

Abstract
Local delivery of IL-12 and GM-CSF to advanced primary tumors results in T- and NK-cell-dependent cure of disseminated disease in a murine spontaneous lung metastasis model. Post-therapy functional dynamics of cytotoxic T- and NK-cells were analyzed in primary and metastatic tumors to determine the specific roles of each subset in tumor eradication. Time-dependent depletion of CD8+ T and NK-cells demonstrated that CD8+ T-cells were critical to eradication of metastatic tumors within 3 days of treatment, but not later. In contrast, NK-cells were found to be essential to tumor regression for at least 10 days after cytokine delivery. Analysis of tumor-infiltrating lymphocyte populations in post-therapy primary tumors demonstrated that treatment resulted in the activation of tumor-associated CD8+ T-cells within 24 h as determined by IFNγ and perforin production. T-cell activity peaked between days 1 and 3 and subsided rapidly thereafter. Activation was not accompanied with an increase in cell numbers suggesting that treatment mobilized pre-existing T-effector/memory cells without inducing proliferation. In contrast, therapy resulted in a ≥3-fold enhancement of both the quantity and the cytotoxic activity of NK-cells in primary and metastatic tumors on day 3 post-therapy. NK-cell activity was also transient and subsided to pre-therapy levels by day 5. Depletion of CD4+ and CD8+ T-cells prior to treatment completely abrogated NK-cell infiltration into primary and metastatic tumors demonstrating the strict dependence of NK-cell recruitment on pre-existing T-effector/memory cells. Treatment failed to induce significant NK-cell infiltration in IFNγ-knockout mice establishing the central role of IFNγ in NK-cell chemotaxis to tumors. These data show that transient activation of tumor-associated T-effector/memory and NK-cells, but not long-term CD8+ T-cell responses, are critical to suppression of metastatic disease in this model; and reveal a novel role for pre-existing adaptive T-cell immunity in the recruitment of innate effectors to tumors.
Keywords: Cytokines, Tumor microenvironment, Metastasis, CD8+ T-cells, NK-cells, Immunotherapy
Introduction
The majority of immune-based tumor therapies focus on induction of tumor-specific cytotoxic T-cells due to the exquisite specificity and long-term memory of T-cell immunity [2, 30]. To this end, numerous studies have demonstrated that tumor vaccination can lead to successful development of measurable tumor-specific CD8+ T-cell activity in cancer patients [20, 21, 23]. On the other hand, vaccine-induced T-cell responses have generally been ineffective in achieving clinical tumor regression [2, 22, 30]. A number of parameters including the quality of the T-cell response, the immune suppressive nature of the tumor microenvironment and the concurrent development of regulatory responses have been proposed as potential factors influencing the clinical efficacy of tumor vaccines [9, 17].
Whereas T-cell-based therapies have taken center stage in the past two decades, recent studies revealed an equally important role for innate effectors including NK, NKT, IKDC and γδT-cells in tumor immune surveillance [10, 24, 26, 27]. It is thus likely that therapeutic strategies that integrate innate and adaptive mechanisms will achieve superior clinical efficacy [25]. Cytokines are major modulators of innate and adaptive immunity and have been extensively evaluated in cancer therapy [5]. Of the numerous cytokines that have been tested, IL-12 is unique in that its manifold pro-inflammmatory activities bridge innate and adaptive immune mechanisms [28]. Studies have demonstrated that IL-12 can directly induce T- and NK/NKT-cell production of IFNγ, which in turn augments T-cell, NK-cell and macrophage cytotoxicity, promotes anti-angiogenic mechanisms and inhibits tumor growth directly [28]. Whereas the ability of IL-12 to stimulate distinct innate and adaptive effector mechanisms is well-established, how these mechanisms are integrated in mediating successful tumor eradication is not fully delineated.
Previous studies in our laboratory demonstrated that a single injection of IL-12-encapsulated sustained-release microspheres into established tumors results in eradication of the treated tumor and the development of anti-tumor T-cell memory [6]. Subsequently, combined administration of IL-12 + GM-CSF microspheres into advanced primary tumors was found to be superior to either cytokine alone in promoting the suppression of primary tumors and the complete eradication of established lung metastases in a murine surgical metastasis model [11]. While preliminary analysis demonstrated that both T- and NK-cells were required for tumor suppression, the specific roles of each subset in long-term cure of metastatic disease were not determined. Here, we investigated the respective roles of CD8+ T and NK-cell subsets in long-term eradication of metastatic lung tumors following IL-12 + GM-CSF therapy.
Materials and methods
Mice and tumor cells
Line-1, a BALB/c lung alveolar carcinoma cell line, was maintained in DMEM/F-12 (Invitrogen Life Technologies) supplemented with 10% heat-inactivated FBS (Equitech-bio), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.1 mM non-essential amino acids, and 0.1 mM sodium pyruvate (Mediatech). Six to 8 weeks old male BALB/c mice were obtained from Taconic Farms. IFNγ knockout C.129S7(B6)-IfngtmlTs/J and the matching wild-type strain BALB/cJ mice were purchased from Jackson Laboratories. All studies were approved by the Institutional Animal Care and Use Committee of University at Buffalo/SUNY.
Cytokines and microspheres
Recombinant murine IL-12 (2.7 × 106 U/mg) was a gift from Wyeth Pharmaceuticals. Recombinant murine GM-CSF (5 × 106 U/mg) was purchased from Pepro Tech. Preparation of cytokine-encapsulated biodegradable polymer microspheres was described in detail previously [13].
Tumor induction and microsphere treatments
Tumors were induced by s.c. injection of 1 × 106 viable tumor cells in 0.1 ml of sterile PBS behind the neck just above the scapula [16]. When tumor volume reached 500–600 mm3 (17–21 days later), mice were treated with GM-CSF and IL-12-encapsulated microspheres (6 mg of each formulation corresponding to 1.5 μg of each cytokine, suspended in 100 μl PBS) via single intratumoral injection [13]. Tumor volume was determined using the formula a 2 × b/2, where a and b are the shortest and longest perpendicular dimensions of the tumor, respectively.
In vivo lymphocyte subset depletions
Purified anti-mouse CD4 monoclonal antibody (mAb) GK1.5 and anti-mouse CD8 mAb 53.6.72 were purchased from Bio express. Depletion of T-cells was performed via intraperitoneal injection of 200 μg anti-CD8 or anti-CD4 mAb in 0.2 ml sterile PBS. Antibody injection was repeated every 3 days for up to five times. Analysis of spleens demonstrated that antibody treatment resulted in the elimination of >95% of the T-cell subsets (11, data not shown). NK-cells were depleted with a single injection of the monoclonal antibody TMβ1 as described previously [11].
Preparation of single-cell suspension from tumors and tumor-draining lymph nodes (TDLN)
At selected time points before/after microsphere treatments, tumors and TDLN were removed from mice. Tumors were weighed, minced into small (1–2 mm3) pieces with a sterile scalpel and immersed in 10 ml of digestion mixture (5% FBS in RPMI-1640 containing 0.5 mg/ml collagenase D (Roche Molecular Systems), 0.02 mg/ml hyaluronidase type V (Sigma-Aldrich), and 0.01 mg/ml DNase I (Sigma-Aldrich). This mixture was incubated at 37°C for 45 min on a rotating platform. The resulting cell suspensions were filtered sequentially through 70- and 40-μm cell strainers (BD Falcon) and washed with 5%FBS in RPMI 1640. TDLN were removed and mechanically disaggregated through 40-μm cell strainers. Red blood cells were lysed by brief incubation in 0.15 M ammonium chloride solution, cells were washed in culture medium and viable lymphocytes were quantified via trypan-blue exclusion.
Clonogenic metastasis assay
Primary tumors were resected 7–8 days after microsphere treatment as described before [16]. Lungs were removed 3 weeks after surgery (or earlier if primary tumors recurred), cut into small (1–2 mm3) pieces and digested in 10 ml of digestion mixture at 37°C for 40 min on a rotating platform. The resulting cell suspensions were filtered through 70 μm cell strainers (BD Falcon) and washed with PBS. The resulting cells were suspended, and serial dilutions (1–10×) were plated in 6-well plates in 10% heat-inactivated FBS DMEM-F12 medium. Tumor cells, which grew rapidly and formed distinct foci within 14 days of plating, were fixed with methanol and stained with 0.03% methylene blue for quantification.
Flow cytometry
Single-cell suspensions obtained from samples were labeled with Abs using standard techniques [4] and analyzed on a four-color FACSCalibur flow cytometer (BD Biosciences) as previously described [13]. The following, directly fluorochrome-conjugated, anti-mouse mAbs were used: CD3-FITC (BD pharmingen, clone 17A2), CD8-Percp cy5.5 (BD Pharmingen, clone 53–6.7), CD49b-PE (BD Pharmingen, clone DX5), NKG2D-APC (eBioscience, clone CX5).
Intracellular cytokine staining
Single-cell suspensions of primary tumors and TDLN were prepared as described above. Tumor-infiltrating lymphocytes (TIL) or TDLN cells were stimulated with PMA (5 ng/ml) and ionomycin (0.5 μg/ml) for 5 h. Two hours before harvesting, 0.5 μl of BD Golgistop (BD Pharmingen) was added to every 1 ml of cell culture (1 × 106 live cells/ml). Intracellular staining for IFNγ was performed using BD Pharmingen Cytofix/cytoperm plus kit. Cells were washed in FACS buffer, stained with surface markers (CD3, CD8, CD49b), fixed, permeabilized and stained with PE-conjugated anti-mouse IFNγ (BD Pharmingen, clone XMG1.2). Perforin staining was performed using PE anti-mouse perforin (eBioscience, clone eBioOMAK-D).
Quantitative real-time PCR analysis
Lungs were removed from each mouse, cut into small (1–2 mm3) pieces and homogenized in 1 ml TRIzol® Reagent (Invitrogen Life Technologies) as described [18]. Total RNA was isolated and reverse-transcribed with Taqman Reverse Transcription Reagents (Applied Biosystems). CD8, CD94 exon 1a, perforin, IFNγ and GADPH mRNA levels were quantified by real-time RT-PCR amplification using the Mx3000PTM Real-Time PCR System (Stratagene). Briefly, cDNA was amplified in a 25 μl reaction mixture containing 12.5 μl 2 × SYBR Green Master Mix (Applied Biosystems), 100 ng of cDNA template, and selected primers (200 nM) using the recommended cycling conditions (denaturation at 95°C for 10 min followed by 40 cycles of 95°C for 15 s, 60°C for 30 s and 72°C for 30 s, 1 cycle of 95°C for 60 s and 55°C for 30 s). The primer sequences, designed with Primer Express software (Applied Biosystems), were as follows: CD8, 5′-GCT ACCACAGGAGCCGAAAG-3′ (forward) and 5′-TGGGCTTGCCTTCCTGTCT-3′ (reverse); CD94 exon 1a, 5′-AATCACTCTTAGGTAAAATAGTCACCTAGACA-3′ (forward) and 5′-TCATACAAGTGTGGGATGT TGATG-3′ (reverse); perforin, 5′-AGG TCT CCC CAC TCT GGT TTC-3′ (forward) and 5′-TTC ACC CTG CCG TGG TTT-3′ (reverse); IFNγ, 5′-GGCACAGTCATTGAAAGC-3′ (forward) and 5′-TGCCAGTTCCTC CAGATA-3′ (reverse); GADPH, 5′-TTGTGGAAGGGCTCATGACC-3′ (forward) and 5′-TCTTCTGGGTGG CAGTGATG-3′ (reverse). Relative quantification of mRNA expression was calculated by the comparative threshold cycle (Ct) method [14]. The relative target quantity, normalized to an endogenous control (GADPH) and relative to the day zero calibrator, is expressed as 2−ΔΔCt (fold), where ΔCt = Ct of the target gene—Ct of endogenous control gene, and ΔΔCt = ΔCt of samples for the target gene—ΔCt of zero day calibrator for the target gene.
Statistical analysis
Student’s t test was utilized to determine the significance of the differences between experimental groups and P ≤ 0.05 was considered significant.
Results
CD8+ T-cells are critical early whereas NK-cells are essential longer-term to eradication of metastatic lung tumors
To define the specific roles of cytotoxic T- and NK-cells in the eradication of metastatic lung tumors, time-dependent depletion of CD8+ T and NK-cells was undertaken. Subcutaneous tumors were induced and allowed to grow to ∼10 mm in diameter to establish metastasis to the lungs. Mice were then depleted of T or NK-cells, starting either prior to (on day -1) or after (on days 4 or 10) IL-12 + GM-CSF microsphere injection into primary tumors. Treated tumors were surgically removed 7–8 days after microsphere injection and mice were monitored for 3 weeks to allow for growth of metastatic tumors. Lungs were removed 3 weeks after surgery and analyzed for tumor burden to determine the short- and long-term requirement for CD8+ T and NK-cells. The results are shown in Fig. 1. All control mice (untreated) had high tumor burden in the lungs demonstrating efficient metastasis. In contrast, IL-12+ GM-CSF microsphere treatment induced complete elimination of established metastatic disease in the majority of mice (no detectable tumor in 14 of 21 animals) confirming therapeutic efficacy. Depletion of CD8+ T-cells prior to treatment on the other hand, resulted in significant loss of tumor suppression demonstrating that CD8+ T-cells were essential to elimination of metastatic disease. However, depletion of CD8+ T-cells starting on day 4 (or day 10) post-therapy did not lead to a significant reduction in anti-tumor efficacy, revealing that CD8+ T-cell activity was important during the first 3 days following treatment but not later. Depletion of NK-cells prior to treatment (on day -1) also resulted in loss of tumor suppression in the lungs confirming previous findings [11]. In contrast to the CD8+ T-cells however, depletion of NK-cells starting on days 4 or 10 post-treatment also led to tumor outgrowth demonstrating a longer-term requirement for NK-cells (Fig. 1B). Collectively, these data establish an early and limited role for CD8+ T-cells but a more extensive requirement for NK-cells in eradication of metastatic lung tumors. The immediate role of CD8+ T-cells in tumor suppression further suggests that treatment primarily activated pre-existing, tumor-associated CD8+ T- effector/memory cells, consistent with our previous findings [13].
Fig. 1.

Time-dependent depletion of CD8+ T- and NK-cells in IL-12+GM-CSF microsphere-treated mice. Mice bearing advanced (500–600 mm3) subcutaneous primary tumors were treated with IL-12+GM-CSF microspheres (day 0) and primary tumors were surgically excised 7–8 days after treatment. Mice were monitored for 3 weeks after surgery and lung tumor burden was determined as described in “Materials and methods”. In some groups CD8+ T- or NK-cells were depleted starting on days -1, 4 or 10 via antibody administration. Tumors of control mice (untreated) were excised upon reaching 500–600 mm3 without treatment and lung tumor burden was analyzed 3 weeks after surgery. a CD8+ T-cell panel. Effect of CD8+ T-cell depletion on lung tumor burden. The differences between untreated control and all other groups were significant (P < 0.005). The difference between no depletion and day −1 groups was significant (P = 0.00034). The differences between no depletion and day 4 or day 10 depletion groups were not significant (P > 0.13). Error bars = S.E., n = 21 for no depletion group and eight for all other groups. b NK-cell panel. Effect of NK-cell depletion on lung tumor burden. The differences between control untreated and all other groups were significant (P < 0.03). The difference between no depletion and days -1, 4 or 10 depletion groups was significant (P < 0.004). Error bars = S.E., n = 20 for no depletion group and 8 for all others
IL-12+GM-CSF microsphere treatment induces immediate but transient CD8+ T-cell activation followed by NK-cell infiltration into primary and metastatic tumors
To understand the cellular basis of the short-term CD8+ T-cell involvement and the extended requirement for NK-cells in tumor regression, post-therapy functional dynamics of each subset was investigated. To this end, single-cell suspensions were prepared from primary tumors and tumor-draining lymph nodes of pre-/post-therapy mice and CD8+ T-/NK-cell populations were analyzed by flow cytometry on days 0 (pre-treatment), 1, 3, 5, 7 and 10. Similarly, total mRNA samples extracted from the lungs of these mice were analyzed for T- and NK-cell activation markers by quantitative real-time PCR. The data are shown in Fig. 2. Consistent with the results of the depletion study, treatment of primary tumors induced an increase in the fraction of IFNγ–producing CD8+ T-cells from a pre-therapy level of 15 to 60% one day after treatment (Fig. 2a). Treatment also enhanced the cytotoxic function of CD8+ T-cells as the fraction of perforin-positive cells increased from 13 to 35% between days 0 and 3. The rapid activation kinetics was consistent with activation of pre-existing CD8+ T-effector memory cells [13]. Analysis of CD8+ T-cell numbers demonstrated that the quantity of intratumoral CD8+ T-cells declined by 4-fold immediately after treatment and remained low for 5 days. The number of intratumoral CD8+ T-cells increased again by 3-fold on day 7 post-therapy, however these cells did not display an activated phenotype. The initial decline in post-therapy CD8+ T-cell numbers and the subsequent increase on day 7 were consistent with our previous findings that activation of tumor-associated CD8+ T-effector/memory cells was followed by rapid apoptotic death and that day 7 cells represented a second wave of CD8+ T-effectors arriving from the TDLN [13]. In contrast to the early decline in CD8+ T-cell numbers, treatment induced a 3-fold increase in intratumoral NK-cell quantity on day 3 (Fig. 2a). The increase in NK-cell numbers was accompanied with a similar rise in the percentage of NK-cells that stained positive for IFNγ and perforin. The NK-cell response subsided on day 5 with both the cell numbers and activity retreating to pre-therapy levels. These data demonstrate that treatment resulted in the rapid activation of pre-existing CD8+ T-cells, which was then followed by infiltration of primary tumors with cytotoxic NK-cells. However both these events were transient and intratumoral lymphocyte activity returned to background levels by day 5.
Fig. 2.
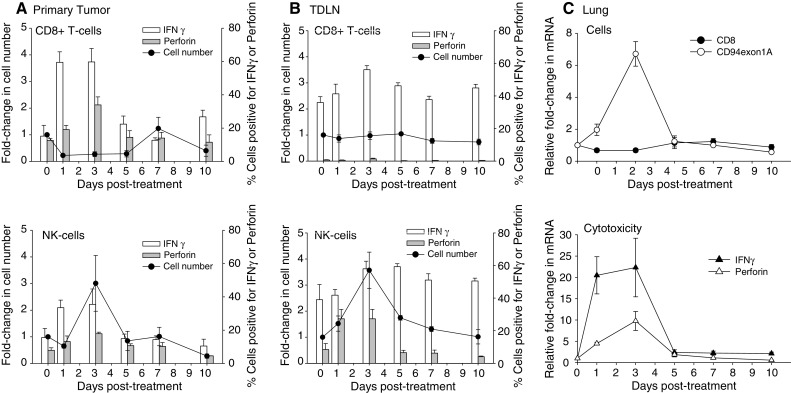
CD8+ T- and NK-cell numbers and activity in post-therapy tumors, TDLN and lungs. Mice bearing advanced (500–600 mm3) primary tumors were treated with a single injection of IL-12+GM-CSF microspheres (day 0). Primary tumors, TDLN and lungs were analyzed for CD8+ T- and NK-cell quantity (cells/gram tumor) and activity (% IFNγ and perforin-positive cells) on indicated days. Day 0 data were obtained prior to treatment. a Primary tumors. Relative fold-change in tumor-infiltrating CD8+ T- and CD3- CD49b + NKG2D + NK-cell numbers is shown on the left ordinate. Percent cells positive for IFNγ or perforin are shown on the right ordinate. The differences between day 0 versus day 1 or 3 IFNγ + CD8+ T-cells were significant (P < 0.005). The differences between day 0 versus day 1 or 3 perforin + CD8+ T-cells were also significant (P < 0.05). The difference between day 0 versus day 3 perforin + NK-cells was significant (P = 0.007). Error bars = S.E., n = 6–13 per group. b TDLN. Cell quantity and activity were determined as in a. Two TDLN were analyzed per mouse. Error bars = S.E., n = 6–13 mice per group. c Lungs. CD8, CD94 exon 1A, IFNγ and perforin mRNA levels in whole lung extracts were quantified by real-time PCR. Relative fold-change compared to pre-therapy (day 0) levels is shown. The increase in CD94 exon 1A mRNA on day 3 was highly significant compared to other days (P < 0.0014). The increases in IFNγ and perforin mRNA (days 1–3) were also significant compared to other time points (P < 0.006). Error bars = S.E., n = 4 mice per group
Analysis of TDLN demonstrated a different activity profile for CD8+ T-cells in comparison to the primary tumor. No significant changes were seen in the overall number of TDLN CD8+ T-cells after treatment (Fig. 2b). Some enhancement of IFNγ expression (1.5-fold) was observed however, it was significantly weaker than that seen in the primary tumor (4-fold). Furthermore, TDLN CD8+ T-cells did not express perforin following treatment, demonstrating that induction of T-cell cytotoxic activity was limited to the tumor. In contrast, post-therapy changes in the quantity and the activity profiles of NK-cells displayed patterns similar to those seen in the primary tumor, revealing the relatively non-specific nature of the NK-cell response. Importantly, CD8+ T and NK-cells isolated from pre-therapy TDLN (Fig. 2b) produced higher (2.5-fold) levels of IFNγ than those obtained from pre-therapy tumors (Fig. 2a), suggesting that immune suppression was more severe in the tumor microenvironment than the TDLN. These observations highlight the tumor-specific nature of post-therapy T-cell activity and the more ubiquitous character of the NK-cell response.
Activation kinetics of CD8+ T and NK-cells in the lungs was monitored via quantitative real-time PCR analysis since direct isolation of lymphocyte populations from lung metastases was not technically feasible. The levels of CD8 and CD94 exon 1A mRNAs were monitored as markers for CD8+ T and NK-cell quantity, respectively. CD94 gene product is a receptor for non-classical MHC molecules and is involved in controlling NK-cell activation [29]. Recent studies demonstrated the existence of two variants of the CD94 gene, exons 1A and B, which are expressed differentially in NK and T-cells [32]. Of the two, exon 1A expression has been shown to be highly specific to NK-cells, making this an ideal marker for molecular quantification of NK-cells [32]. In addition to the quantitative markers, IFNγ and perforin mRNA levels were monitored as common markers for activation of both T- and NK-cells. The results are shown in Fig. 2c. These data demonstrate that CD8+ T and NK-cell activation kinetics in metastatic tumors were similar to that observed in primary tumors. An initial decrease in CD8 mRNA expression (days 1–3) was followed by a modest recovery on day 7 (1.8-fold) whereas CD94 exon 1A expression increased by 6-fold on day 3 post-therapy. The NK-cell numbers declined rapidly thereafter, reaching pre-therapy levels on day 5. Treatment also strongly enhanced the expression of IFNγ and perforin in the lungs with increases of 20- and 10-fold in mRNA levels on day 3, respectively. IFNγ and perforin expression in the lungs returned to background levels on day 5, duplicating the pattern observed in primary tumors. These results establish that treatment of primary tumors with IL-12 and GM-CSF microspheres induced similar lymphocyte activity patterns in primary and metastatic tumors.
NK-cell recruitment to primary and metastatic tumors is dependent on pre-existing T-cells and IFNγ
The 24–48 h delay between CD8+ T-cell activation and NK-cell infiltration, coupled with the transient nature of both events raised the possibility that NK-cell infiltration was functionally linked to T-effector/memory cell activation. To address this question, infiltration of NK-cells into primary and metastatic tumors was monitored in control (non-depleted), CD8+ T-cell-depleted and CD4+/CD8+ T-cell-depleted mice. The results are shown in Fig. 3. Treatment resulted in a 2.5-fold increase in intratumoral NK-cell numbers in the primary tumors of control mice on day 3 confirming earlier observations (Fig. 3, primary tumor). Depletion of CD8+ T-cells on the other hand, resulted in the complete loss of day 3 NK-cell infiltration suggesting that CD8+ T-cells were critical to NK-cell recruitment to primary tumors. Depletion of both CD8+ and CD4+ T-cells further reduced NK-cell numbers but this effect was not statistically significant. In contrast, depletion of CD8+ T-cells led to a modest decline in NK-cell infiltration into the TDLN (Fig. 3, TDLN). However, co-depletion of CD8+ and CD4+ T-cells resulted in the complete abrogation of NK-cell recruitment in the TDLN demonstrating that both subsets were important to NK-cell recruitment to the TDLN. Analysis of NK-cell infiltration in the lungs displayed a similar pattern further confirming that both CD8+ and CD4+ T-cells were important to systemic NK-cell recruitment (Fig. 3, Lungs). In reverse experiments, depletion of NK-cells did not significantly alter the activation kinetics of tumor-associated CD8+ T-cells suggesting that NK-cells did not play a significant role in the promotion of intratumoral T-cell activity (Table 1).
Fig. 3.

Effect of T-cell depletion on post-therapy NK-cell infiltration. Mice bearing advanced (500–600 mm3) primary tumors were treated with IL-12 + GM-CSF microspheres. The depletion groups received the relevant antibodies on days -4 and -1 prior to treatment. Relative fold-changes in CD3- CD49b + NKG2D + NK-cell numbers were quantified in pre-therapy (day 0) and post-therapy (day 3) primary tumors (cells/gram tumor) and TDLN (cells/lymph node) by flow cytometry. Relative fold-changes in lung CD94 exon 1A mRNA was quantified by real-time PCR analysis. The difference between days 0 and 3 NK-cell numbers in primary tumors was significant in the no depletion group (P = 0.021). The differences between days 0 and 3 in CD8 or CD8+ CD4 depletion groups were not significant (P > 0.7). The differences between days 0 and 3 in no depletion and CD8+ depletion groups were significant in the TDLN and lungs (P < 0.05). Error bars = S.E., n = 5–6 mice per group
Table 1.
Effect of NK-cell depletion on post-therapy intra-tumoral CD8+ T-cell activity
| Day | CD8+ T-cell numbersa | CD8+ T-cell activationb | ||
|---|---|---|---|---|
| Control | NK-cell depletion | Control | NK-cell depletion | |
| 0 | 2,900 ± 1,550 | 2,731 ± 692 | 14.8 ± 2.1 | 10.4 ± 2.2 |
| 3 | 422 ± 260 | 372 ± 66 | 19.2 ± 5.3 | 11.7 ± 5.4 |
| 7 | 1,579 ± 662 | 806 ± 457 | 13.4 ± 3.7 | 8.5 ± 4.5 |
aDisplayed as average cells/gram tumor ± SE. The differences between control (no depletion) and NK-cell-depleted groups were not statistically significant at any given time point (P ≥ 0.55; n = 4–6 per group)
bDetermined as percent CD8+ T-cells that are double-positive for activation markers CD43 and CD69. The differences between control (no depletion) and NK-cell-depleted groups were not statistically significant at any given time point (P ≥ 0.13; n = 4–6 per group)
The central role of IFNγ in IL-12-mediated tumor eradication is well established [28]. IFNγ is a pluripotent cytokine with diverse anti-tumor activities including stimulation of T/NK-cell cytotoxicity and macrophage activation, augmentation of antigen-processing/presentation, direct tumor suppression and induction of anti-angiogenic mechanisms [12]. A recent study also demonstrated a role for IFNγ in NK-cell recruitment to the periphery [31]. To this end, the requirement for IFNγ in T-effector/memory cell-dependent NK-cell infiltration of post-therapy tumors was evaluated in IFNγ knockout (GKO) mice. Subcutaneous primary tumors were induced in wild-type and GKO Balb/C mice and were allowed to grow to 10 mm in diameter. The mice were then treated with a single intratumoral injection of IL-12+GM-CSF microspheres. NK-cell infiltration into primary tumors, TDLN and the lungs were then monitored on days 0 (pre-therapy) and 3 (post-therapy) by flow cytometric analysis of single-cell suspensions or by real-time PCR quantification. The results are shown in Fig. 4. These data demonstrate that NK-cell recruitment to primary tumors was significantly reduced in GKO mice (Fig. 4a) and was essentially eliminated in the corresponding TDLN (Fig. 4b). Analysis of the lungs also demonstrated a 2-fold reduction in NK-cell numbers in post-therapy GKO mice similar to that seen in the primary tumors. Finally, analysis of perforin production in the lungs revealed a near-complete elimination of cytotoxic activity in the lungs of post-treatment GKO mice as compared to the wild-type animals linking NK-cell infiltration to enhanced cytotoxic activity (Fig. 4b). These data establish a critical role for IFNγ in the therapy-induced NK-cell infiltration and cytotoxicity in primary and metastatic tumors.
Fig. 4.

Post-therapy NK-cell infiltration into tumors, TDLN and lungs of IFNγ-knockout mice. Primary tumors (500–600 mm3), TDLN and lungs were analyzed for NK-cell infiltration prior to (day 0) and post-treatment (day 3) in wild-type and IFNγ-knockout (GKO) mice. a Fold-change in tumor-associated CD3- CD49b + NKG2D + NK-cells between days 0 and 3 was significant in both wild-type and GKO mice (P < 0.046). The difference between wild-type versus GKO mice on day 3 was not significant (P = 0.16). b Fold-change in TDLN NK-cells was significant between days 0 and 3 in both wild-type and GKO mice (P < 0.046). The difference between wild-type and GKO mice on day 3 was also significant (P = 0.0048). c The differences between days 0 and 3 in wild-type as well as GKO mice were significant for both CD94 exon 1A and perforin (P < 0.0043). The differences between wild-type and GKO mice on day 3 with regard to CD94 exon 1A and perforin expression were also significant (P < 0.034). Error bars = S.E., n = 5–8 mice per group
Discussion
The goal of this study was to define the individual roles of CD8+ T- and NK-cells in the eradication of established lung metastases following local delivery of IL-12 and GM-CSF to advanced primary tumors. The results show that both CD8+ T and NK-cells were required for elimination of metastatic disease and that each subset displayed distinct functional dynamics. The studies revealed an early and limited involvement for CD8+ T-cells, which was followed by longer-term NK-cell activity. The immediate yet transient activation kinetics of CD8+ T-cells suggested that treatment specifically induced the activation of tumor-associated T-effector/memory cells [13]. Maintenance of partial tumor suppression in the absence of NK-cells supported a direct cytotoxic role for CD8+ T-effector/memory cells. At the same time, the absolute requirement for CD8+ T-cells in the recruitment of NK-cells to primary tumors demonstrated an additional and indirect anti-tumor role for pre-existing CD8+ T-cells. Collectively, these data delineate the distinct functions of tumor-associated T-effector/memory and NK-cells in the eradication of established systemic disease and reveal an important link between pre-existing adaptive immunity and innate effectors in tumor suppression.
An unexpected result here was the lack of CD8+ T-cell involvement in the suppression of lung tumors beyond day 3 post-treatment. This was not due to an inability to induce long-term T-effector cells since previous studies established that IL-12/GM-CSF microsphere treatment promotes potent long-term, tumor specific T-cell immunity [1, 6, 11, 18]. In contrast, it is possible that the absence of CD8+ T-effector involvement in long-term tumor eradication was associated with the low MHC Class I phenotype of Line-1 cells [3]. However, MHC Class I expression on Line-1 cells is IFNγ-inducible, exposure to which can result in dramatically enhanced sensitivity to cytotoxic T-cell killing [3]. Since IL-12 microsphere treatment induces high systemic IFNγ [11], a primary role for MHC Class I loss-dependent tumor escape here is unlikely. Alternatively, other mechanisms such as tumor-mediated immune suppression [9] and/or the development of homeostatic regulatory mechanisms [33] could be responsible for the observed inefficacy of post-day 3 CD8+ T-effectors. To this end, our previous studies demonstrated that IL-12+GM-CSF microsphere treatment is ultimately followed by CD4+ CD25+ Foxp3+ T-suppressor cell infiltration of tumors on day 7 post-therapy [18].
Recent studies demonstrated a role for NK-cells in T-cell recruitment [19] and in shaping the phenotype of T-helper cells [15]. Conversely, our data reveals a novel function for tumor-associated CD8+ T-effector/memory cells in the recruitment of NK-cells to tumors. Furthermore, the results obtained in the TDLN and the lungs of CD8+/CD4+ T-cell-depleted mice suggest that this may be part of a general mechanism that requires T-cell participation in NK-cell mobilization. To this end, a role for T-cells in mobilization of NK-cells from bone marrow and spleen was reported recently [31]. Rapid and abundant production of IFNγ by post-therapy T-cells and the demonstrated ability of IFNγ to induce T- and NK-cell chemokines [8] further suggested that T-cell-dependent NK-cell chemotaxis could be mediated by IFNγ. In this context, a requirement for IFNγ in IL-12-mediated NK-cell infiltration into liver has been demonstrated [7]. The loss of NK-cell recruitment in IL-12+GM-CSF-treated GKO mice here confirmed the critical role of IFNγ in NK-cell mobilization. Collectively, these data are consistent with the notion that T-cells promoted NK-cell infiltration via IFNγ in our model.
The long-term requirement for NK-cells (i.e. for at least 10 days after treatment) in the suppression of metastatic lung tumors was surprising since therapy-induced anti-tumor NK-cell activity subsided within 5 days of treatment in all tissues tested. One possible explanation for the continued requirement for NK cells could be the persistence of low-level NK-cell activity beyond day 5 in residual metastatic tumors. To this end, studies performed in NK-cell depleted and immune-deficient/mutant mice have established a significant role for NK and NKT-cells in surveillance of spontaneously arising and/or chemically induced tumors [26]. These observations support the hypothesis that post-peak basal NK-cell activity was sufficient for continued suppression of residual metastatic tumors in our model.
The lack of any significant contribution by long-term T-effector cells to elimination of lung metastases here is consistent with others’ findings that vaccine-induced anti-tumor CD8+ T-effector cell activity does not correlate well with clinical tumor regression [21, 22]. It will, therefore, be important to determine whether the failure of long-term CD8+ T-effector cells to mediate tumor suppression in this model involves a unique case of immune evasion or other more universal mechanisms including active suppression of T-cell function by tumors and/or development of homeostatic regulatory mechanisms in response to immune stimulation. The clinically relevant surgical metastasis model utilized here represents an ideal setting for such studies.
Acknowledgments
The authors thank Dr. Stan Wolf of Wyeth Pharmaceuticals for providing the recombinant IL-12 and for his continued support of our studies.
Footnotes
This work was supported by NIH/NCI grant R01-CA100656-01A1 to N.K.E.
References
- 1.Arora A, Su G, Mathiowitz E, Reineke J, Chang AE, Sabel MS. Neoadjuvant intratumoral cytokine-loaded microspheres are superior to postoperative autologous cellular vaccines in generating systemic anti-tumor immunity. J Surg Oncol. 2006;94(5):403–412. doi: 10.1002/jso.20572. [DOI] [PubMed] [Google Scholar]
- 2.Berzofsky JA, Terabe M, Oh S, Belyakov IM, Ahlers JD, Janik JE, Morris JC. Progress on new vaccine strategies for the immunotherapy and prevention of cancer. J Clin Invest. 2004;113(11):1515–1525. doi: 10.1172/JCI21926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blieden TM, McAdam AJ, Frelinger JG, Lord EM. Mechanism of cytolytic T lymphocyte killing of a low class I-expressing tumor. J Immunol. 1991;147(4):1433–1438. [PubMed] [Google Scholar]
- 4.Clevenger CV, Shankey TV. Preparation of cells and reagents for flow cytometry. In: Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W, editors. Current protocols in immunology. 2001 edn. New York: Wiley; 1993. pp. 531–5324. [DOI] [PubMed] [Google Scholar]
- 5.Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer. 2004;4:11–22. doi: 10.1038/nrc1252. [DOI] [PubMed] [Google Scholar]
- 6.Egilmez NK, Jong YS, Sabel MS, Jacob JS, Mathiowitz E, Bankert RB. In situ tumor vaccination with Interleukin-12 encapsulated biodegradable microspheres: induction of tumor regression and potent antitumor immunity. Cancer Res. 2000;60:3832–3837. [PubMed] [Google Scholar]
- 7.Fogler WE, Volker K, Watanabe M, Wigginton JM, Roessler P, Brunda MJ, Ortaldo JR, Wiltrout RH. Recruitment of hepatic NK cells by IL-12 is dependent on IFN-gamma and VCAM-1 and is rapidly down-regulated by a mechanism involving T cells and expression of Fas. J Immunol. 1998;161:6014–6021. [PubMed] [Google Scholar]
- 8.Fraticelli P, Sironi M, Bianchi G, D’Ambrosio D, Albanesi C, Stoppacciaro A, Chieppa M, Allavena P, Ruco L, Girolomoni G, Sinigaglia F, Vecchi A, Mantovani A. Fractalkine (CX3CL1) as an amplification circuit of polarized Th1 responses. J Clin Invest. 2001;107:1173–1181. doi: 10.1172/JCI11517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gajewski TF, Meng Y, Harlin H. Immune suppression in the tumor microenvironment. J Immunother. 2006;29(3):233–240. doi: 10.1097/01.cji.0000199193.29048.56. [DOI] [PubMed] [Google Scholar]
- 10.Girardi M, Oppenheim DE, Steele CR, Lewis JM, Glusac E, Filler R, Hobby P, Sutton B, Tigelaar RE, Hayday AC. Regulation of cutaneous malignancy by gammadelta T cells. Science. 2001;294(5542):605–609. doi: 10.1126/science.1063916. [DOI] [PubMed] [Google Scholar]
- 11.Hill HC, Conway TF, Sabel MS, Jong YS, Mathiowitz E, Bankert RB, Egilmez NK. Cancer immunotherapy with interleukin-12 and granulocyte–macrophage colony-stimulating factor-encapsulated microspheres: coinduction of innate and adaptive immunity and cure of disseminated disease. Cancer Res. 2002;62:7254–7263. [PubMed] [Google Scholar]
- 12.Ikeda H, Old LJ, Schreiber RD. The roles of IFNγ in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002;13:95–109. doi: 10.1016/S1359-6101(01)00038-7. [DOI] [PubMed] [Google Scholar]
- 13.Kilinc MO, Aulakh KS, Nair RE, Jones SA, Alard P, Kosiewicz MM, Egilmez NK. Reversing tumor immune suppression with intra-tumoral IL-12: activation of tumor-associated Tem, induction of T-Suppressor apoptosis and infiltration of CD8+ T-effectors. J Immunol. 2006;177(10):6962–6973. doi: 10.4049/jimmunol.177.10.6962. [DOI] [PubMed] [Google Scholar]
- 14.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 15.Martin-Fontecha A, Thomsen LL, Brett S, Gerard C, Lipp M, Lanzavecchia A, Sallusto F. Induced recruitment of NK cells to lymph nodes provides IFN-γ for TH1 priming. Nat Immunol. 2006;5(12):1260–1265. doi: 10.1038/ni1138. [DOI] [PubMed] [Google Scholar]
- 16.McLean M, Wallace HL, Sharma A, Hill HC, Sabel Egilmez MS NK. A BALB/c murine lung alveolar carcinoma used to establish a surgical spontaneous metastasis model. Clin Exp Met. 2004;21(4):363–369. doi: 10.1023/B:CLIN.0000046176.33867.c5. [DOI] [PubMed] [Google Scholar]
- 17.Monsurro V, Wang E, Yamano Y, Migueles SA, Panelli MC, Smith K, Nagorsen D, Connors M, Jacobson S, Marincola FM. Quiescent phenotype of tumor-specific CD8+ T-cells following immunization. Blood. 2004;104(7):1970–1978. doi: 10.1182/blood-2004-02-0525. [DOI] [PubMed] [Google Scholar]
- 18.Nair RE, Kilinc MO, Jones SA, Egilmez NK. Chronic immune therapy induces a progressive increase in intra-tumoral T-suppressor activity and a concurrent loss of tumor-specific CD8+ T-effectors in her-2/neu transgenic mice bearing advanced spontaneous tumors. J Immunol. 2006;176(12):7325–34. doi: 10.4049/jimmunol.176.12.7325. [DOI] [PubMed] [Google Scholar]
- 19.Roda JM, Parihar R, Magro C, Nuovo GJ, Tridandapani S, Carson WE., III Natural killer cells produce T cell-recruiting chemokines in response to antibody-coated tumor cells. Cancer Res. 2006;66(1):517–526. doi: 10.1158/0008-5472.CAN-05-2429. [DOI] [PubMed] [Google Scholar]
- 20.Rosenberg SA. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med. 1998;4:321–327. doi: 10.1038/nm0398-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenberg SA, Sherry RM, Morton KE, Scharfman WJ, Yang JC, Topalian SL, Royal RE, Kammula U, Restifo NP, Hughes MS, Schwartzentruber D, Berman DM, Schwarz SL, Ngo LT, Mavroukakis SA, White DE, Steinberg SM. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol. 2006;175(9):6169–6176. doi: 10.4049/jimmunol.175.9.6169. [DOI] [PubMed] [Google Scholar]
- 22.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slingluff CL., Jr Clinical and immunologic results of a randomized phase II trial of vaccination using four melanoma peptides either administered in granulocyte-macrophage colony-stimulating-factor in adjuvant or pulsed on dendritic cells. J Clin Oncol. 2003;21:4016–4026. doi: 10.1200/JCO.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 24.Smyth MJ, Thia KY, Street SE, Cretney E, Trapani JA, Taniguchi M, Kawano T, Pelikan SB, Crowe NY, Godfrey DI. Differential tumor immune surveillance by natural killer (NK) and NKT cells. J Exp Med. 2000;191:661–668. doi: 10.1084/jem.191.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stagg J, Smyth MJ. NK-cell based cancer immunotherapy. Drug News Perspect. 2007;20(3):155–163. doi: 10.1358/dnp.2007.20.3.1092096. [DOI] [PubMed] [Google Scholar]
- 26.Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest. 2007;117(5):1137–1146. doi: 10.1172/JCI31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taieb J, Chaput N, Ménard C, Apetoh L, Ullrich E, Bonmort M, Péquignot M, Casares N, Terme M, Flament C, Opolon P, Lecluse Y, Métivier D, Tomasello E, Vivier E, Ghiringhelli F, Martin F, Klatzmann D, Poynard T, Tursz T, Raposo G, Yagita H, Ryffel B, Kroemer G, Zitvogel L. A novel dendritic cell subset involved in tumor immunosurveillance. Nat Med. 2006;12(2):214–219. doi: 10.1038/nm1356. [DOI] [PubMed] [Google Scholar]
- 28.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nature Rev. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 29.Vance RE, Kraft JR, Altman JD, Jensen PE, Raulet DH. Mouse CD94/NKG2A is a natural killer cell receptor for the nonclassical major histocompatibility complex (MHC) class I molecule Qa-1(b) J Exp Med. 1998;188:1841–1848. doi: 10.1084/jem.188.10.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vieweg J. Future directions for vaccine-based therapies. Urol Oncol. 2006;24:448–455. doi: 10.1016/j.urolonc.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 31.Wald O, Weiss ID, Wald H, Shoham H, Bar-Shavit Y, Beider K, Galun E, Weiss L, Flaishon L, Shachar I, Nagler A, Lu B, Gerard C, Gao J-L, Mishani E, Farber J, Peled A. IFN-γ acts on T cells to induce NK cell mobilization and accumulation in target organs. J Immunol. 2006;176:4716–4729. doi: 10.4049/jimmunol.176.8.4716. [DOI] [PubMed] [Google Scholar]
- 32.Wilhelm BT, Josette-Renee L, Takei F, Mager DL. Transcriptional control of murine CD94 gene: differential usage of dual promoters by lymphoid cell types. J Immunol. 2003;171:4219–4226. doi: 10.4049/jimmunol.171.8.4219. [DOI] [PubMed] [Google Scholar]
- 33.Zhou G, Drake CG, Levitsky HI. Amplification of tumor-specific regulatory T cells following therapeutic cancer vaccines. Blood. 2006;107(2):628–636. doi: 10.1182/blood-2005-07-2737. [DOI] [PMC free article] [PubMed] [Google Scholar]