–2846. doi: 10.1002/eji.200737112. [DOI] [PubMed] [Google Scholar]
- 17.Bohnhorst J, Rasmussen T, Moen SH, et al. Toll-like receptors mediate proliferation and survival of multiple myeloma cells. Leukemia. 2006;20:1138–1144. doi: 10.1038/sj.leu.2404225. [DOI] [PubMed] [Google Scholar]
- 18.Spaner DE, Masellis A. Toll-like receptor agonists in the treatment of chronic lymphocytic leukemia. Leukemia. 2007;21:53–60. doi: 10.1038/sj.leu.2404456. [DOI] [PubMed] [Google Scholar]
- 19.Jego G, Bataille R, Geffroy-Luseau A, et al. Pathogen-associated molecular patterns are growth and survival factors for human myeloma cells through Toll-like receptors. Leukemia. 2006;20:1130–1137. doi: 10.1038/sj.leu.2404226. [DOI] [PubMed] [Google Scholar]
- 20.Pellacani A, Tosi P, Zinzani PL, et al. Cytotoxic combination of loxoribine with fludarabine and mafosfamide on freshly isolated B-chronic lymphocytic leukemia cells. Leuk Lymphoma. 1999;33:147–153. doi: 10.3109/10428199909093736. [DOI] [PubMed] [Google Scholar]
- 21.Spaner DE, Shi Y, White D, et al. Immunomodulatory effects of Toll-like receptor-7 activation on chronic lymphocytic leukemia cells. Leukemia. 2006;20:286–295. doi: 10.1038/sj.leu.2404061. [DOI] [PubMed] [Google Scholar]
- 22.Smits EL, Ponsaerts P, Van de Velde AL, et al. Proinflammatory response of human leukemic cells to dsRNA transfection linked to activation of dendritic cells. Leukemia. 2007;21:1691–1699. doi: 10.1038/sj.leu.2404763. [DOI] [PubMed] [Google Scholar]
- 23.Ponsaerts P, Van Tendeloo VF, Cools N, et al. mRNA-electroporated mature dendritic cells retain transgene expression, phenotypical properties and stimulatory capacity after cryopreservation. Leukemia. 2002;16:1324–1330. doi: 10.1038/sj.leu.2402511. [DOI] [PubMed] [Google Scholar]
- 24.Fischer K, Andreesen R, Mackensen A. An improved flow cytometric assay for the determination of cytotoxic T lymphocyte activity. J Immunol Methods. 2002;259:159–169. doi: 10.1016/S0022-1759(01)00507-5. [DOI] [PubMed] [Google Scholar]
- 25.Caron G, Duluc D, Fremaux I, et al. Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-gamma production by memory CD4+ T cells. J Immunol. 2005;175:1551–1557. doi: 10.4049/jimmunol.175.3.1551. [DOI] [PubMed] [Google Scholar]
- 26.Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human toll-like receptors and related genes. Biol Pharm Bull. 2005;28:886–892. doi: 10.1248/bpb.28.886. [DOI] [PubMed] [Google Scholar]
- 27.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 28.Mazzoni A, Segal DM. Controlling the Toll road to dendritic cell polarization. J Leukoc Biol. 2004;75:721–730. doi: 10.1189/jlb.1003482. [DOI] [PubMed] [Google Scholar]
- 29.Corthals SL, Wynne K, She K, et al. Differential immune effects mediated by Toll-like receptors stimulation in precursor B-cell acute lymphoblastic leukaemia. Br J Haematol. 2006;132:452–458. doi: 10.1111/j.1365-2141.2005.05893.x. [DOI] [PubMed] [Google Scholar]
- 30.Maratheftis CI, Giannouli S, Spachidou MP, et al. RNA interference of interferon regulatory factor-1 gene expression in THP-1 cell line leads to Toll-like receptor-4 overexpression/activation as well as up-modulation of annexin-II. Neoplasia. 2007;9:1012–1020. doi: 10.1593/neo.07640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maratheftis CI, Andreakos E, Moutsopoulos HM, et al. Toll-like receptor-4 is up-regulated in hematopoietic progenitor cells and contributes to increased apoptosis in myelodysplastic syndromes. Clin Cancer Res. 2007;13:1154–1160. doi: 10.1158/1078-0432.CCR-06-2108. [DOI] [PubMed] [Google Scholar]
- 32.Burns RP, Jr, Ferbel B, Tomai M, et al. The imidazoquinolines, imiquimod and R-848, induce functional, but not phenotypic, maturation of human epidermal Langerhans’ cells. Clin Immunol. 2000;94:13–23. doi: 10.1006/clim.1999.4804. [DOI] [PubMed] [Google Scholar]
- 33.Bernstein DI, Harrison CJ, Tomai MA, et al. Daily or weekly therapy with resiquimod (R-848) reduces genital recurrences in herpes simplex virus-infected guinea pigs during and after treatment. J Infect Dis. 2001;183:844–849. doi: 10.1086/319262. [DOI] [PubMed] [Google Scholar]
- 34.Schon M, Bong AB, Drewniok C, et al. Tumor-selective induction of apoptosis and the small-molecule immune response modifier imiquimod. J Natl Cancer Inst. 2003;95:1138–1149. doi: 10.1093/jnci/djg016. [DOI] [PubMed] [Google Scholar]
- 35.Sun R, Tian Z, Kulkarni S, et al. IL-6 prevents T cell-mediated hepatitis via inhibition of NKT cells in CD4+ T cell- and STAT3-dependent manners. J Immunol. 2004;172:5648–5655. doi: 10.4049/jimmunol.172.9.5648. [DOI] [PubMed] [Google Scholar]
- 36.Vredevoe DL, Widawski M, Fonarow GC, et al. Interleukin-6 (IL-6) expression and natural killer (NK) cell dysfunction and anergy in heart failure. Am J Cardiol. 2004;93:1007–1011. doi: 10.1016/j.amjcard.2003.12.054. [DOI] [PubMed] [Google Scholar]
- 37.Huang B, Zhao J, Li H, et al. Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res. 2005;65:5009–5014. doi: 10.1158/0008-5472.CAN-05-0784. [DOI] [PubMed] [Google Scholar]
- 38.Averett DR, Fletcher SP, Li W, et al. The pharmacology of endosomal TLR agonists in viral disease. Biochem Soc Trans. 2007;35:1468–1472. doi: 10.1042/BST0351468. [DOI] [PubMed] [Google Scholar]
- 39.Diebold SS, Kaisho T, Hemmi H, et al. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 40.Kariko K, Buckstein M, Ni H, et al. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 41.Linn YC, Hui KM. Cytokine-induced killer cells: NK-like T cells with cytotolytic specificity against leukemia. Leuk Lymphoma. 2003;44:1457–1462. doi: 10.1080/1042819031000083082. [DOI] [PubMed] [Google Scholar]
- 42.Passweg JR, Tichelli A, Meyer-Monard S, et al. Purified donor NK-lymphocyte infusion to consolidate engraftment after haploidentical stem cell transplantation. Leukemia. 2004;18:1835–1838. doi: 10.1038/sj.leu.2403524. [DOI] [PubMed] [Google Scholar]
- 43.Tanaka J, Asaka M, Imamura M. Potential role of natural killer cell receptor-expressing cells in immunotherapy for leukemia. Int J Hematol. 2005;81:6–12. doi: 10.1532/IJH97.04152. [DOI] [PubMed] [Google Scholar]
- 44.Ruggeri L, Mancusi A, Perruccio K. Natural killer cell alloreactivity for leukemia therapy. J Immunother. 2005;28:175–182. doi: 10.1097/01.cji.0000161395.88959.1f. [DOI] [PubMed] [Google Scholar]
- 45.Seino K, Motohashi S, Fujisawa T, et al. Natural killer T cell-mediated antitumor immune responses and their clinical applications. Cancer Sci. 2006;97:807–812. doi: 10.1111/j.1349-7006.2006.00257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalinski P, Giermasz A, Nakamura Y, et al. Helper role of NK cells during the induction of anticancer responses by dendritic cells. Mol Immunol. 2005;42:535–539. doi: 10.1016/j.molimm.2004.07.038. [DOI] [PubMed] [Google Scholar]
- 47.Ruggeri L, Mancusi A, Burchielli E, et al. Natural killer cell alloreactivity and haplo-identical hematopoietic transplantation. Cytotherapy. 2006;8:554–558. doi: 10.1080/14653240601078721. [DOI] [PubMed] [Google Scholar]
- 48.Moretta L, Locatelli F, Moretta A. Alloreactive natural killer cells in targeting high-risk leukaemias. Ann Rheum Dis. 2008;67(Suppl 3):iii39–iii43. doi: 10.1136/ard.2008.097980. [DOI] [PubMed] [Google Scholar]
- 49.Costello RT, Fauriat C, Rey J, et al. Immunobiology of haematological malignant disorders: the basis for novel immunotherapy protocols. Lancet Oncol. 2004;5:47–55. doi: 10.1016/S1470-2045(03)01323-8. [DOI] [PubMed] [Google Scholar]
- 50.Hart OM, Athie-Morales V, O’Connor GM, et al. TLR7/8-mediated activation of human NK cells results in accessory cell-dependent IFN-gamma production. J Immunol. 2005;175:1636–1642. doi: 10.4049/jimmunol.175.3.1636. [DOI] [PubMed] [Google Scholar]
- 51.Gorski KS, Waller EL, Bjornton-Severson J, et al. Distinct indirect pathways govern human NK-cell activation by TLR-7 and TLR-8 agonists. Int Immunol. 2006;18:1115–1126. doi: 10.1093/intimm/dxl046. [DOI] [PubMed] [Google Scholar]
- 52.Sawaki J, Tsutsui H, Hayashi N, et al. Type 1 cytokine/chemokine production by mouse NK cells following activation of their TLR/MyD88-mediated pathways. Int Immunol. 2007;19:311–320. doi: 10.1093/intimm/dxl148. [DOI] [PubMed] [Google Scholar]
- 53.Alter G, Suscovich TJ, Teigen N, et al. Single-stranded RNA derived from HIV-1 serves as a potent activator of NK cells. J Immunol. 2007;178:7658–7666. doi: 10.4049/jimmunol.178.12.7658. [DOI] [PubMed] [Google Scholar]
- 54.Girart MV, Fuertes MB, Domaica CI, et al. Engagement of TLR3, TLR7, and NKG2D regulate IFN-gamma secretion but not NKG2D-mediated cytotoxicity by human NK cells stimulated with suboptimal doses of IL-12. J Immunol. 2007;179:3472–3479. doi: 10.4049/jimmunol.179.6.3472. [DOI] [PubMed] [Google Scholar]
- 55.Degli-Esposti MA, Smyth MJ. Close encounters of different kinds: dendritic cells and NK cells take centre stage. Nat Rev Immunol. 2005;5:112–124. doi: 10.1038/nri1549. [DOI] [PubMed] [Google Scholar]
- 56.Biassoni R. Natural killer cell receptors. Adv Exp Med Biol. 2008;640:35–52. doi: 10.1007/978-0-387-09789-3_4. [DOI] [PubMed] [Google Scholar]
- 57.Verheyden S, Demanet C. NK cell receptors and their ligands in leukemia. Leukemia. 2008;22:249–257. doi: 10.1038/sj.leu.2405040. [DOI] [PubMed] [Google Scholar]
- 58.Ogasawara K, Lanier LL. NKG2D in NK and T cell-mediated immunity. J Clin Immunol. 2005;25:534–540. doi: 10.1007/s10875-005-8786-4. [DOI] [PubMed] [Google Scholar]
- 59.Moretta A, Bottino C, Vitale M, et al. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol. 2001;19:197–223. doi: 10.1146/annurev.immunol.19.1.197. [DOI] [PubMed] [Google Scholar]