

Abstract
Radio-labelling of blood cells is an established technique for evaluating in vivo migration of normal cells to sites of pathology such as infection and haemorrhage. A limitation of cellular immunotherapies to induce anti-tumour responses is in part due to the uncertain ability of cellular effectors to reach their intended target. We extended the approach of cell radiolabelling to accurately examine the in vivo distribution of cellular immunotherapy with ex-vivo macrophage activated killer (MAK) cells. We describe the use of two methods of cell labelling for tracking the destination of autologous-derived macrophage activated killer (MAK®) cells linked to the bi-specific antibody MDX-H210 delivered either by intravenous (i.v.) or intraperitoneal (i.p.) injection in ten patients with peritoneal relapse of epithelial ovarian carcinoma. Our results demonstrate the feasibility of generating high numbers and purity of GMP quality MAK cells, which can be radiolabelled with 18F-FDG or 111In-oxime. MAK cell administration produced minimal infusional toxicity and demonstrated a reproducible pattern of in vivo distribution and active in vivo tracking to sites of known tumour following 8 of 16 i.v. infusions or 4 of 6 i.p. infusions. However, the leakage of 18F-FDG limited the ability to confidently confirm the tracking of MAK cells to tumour in all cases and improved PET labels are required. The addition of MDX-H210 bispecific antibody did not alter the distribution of cells to tumour sites, but did accelerate the clearance of i.v. administered MAK cells from the pulmonary circulation. This data demonstrates that cellular cancer immunotherapies may be successfully delivered to the sites of active tumour following either i.v. or i.p. injection in a proportion of patients with metastatic cancer. Incorporation of tracking studies in early cycles of cellular immunotherapy may allow selection of patients who demonstrate successful targeting of the immunotherapy for ongoing treatment.
Keywords: Immunotherapy, Macrophage, Radiolabel, PET
Introduction
Early phase clinical studies of adoptive cellular immunotherapy for cancer suggest that anti-tumour activity can be induced in a range of malignancies [1]. In those patients who fail to achieve an anti-tumour response, there is uncertainty as to whether this is due to a failure of migration of cellular therapies to the intended target, or alternatively a resistance of the tumour to cytotoxicity mechanisms. There have been few clinical studies investigating the ability of cell-based therapies to reach their tumour target in vivo, although the clinical feasibility of in vivo tracking of stem cell populations has been used in the cardiovascular disease clinical trials [2].
Macrophages, activated by interferon-gamma (IFN-γ), attain an activated killer phenotype. These macrophage activated killer (MAK®) cells have been demonstrated to have tumour killing capacity both in vitro [3–7] and in animal tumour models and in early phase human trials [8–13].
The half-humanized monoclonal anti-HER2/neu bi-specific antibody MDX-H210 binds to both the high-affinity Fc receptor (FcγRI; CD64) on macrophages, and HER2/neu, which is over-expressed in 5–10% of ovarian cancers [14]. Pre-clinical studies demonstrate that MDX-H210 bound to macrophages enhances the phagocytosis or lysis of tumour cells that over-express HER2 [15]. A phase II trial of ovarian cancer patients treated with MAK cells combined with MDX-H210, demonstrated anti-tumour activity following intraperitoneal (i.p.) administration of these cells [16]. Tracking of these i.p. administered cells to sites of tumour was, however, not addressed.
In mice, SPECT tracking of i.v. injected indium-oxine-labelled murine macrophages reveals the initial margination of macrophages in the lungs, then eventual migration to liver, spleen and kidneys before accumulating within sites of tumour. However, little is known about the in vivo distribution of these cells in humans [17].
In this present study we tracked the destination of MAK cells combined with MDX-H210 in vivo in women with recurrent ovarian epithelial cancer. In addition, we wished to compare the anatomy and kinetics of distribution of cells delivered by two different methods of cell labelling and two routes of administration. Firstly, we used positron emission tomography (PET) to track 18fluorine fluorodeoxyglucose (18F-FDG)-labelled cells injected by either i.v. or i.p. routes. This technique has been demonstrated with other cell-based therapies in both in vitro and in vivo mouse studies [18], and in the tracking of bone marrow cells to infarcted human myocardium [19].
In the second method we tracked MAK cells labelled with 111indium-oxime by whole body planar gamma camera imaging.
Patients, materials and methods
Patients
Patients with recurrent epithelial ovarian carcinoma with peritoneal disease that was measurable by RECIST criteria or peritoneal disease that was non-measurable on CT but PET positive in association with an elevated CA-125 level were eligible for study enrolment. The study protocol was approved by the Peter MacCallum Cancer Centre Ethics Committee and all patients gave informed written consent. All patients underwent baseline imaging with combined PET/CT scan (Discovery LS, GE Medical Systems, Milwaukee, WI, USA) to detect the sites of recurrent disease for later comparison to tracking images. As this was primarily a study of in vivo cell tracking, disease responses to MAK cell administration was not included in the formal assessment. Ten patients were enroled and a total of 22 MAK cell infusions delivered as described below.
Schedule of MAK cell administration
Patients were enroled onto the study in a series of cohorts of up to three patients (Table 1). The initial four cohorts received MAK cells conjugated with MDX-H210 and were designed to compare the in vivo patterns of distribution of MAK cells when administered by either i.v. or i.p. route, labelled with either of the two radiolabels (18F-FDG or 111In-oxime). Patients could enter more than one cohort; however, when patients were enroled into different routes of administration, the same radiolabel was used in each instance but separated by at least 7 days after the completion of the first tracking study to allow clearance of any visible radiolabel (in the case of In-111). Cohorts 5, 6 and 7 were designed to compare the effect of the presence or absence of MDX-H210 on MAK cell migration. Patients 7 to 10 inclusive each received at least one MAK cell infusion in the presence of MDX-H210 and one MAK cell infusion in the absence of MDX-H210.
Table 1.
Cohort design for the administration of MAK cells radiolabelled with one of two radiotracers and co-administered with MDX-H210 anti-Her2neu antibody
| Cohort | Route of injection | Radiolabel | Addition of MDX-H210 | Patients enroled |
|---|---|---|---|---|
| 1 | i.v. | FDG | Yes | 1, 3, 5, 7, 8 |
| 2 | i.v. | 111In | Yes | 2, 4, 6, 10, 9 |
| 3 | i.p. | FDG | Yes | 1, 3, 7 |
| 4 | i.p. | 111In | Yes | 2, 4 |
| 5 | i.v. | FDG | No | 7, 8, 9 |
| 6 | i.v. | 111In | No | 10 |
| 7 | i.p. | 111In | No | 9 |
MAK cell preparation and radio-isotope labelling
All cell processing was performed under Good Manufacturing Practice conditions according to Australian legislation [20]. Autologous mononuclear cells were collected via steady-state apheresis (COBE Spectra Apheresis System, Gambro BCT, Lakewood, CO, USA). Cells subsequently underwent in vitro differentiation using the MAK® Cell Processor (IDM, Paris, France) for 6 days in the presence of 500 U/ml granulocyte-macrophage colony stimulating factor (GM-CSF) (Leucomax, Novartis, Basel, Switzerland). Resulting macrophages were then stimulated for 24 h with 166 U/ml IFN-γ (Imukin, Boehringer Ingelheim GmbH, Germany), and the cell product purified by counterflow elutriation using the Beckman Avanti elutriator (Beckman Coulter, Fullerton, Ca, USA) yielding a median purity of >91% (Table 2).
Table 2.
Details of starting mononuclear cells counts and the MAK cell dose, purity, viability and radiolabelling efficiency obtained for each product
| Patient number | Pre-culture mononuclear cells (×1010) | MAK cells elutriation (%) | Isotope added | Viability (%) | Number of MAK cells injected (×109) | Cell labelling efficiency (%) | Route of injection |
|---|---|---|---|---|---|---|---|
| 1 | 6.58 | 96.5 | 18F-FDG | 95.6 | 2.88 | 90.6 | i.v. |
| 3.44 | 96.2 | 18F-FDG | 91.5 | 2.5 | 88 | i.v. | |
| 2 | 4.69 | 93.7 | 111In | 78.4 | 0.71 | 48 | i.v. |
| 6.1 | 82 | 111In | 98.1 | 1.2 | 71 | i.p. | |
| 3 | 8.42 | 90.2 | 18F-FDG | 96.5 | 0.93 | 90 | i.v. |
| 2.72 | 75 | 18F-FDG | 99 | 0.62 | 42 | i.p. | |
| 4 | 7.96 | 86 | 111In | 91.9 | 2.51 | 64 | i.v. |
| 1.26 | 90.4 | 111In | 95 | 2.87 | 76 | i.p. | |
| 5 | 1.43 | 85.3 | 18F-FDG | 94.3 | 1.77 | 87.6 | i.v. |
| 6 | 1.2 | 84.3 | 111In | 98 | 1.42 | 85 | i.v. |
| 7 | 0.78 | 92.1 | 18F-FDG | 96.3 | 1.55 | 84.9 | i.v. |
| 0.75 | 91.9 | 18F-FDG | 94.5 | 1.81 | 83 | i.v. | |
| 0.92 | 91.2 | 18F-FDG | 95.2 | 1.28 | 88 | i.p. | |
| 8 | 1.17 | 93.1 | 18F-FDG | 91.9 | 1.82 | 93 | i.v. |
| 1.26 | 93.6 | 18F-FDG | 97.9 | 2.81 | NR | i.v. | |
| 1.23 | 93.9 | 18F-FDG | 97 | 2.14 | 88 | i.v. | |
| 0.51 | 91.2 | 18F-FDG | 94.7 | 1.25 | 93.5 | i.v. | |
| 9 | 0.79 | 94.5 | 111In | 95 | 2.56 | 64 | i.v. |
| 0.63 | 85.5 | 18F-FDG | 70.4 | 0.63 | 89 | i.v. | |
| 1.07 | 91.9 | 111In | 95.6 | 3.58 | 77.6 | i.p. | |
| 10 | 0.59 | 98.3 | 111In | 95.5 | 2.18 | 77 | i.v. |
| 0.39 | 94.7 | 111In | 100 | 2.01 | 90.6 | i.v. |
In treatment cohorts 1–4, MAK cells were then linked with the half-humanized anti- HER2/neu antibody MDX-H210 (1.5 mg; Medarex, Princeton, NJ, USA) and suspended in 15 ml of 4% Human Albumin (Albumex® 4, CSL Limited, Parkville, Australia). For PET tracking, the suspended cells were labelled with 84.7 MBq 18F-FDG by incubating for 60 min at room temperature. For gamma camera tracking, MAK cells + MDX-H210 were labelled with 39 MBq 111indium for 50 min at room temperature. The cells were then washed, re-suspended and injected into the patient within 20 min.
Tracking studies
For 18F-FDG-labelled MAK cell tracking, serial PET scans were performed at 1, 2 and 4 h following injection using a dedicated PET scanner based on sodium iodide detectors (GE Quest, GE Medical Systems Inc, Milwaukee, WI, USA). The resulting images were processed with and without attenuation correction, and the results compared to known sites of recurrent carcinoma detected on a baseline combined PET/CT scanner. Where 111indium-labelled cells were being tracked, serial planar whole body scans were performed after 4, 21, 45 and 70 h, and the results compared to a baseline PET scan displayed as a projection image as demonstrated in Fig. 1.
Fig. 1.

Baseline and serial PET images in patient 7 after i.v. injection of MAK cells labelled with18F-FDG+ MDX-H210 antibody. a Baseline PET images obtained on a combined PET/CT scanner 2 weeks prior to MAK cell injection following direct intravenous administration of 111MBq 18F-FDG demonstrating multi-focal peritoneal metastases predominantly in the pelvis and additional lesions in the serosal peritoneum over the liver and anterior superior tip of the spleen. b Scans 1 h after injection: whole body projection image (left), and orthogonal axial through pelvis (second from left), sagittal (second from right) and coronal (right) planes demonstrate MAK cells predominantly within the lungs with early uptake in liver, spleen and pelvic tumor. c Scans 4 h after injection: whole body images (left), axial through pelvis (second from left), sagittal (second from right) and coronal (right) demonstrate activity predominantly within the liver, spleen and localized to sites of pelvic peritoneal tumor deposits
Quantitation of the biodistribution of 111In-labelled MAK cells was performed using the region of interest (ROI) method. In brief, regions were drawn around individual organs (lungs or spleen or liver) on both anterior and posterior whole body images. Total counts within these regions were corrected for background, signal decay and image acquisition rates. The geometric mean of the anterior and posterior images signal contained within each organ is expressed as a fraction of the whole body geometric mean.
Results
MAK cell generation and radiolabelling
MAK cells were purified by continuous counter-flow centrifugal elutriation yielding a median of 1.8×109 (range 0.63–3.58×109) MAK cells with >70% viability (Table 2). A labelling efficiency for each tracer was calculated as the percentage of signal retained within a spun pellet of cells relative to the signal obtained within the unspun specimen. Co-culture of MAK cells with 111indium or 18F-FDG results in a median cellular labelling efficiency of 76% (range 48–91%) and 88% (range 42–93%), respectively.
Clinical administration
The details of the ten enroled patients are outlined in Table 3. A total of 22 MAK cell infusions were completed. The distribution of injection by route, tracer and tumour localization is also given in Table 3.
Table 3.
Demographic, toxicity and tracking data in all ten enroled patients
| Patient | Age | Tumor histology and stage | Number of previous chemotherapy treatments | Cohort | MAK cell dose (×109) | Infusional toxicity | 18FDG PET | 111In SPECT | Tracking to sites of tumor | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| i.v. | i.p. | i.v. | i.p. | ||||||||
| 1 | 61 |
Serous IIIC Relapse |
3 | 1 | 2.88 | II | X | Yes | |||
| 3 | 2.5 | II | X | Yes | |||||||
| 2 | 52 |
Serous/clear IIIC Ref |
2 | 2 | 0.71 | 0 | X | Yes | |||
| 4 | 1.2 | 0 | X | Cells trapped in peritoneal adhesions | |||||||
| 3 | 51 |
Serous IIIC Relapse |
2 | 1 | 0.93 | 0 | X | No | |||
| 3 | 0.62 | 0 | X | No | |||||||
| 4 | 39 |
Serous IIIC Relapse |
2 | 2 | 2.51 | II | X | Yes (Fig. 2) | |||
| 4 | 2.87 | II | X | Yes (Fig. 4) | |||||||
| 5 | 50 |
Serous IIIC Relapse |
2 | 1 | 1.77 | I | X | No | |||
| 6 | 52 |
Serous IIIC Refractory |
2 | 2 | 1.42 | I | X | No | |||
| 7 | 24 |
Serous IIIC Relapse |
2 | 1 | 1.55 | 0 | X | Yes (Fig. 1) | |||
| 5 | 1.81 | 0 | X | Yes (Fig. 3) | |||||||
| 3 | 1.28 | 0 | X | Yes | |||||||
| 8 | 43 |
Serous IIIC Refractory |
2 | 1 | 1.82 | I | X | No | |||
| 1 | 2.81 | I | X | No | |||||||
| 1 | 2.14 | I | X | No | |||||||
| 5 | 1.25 | 0 | X | Yes | |||||||
| 9 | 48 |
Endometrial IIIC Relapse |
1 | 2 | 2.56 | 0 | X | No | |||
| 5 | 0.63 | 0 | X | No | |||||||
| 7 | 3.58 | 0 | X | Yes | |||||||
| 10 | 53 |
Serous IIIC Relapse |
1 | 6 | 2.18 | I | X | Yes | |||
| 2 | 2.01 | 0 | X | Yes | |||||||
All infusions were well tolerated and could be completed without acute toxicity. Three episodes of grade I toxicity (transient lymphadenopathy and two episodes of fever/chills) were thought to be directly related to MAK cell infusion. In addition, three patients experienced grade II nausea and grade I abdominal cramps due to peritoneal disease. One of these patients subsequently developed a small bowel obstruction. No grade 3 toxicity was observed due to the administration of MAK cells.
Distribution of MAK cells following intravenous injection
A typical pattern of distribution for i.v. administered cells was observed in all patients independent of the radiolabel used and is shown for patient 7 in Fig. 1. 18F-FDG-labelled MAK cells were initially observed to accumulate within the lungs, with lung clearance commencing within 4 h and as early as 1 h after injection; signal was also seen to accumulate within the liver and spleen.
The distribution pattern of i.v. injected 111In-labelled MAK cells was similar to that observed with18F-FDG-labelled MAK cells. By 4 h after i.v. injection, residual activity was seen throughout both lung fields, consistent with white cell margination with a progressive increase in activity seen in the liver, spleen and bone marrow over the next 24 h, which remained visible for more than 72 h. The time course for patient four is shown in Fig. 2.
Fig. 2.

a Baseline PET images and serial planar whole body imaging in patient 4 after i.v. injection of 111indium labelled MAK cells + MDX-H210 antibody. Far left panel: baseline PET images demonstrating mildly increased radiotracer uptake through out the peritoneal cavity with nodular areas of moderately increased radiotracer uptake in the left upper quadrant, right iliac fossa and the presacral space consistent with peritoneal deposits. Other upper panels: serial whole body images of 111indium labelled MAK cells at 4, 21, 45 and 70 h post-injection. At 4 h, diffuse activity was seen throughout both lung fields, consistent with normal white cell margination. Three foci of moderately intense activity in the left lung were consistent with micro-emboli of radiolabelled cells to this lung. At 21 h there was progressive clearance of activity from the lungs and increasing activity in the liver, spleen and bone marrow. At 21 h there was faint visualization of the peritoneal space. This became more obvious by 70 h where there was clearly a diffuse pattern of uptake in the abdomen with most marked uptake in the left mid abdomen corresponding to the area of most marked abnormality on the baseline 18F-FDG scan. b Distribution of injected MAK cells by organ of interest in patient 4. Signals present in each organ at each time point are expressed as a percentage of the whole body signal at that time. Data presented are typical for that obtained in six separate administrations
We then studied if the presence of the HER2/neu-targeting antibody, MDX-H210, affected MAK cell biodistribution. In those patients who received both MDX-H210-labelled and naked MAK cells (patients 7–10) clearance of signal from the lungs was considerably slowed, and liver and spleen distribution was similarly delayed in the absence of MDX-H210 (Fig. 3).
Fig. 3.
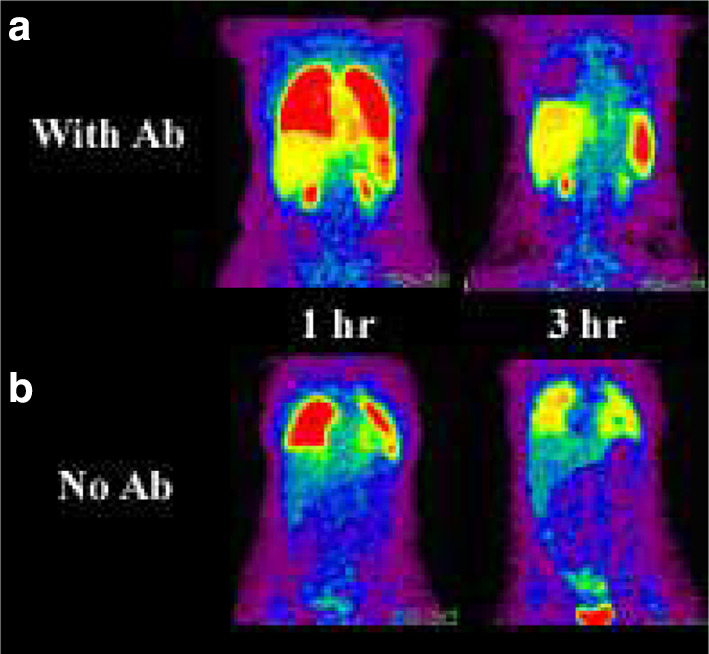
Comparison of 18F-FDG-labelled MAK cell biodistribution with (a) and without (b) antibody. Images obtained at 1 h after reinfusion of radiolabelled cells (left panels) demonstrates significantly greater liver, splenic and renal activity when antibody is attached to the cell. This likely reflects more rapid demargination of cells from the pulmonary vasculature. By 3 h the antibody-labelled cells had almost completely cleared from the lungs whereas the cells without antibody remained to a greater extent marginated in the lungs with relatively less liver and particularly splenic localization.There is a reciprocal earlier appearance of antibody labelled MAK cells within the liver and spleen and in the areas of known metastatic tumor in the right iliac fossa
MAK cell tracking to tumour following i.v. injection
Overall, tracking of intravenously injected MAK cells to areas of known disease was seen following eight of the 16 i.v. infusions (four each of 18F-FDG and 111In). Following administration of 18F-FDG-labelled MAK cells, tracking to the tumour was observed as early as 4 h, as illustrated in patient 7, with increasing activity in the right iliac fossa (Fig. 1b), corresponding to the site of known disease on the baseline PET scan (Fig. 1a).
In three cases (patients 2, 4 and 10) who received i.v. 111In-labelled MAK cells, a pattern of faint visualization of the peritoneal space was observed at 24 h, which became progressively more intense over the next 48 h so that by 70 h, diffuse uptake in the abdomen was seen with most marked uptake in the known sites of peritoneal disease. This is demonstrated in patient 4 in the left mid-abdomen and left upper quadrant consistent with quantified tracking of the labelled MAK cells to the peritoneum (Fig. 2). In the only patient (patient 10) who received separate i.v. infusions of MDX-H210-labelled and naked MAK cells, localization to tumour sites was seen with each infusion, irrespective of the addition of the antibody.
Intraperitoneal injection
A total of six i.p. infusions were performed; three of either 18F-FDG-labelled MAK cells or 111In-labelled MAK cells. Evidence for active MAK cell tracking to sites of known disease was seen on four occasions in three patients. A representative example of 111In-labelled MAK cell tracking is shown in Fig. 4.
Fig. 4.

Progressive accumulation of 111In-labelled MAK cells + MDX-H210 antibody in the left upper quadrant area of known metastatic tumor in patient 4. a A pre-MAK cell administration PET scan reveals radiotracer uptake in the left upper quadrant of the abdomen. b Following i.p. administration of In-labelled MAK cells intraperitoneal distribution throughout the peritoneum is seen with early and then progressive tracking of MAK cells to the known tumor site at 3.5 h. c Progressive tracking observed at 18 h
No appreciable difference in tracking was seen between the two different radiotracers used. Patient 2 demonstrated intense localization in the right iliac fossa and less intense activity extending along the paracolic gutter on whole body 111In- gamma camera imaging. This biodistribution remained largely unchanged at 48 and 72 h after radiotracer administration consistent with poor peritoneal space diffusion of cells reflecting peritoneal adhesions. By 48 and 72 h there was some visualization of bone marrow, liver and spleen suggesting some margination of radiolabelled white cells into the systemic circulation, but no definite localization of peritoneal deposits was identified. No comparison was possible between the effect of linking i.p. administered MAK cells to MDX-H210.
Discussion
The in vivo fate, anatomical distribution and biological activity of cellular therapies in vivo following injection into human subjects has to date remained largely unknown. We demonstrate that radio-labelling and serial tracking studies provide a means of elucidating these issues. Here we successfully generated, labelled and tracked autologous MAK cells.
In this study we were able to generate cytokine-activated MAK cells in adequate numbers and with excellent purity and viability to allow clinical administration with minimal toxicity. Further, the in vitro radiotracer labelling had no substantive effect on cell viability and was sufficiently stable to allow sequential images to be obtained for cell tracking purposes. The patterns of tracking observed after i.v. cell administration were similar with both 111In and 18F-FDG labelled cells. Importantly, although cells were initially observed to pool within the pulmonary vasculature, the vast majority of cells had migrated out of the lungs by 4 h (and faster when cells were labelled with MDX-H210), suggesting that despite the normally adherent nature of monocyte/macrophages, these cells are capable of surviving passage through the lungs and subsequently migrate to the liver, spleen and distant tumour sites. Early (<4 h) differences in MAK cell distribution between 18F-FDG labelled cells and 111In labelled cells cannot be fully discounted as the first scanning time point during 111In studies was at 4 h, in order to allow later time points to be included.
Tracking of MAK cells to known tumour sites was also observed in four patients following i.p. administration. This may be a consequence of peritoneal inflammation at sites of tumour involvement or active migration of cells to tumour sites. In either case, this data supports the notion that cellular immunotherapies may successfully track to sites of tumour via either i.v. or i.p. routes. Furthermore, slow clearance of the radiolabelled cell into the general circulation following i.p. administration may be of benefit for patients with primarily small volume peritoneal deposits in whom high concentration of cellular therapies are required.
Radio-labelling has, however, a number of potential limitations including anatomical resolution of the images obtained. PET scanning using 18F isotope can provide high-resolution three-dimensional anatomical imaging with the potential for quantification of labelled cell migration to anatomical sites within 10 cm of subcutaneous or intradermal administration (manuscript in preparation). Whilst 18F-FDG can provide excellent spatial resolution, its relatively short half-life of 109.7 min provides a limited window of only 4–6 h for obtaining images following injection. This timeframe may be insufficient to fully appreciate the migration of labelled cells in vivo particularly when tracking small numbers of cells. An alternative (and complementary) strategy of using a radiotracer such as 111indium-oxime detected by planar imaging allows cell tracking to be followed over a more extended period (up to 96 h), but at the cost of lower resolution images. The benefits of sustained signal without loss of resolution may potentially be obtained by the use of alternative radiolabels such as 64Cu, studies of which are on going in our laboratory [21]. Others have reported inconclusive data for successful tracking of radiolabelled macrophage cellular therapies to sites of metastatic renal cell carcinoma [22] and melanoma [23]. Our reproducible tracking findings in patients with metastatic ovarian cancer may represent the ready access of circulating cells to tumour beds via the inflamed peritoneum.
One important finding from our study was a major limitation with the use of 18F-FDG, namely the demonstration of free 18F-FDG ‘leakage’ out of the MAK cells. This is demonstrated by renal and bladder activity observed even as early as 1 h post-infusion (Figs. 1 and 3). This implies in vivo dephosphorylation of 18F-FDG, leading to ‘back diffusion’ into serum and subsequent uptake in kidneys, heart and possibly tumour. However, this phenomenon does not occur with 111In-oxime. Although there appears to be free 18F-FDG appearing within a short time after MAK cell administration, we believe there is sufficient retained label in the cells to provide useful information of MAK cell tracking. Firstly, 18F-FDG does not normally appear in the lungs and so the early images must indicate retention in cells, with subsequent cell clearance from the lungs. Similarly, the intensity of 18F-FDG in the liver and spleen is above normally observed values and so in the absence of tumour at these sites the high levels indicate subsequent ‘tracking’ to the reticuloendothelial system. Secondly, the similarity of tracking patterns achieved with both PET and SPECT strongly suggest that cell tracking to tumour, and not free radiotracer, was the main mechanism of enhancement of signal from sites of known disease and lastly, when the pre-tracking PET scans using free 18F-FDG are compared to those following labelled MAK cells the liver and tumour images are not identical suggesting that tissue distribution after MAK cell administration is dependent on label retained within administered cells rather than by free FDG. Of note, we have found that the phenomenon of rapid 18F-FDG efflux occurs to a much greater extent in other cell types such as monocyte-derived dendritic cells (manuscript in preparation) and it can therefore not be extrapolated that labelling with 18F-FDG will be suitable for all cell types, and improved labels are required. It is of interest that others have suggested that 18F-FDG is a useful marker for labelling immature hematopoietic cells but it seems possible that ‘leakage’ may also occur in that setting [19].
Our findings also show that ex vivo modifications of cells, such as the addition of monoclonal antibodies, can significantly alter their in vivo kinetics as observed by the faster clearance of cells from the lungs in the presence of MDX-H210. The mechanism for this observed effect is not clear. The bispecific antibody may potentially alter the adhesion molecule expression of the local pulmonary vasculature, or alternatively, adhesion molecules expressed by the MAK cells them selves may be blocked in the presence of MDX-H210.
Of interest, we observed that the addition of the HER2 specific monoclonal antibody was not an absolute requirement for the migration of MAK cells to sites of disease, but we cannot discount the possibility that over expression of HER2 may lead to improved cell migration as none of the enroled patients had over-expression of HER2 by immunohistochemistry on their original histology specimens.
Also, in five of six patients in whom both i.v. and i.p. routes of administration were comparable, either route gave concordant results (failure of tracking, or evidence of tracking), indicating that direct i.p. administration was not necessary for delivery of this form of cellular immunotherapy.
In summary, we demonstrate that use of the imaging techniques displayed here provide valuable insight into the fate of infused ex vivo manipulated cells in addition to a means by which patients can be pre-selected for immunotherapies depending on the demonstration of successful tracking and determination of the optimal route of injection to obtain tumour targeting.
Statement of responsibility
The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written.
Acknowledgments
This work was supported, in part, by the European Commission under the 6th Framework Programme for Research, Technological Development and Demonstration.
References
- 1.Ribas A, Butterfield LH, Glaspy JA, Economou JS. Current developments in cancer vaccines and cellular immunotherapy. J Clin Oncol. 2003;21:2415–2432. doi: 10.1200/JCO.2003.06.041. [DOI] [PubMed] [Google Scholar]
- 2.Frangioni J, Hajjar R. In vivo tracking of stem cells for clinical trials in cardiovascular disease. Circulation. 2004;110:3378–3384. doi: 10.1161/01.CIR.0000149840.46523.FC. [DOI] [PubMed] [Google Scholar]
- 3.Chokri M, Lopez M, Oleron C, Girard A, Martinache C, Siffert JC, Canepa S, Bartholeyns J. Production of macrophages with potent antitumoral properties (MAK) by culture of monocytes in the presence of GM-CSF and 1,25 (OH)2 vitD3. Anticancer Res. 1992;12:2257–2260. [PubMed] [Google Scholar]
- 4.Munn D, Cheung N. Phagocytosis of tumor cells by human monocytes cultured in GM-CSF. J Exp Med. 1990;172:231–237. doi: 10.1084/jem.172.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fidler I, Kleinerman E. Therapy of cancer metastases by systemic activation of macrophages. Res Immunol. 1992;144:284–287. doi: 10.1016/0923-2494(93)80109-C. [DOI] [PubMed] [Google Scholar]
- 6.Dumont S, Hartmann D, Poindron P, Faradji A, Oberling F, Bartholeyns J. Control of the antitumoral activity of human macrophages produced in large amount in view of adoptive transfer. Eur J Cancer Clin Oncol. 1988;24:1691–1698. doi: 10.1016/0277-5379(88)90069-7. [DOI] [PubMed] [Google Scholar]
- 7.Baron-Bodo V, Doceur P, Lefebvre ML, Labroquère K, Defaye C, Cambouris C, Prigent D, Salcedo M, Boyer A, Nardin A. Anti-tumor properties of human-activated macrophages produced in large scale for clinical application. Immunobiol. 2005;210:267–277. doi: 10.1016/j.imbio.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 8.Faradji A, Bohbot A, Schmitt M, Siffert J, Dumont S, Wiesel M, Piemont Y, Eischen A, Bergerat J, Bartholeyns J, Poindron P, Witz JP, Oberling F. Large scale isolation of human blood monocytes by continuous flow centrifugation leukapheresis and counterflow centrifugation elutriation for adoptive cellular immunotherapy in cancer patients. J Immunol Methods. 1994;174:297–309. doi: 10.1016/0022-1759(94)90033-7. [DOI] [PubMed] [Google Scholar]
- 9.Bartholeyns J, Lombard Y, Dumont S, Hartmann D, Chokri M, Giaimis J, Kaufmann S, Poindron P. Immunotherapy of cancer: experimental approach with activated macrophages proliferating in culture. Cancer Detect Prev. 1988;12:413–420. [PubMed] [Google Scholar]
- 10.Chokri M, Freudenberg M, Galanos C, Poindron P, Bartholeyns J. Compared antitumoral effects of LPS, TNF, interferon and activated macrophages on experimental tumors. Synergism and tissue distribution. Anticancer Res. 1989;9:1185–1190. [PubMed] [Google Scholar]
- 11.Lopez M, Fechtenbaum J, David B, Martinache C, Chokri M, Canepa S, De Gramont A, Louvet C, Gorin I, Mortel O, Bartholeyns J. Adoptive immunotherapy with activated macrophages grown in vitro from blood monocytes in cancer patients: a pilot study. J Immunotherapy. 1992;11:209–217. doi: 10.1097/00002371-199204000-00008. [DOI] [PubMed] [Google Scholar]
- 12.Faradji A, Bohbo A, Frost H, Schmitt-Goguel M, Siffert JC, Dufour P, Eber M, Lallot C, Wiesel ML, Bergerat JP, Oberling F. Phase I study of liposomal PTP-PE-activated autologous monocytes administered intraperitoneally to patients with peritoneal carcinomatosis. J Clin Oncol. 1991;9:1251–1260. doi: 10.1200/JCO.1991.9.7.1251. [DOI] [PubMed] [Google Scholar]
- 13.Valone F, Kaufman P, Guyre P. Phase I trial of bispecific antibody MDX-H210 in patients with advanced breast or ovarian cancer that overexpresses the proto-oncogene HER-a neu. J Clin Oncol. 1995;13:2281–2292. doi: 10.1200/JCO.1995.13.9.2281. [DOI] [PubMed] [Google Scholar]
- 14.Mano MS, Awada A, Di Leo A, Durbecq V, Paesmans M, Cardoso F, Larsimont D, Piccart M. Rates of topoisomerase II-alpha and HER-2 gene amplification and expression in epithelial ovarian carcinoma. Gynecol Oncol. 2004;92(3):887–895. doi: 10.1016/j.ygyno.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 15.Curnow RT. Clinical experience with CD64-directed immunotherapy. An overview. Cancer Immunol Immunother. 1997;45:210–215. doi: 10.1007/s002620050435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Gramont A, Gangji D, Louvet C, Garcia ML, Tardy D, Romet-Lemonne JL. Adoptive immunotherapy of ovarian carcinoma. Gynecol Oncol. 2002;86:102–103. doi: 10.1006/gyno.2002.6667. [DOI] [PubMed] [Google Scholar]
- 17.Chokri M, Lallot C, Ebert M, Poindron P, Bartholeyns J. Biodistribution of indium-labelled macrophages in mice bearing solid tumors. Int J Immunother. 1990;6:79–84. [Google Scholar]
- 18.Forstrom LA, Mullan BP, Hung JC, Lowe VJ, Thorson LM. 18F-FDG labelling of human leucocytes. Nucl Med Commun. 2000;21:691–694. doi: 10.1097/00006231-200007000-00014. [DOI] [PubMed] [Google Scholar]
- 19.Hofmann M, Wollert KC, Meyer GP, Menke A, Arseniev L, Hertenstein B, Ganser A, Knapp WH, Drexler H. Monitoring of bone marrow cell homing into the infarcted human myocardium. Circulation. 2005;111(17):2198–2202. doi: 10.1161/01.CIR.0000163546.27639.AA. [DOI] [PubMed] [Google Scholar]
- 20.Wall DM, Prince HM. Regulation of cellular therapies: the Australian perspective. Cytotherapy. 2003;5(4):284–288. doi: 10.1080/14653240310002207. [DOI] [PubMed] [Google Scholar]
- 21.Adonai N, Nguyen KN, Walsh J, Iyer M, Toyokuni T, Phelps ME, McCarthy T, McCarthy DW, Gambhir SS. Ex vivo cell labeling with 64Cu-pyruvaldehyde-bis(N4-methylthiosemicarbazone) for imaging cell trafficking in mice with positron-emission tomography. Proc Natl Acad Sci USA. 2002;99:3030–3035. doi: 10.1073/pnas.052709599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quillien V, Moisan A, Lesimple T, Leberre C, Toujas L. Biodistribution of 111indium-labeled macrophages infused intravenously in patients with renal carcinoma. Cancer Immunol Immunother. 2001;50(9):477–482. doi: 10.1007/s002620100224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lesimple T, Moisan A, Carsin A, Ollivier I, Mousseau M, Meunier B, Leberre C, Collet B, Quillien V, Drenou B, Lefeuvre-Plesse C, Chevrant-Breton J, Toujas L. Injection by various routes of melanoma antigen-associated macrophages: biodistribution and clinical effects. Cancer Immunol Immunother. 2003;52(7):438–444. doi: 10.1007/s00262-003-0390-y. [DOI] [PMC free article] [PubMed] [Google Scholar]