

Abstract
Tumor-targeted delivery of a potent cytotoxic agent, calicheamicin, using its immunoconjugates is a clinically validated therapeutic strategy. Rituximab is a human CD20-specific chimeric antibody extensively used in B-NHL therapy. We investigated whether conjugation to calicheamicin can improve the anti-tumor activity of rituximab against human B-cell lymphoma (BCL) xenografts in preclinical models. BCL cells were cultured with rituximab or its calicheamicin conjugates and their in vitro growth was monitored. BCL cells were injected s.c. to establish localized xenografts in nude mice or i.v. to establish disseminated BCL in severe combined immunodeficient (scid) mice. I.p. treatment with rituximab or its calicheamicin conjugates was initiated and its effect on s.c. BCL growth or survival of mice with disseminated BCL was monitored. Conjugation of calicheamicin to rituximab vastly enhanced its growth inhibitory activity against BCL in vitro. Conjugation to calicheamicin had no deleterious effect on the effector functional activity of rituximab. Calicheamicin conjugated to rituximab with an acid-labile linker exhibited greater anti-tumor activity against s.c. BCL xenografts and improved survival of mice with disseminated BCL over that of unconjugated rituximab. Anti-tumor activities of rituximab conjugated to calicheamicin via an acid-stable linker were similar to that of unconjugated rituximab. Superior anti-tumor efficacy exhibited by a calicheamicin immunoconjugate of rituximab with an acid-labile linker over that of rituximab demonstrates the therapeutic potential of CD20-specific antibody-targeted chemotherapy strategy in the treatment of B-NHL.
Keywords: Calicheamicin, Rituximab, CD20, Immunoconjugates, Anti-tumor
Introduction
Monoclonal antibodies (mAbs) against tumor-associated antigens (TAA) are now widely used in cancer therapy. Tumor targeted mAb can exert its anti-tumor effects via its ability to directly cause tumor-cell apoptosis, recruit a host’s own FcR-expressing effector cells to mediate antibody-dependent cellular cytotoxicity (ADCC) or fix complement to bring about complement-dependent cytotoxicity (CDC). In addition, radioimmunoconjugates of tumor-targeted mAbs have been used to deliver radioactive isotopes to tumor cells. Immunotoxins generated by either chemically conjugating or genetically fusing bacterial or plant-derived toxins to tumor-targeted mAb have also been explored in cancer therapy.
Antibody-targeted chemotherapy (ATC) is a therapeutic strategy in which a TAA-specific mAb is covalently linked (conjugated) to a cytotoxic agent to deliver it specifically to the tumor cells expressing the internalizable TAA [1]. The main advantage of this strategy is the preferential delivery of the conjugated cytotoxic agent within the TAA-expressing intratumoral compartment while minimizing the exposure of tissues that do not express the TAA to the conjugated cytotoxic agent. These two attributes maximize anti-tumor efficacy and minimize toxicity of the targeted cytotoxic agent and thus, improve its therapeutic index.
Tumor-targeted chemotherapy as a therapeutic strategy has now been clinically validated using gemtuzumab ozogamicin (Mylotarg® or CMA-676), which is the first and currently the only antibody-targeted chemotherapy approved for use [2]. Gemtuzumab ozogamicin is indicated for the treatment of elderly patients with acute myeloid leukemia in first relapse and who are not candidates for standard combination chemotherapy [3, 4]. Gemtuzumab ozogamicin is an immunoconjugate of calicheamicin in which N-acetyl gamma calicheamicin dimethylhydrazide (CalichDMH) is covalently linked via an acid-labile AcBut linker to a humanized anti-CD33 antibody, hP67.6 [5]. It binds human CD33 with high affinity and upon internalization, exerts potent cytotoxic activity against CD33+ myeloid cells. CalichDMH is a derivative of gamma calicheamicin, a natural product of the enediyne class, that binds DNA in the minor groove and brings about double-strand DNA breaks leading to cellular apoptosis [6, 7]. The cytotoxic activity of CalichDMH is at least 10–100 fold more potent than conventional cytotoxic agents currently used in cancer chemotherapy and is ideally suited for the targeted chemotherapy strategy.
B lymphoid lineage-specific molecules CD20 and CD22 have been the focus of preclinical and clinical investigations with mAbs, immunotoxins and radioimmunoconjugates. The anti-CD20 mAb, rituximab (Rituxan®), is now the most extensively used antibody therapeutic in the treatment of non-Hodgkin’s B-cell lymphomas (B-NHL) [8]. In addition, CD20-targeted radioimmunotherapeutics Bexxar® and Zevalin® have also been approved for patients with B-NHL refractory to combination chemotherapy and rituximab [9]. Rituximab can mediate its anti-tumor effects by multiple mechanisms, including direct induction of apoptosis of BCL, effector cell-mediated ADCC, CDC and effector cell-mediated complement-dependent cellular cytotoxicity (CDCC) against BCL targets [10–12]. Clinically, rituximab is often used in B-NHL patients either alone or combined with cytoreductive chemotherapy [13]. In addition to its extensive usage in the treatment of B-NHL, rituximab is also being actively investigated as a B cell-depleting agent in inflammatory diseases such as rheumatoid arthritis and systemic lupus erythematosus [14].
The clinical success of gemtuzumab ozogamicin in treating malignancies of the myeloid lineage has provided impetus to apply the ATC strategy to other hematological malignancies including B-cell lymphomas and leukemias. A similar CD22-targeted immunoconjugate of calicheamicin, inotuzumab ozogamicin [15] is presently being investigated as a targeted chemotherapeutic agent in B-NHL patients [16] and has been shown preclinically to be more efficacious in combination with rituximab [17]. The expression of CD20 on the surface of most B cells and their malignant counterparts is often higher than that of CD22. We questioned whether this higher expression of CD20 over that of CD22 on the surface of BCL can be utilized to derive greater anti-tumor activity for the delivery of CD20 targeted-CalichDMH. The present study shows that calicheamicin conjugated to CD20-specific antibodies may be used as a potential therapeutic agent in the treatment of B lymphoid malignancies.
Materials and methods
Cell lines
B-cell lymphoma (BCL) lines Raji (CCL-86), Ramos (CRL-1923), Daudi (CCL-213) and RL (CRL-2261), and the multiple myeloma cell lines ARH-77 (CRL-1621) and RPMI-8226 (CRL-155) were all obtained from the American Type Culture Collection (ATCC, Manassas, VA). The cell lines were determined to be mycoplasma free as determined by a polymerase chain reaction mycoplasma detection assay (ATCC, Manassas, VA). The cell lines were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES, HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid), 1 mM sodium pyruvate, 0.2% glucose, penicillin G sodium 100 U/ml, and streptomycin sulfate 100 μg/ml. Before use, viable cells were isolated by centrifugation (30 min at 1,000g) using Lymphoprep (Nycomed, Oslo, Norway) density gradient.
Mice
Female, BALB/c, nu/nu (nude) mice (18–23 g) and male scid mice (CB17 scid, 20–25 g) were obtained from Charles River Laboratories, Wilmington, MA. All mice were housed in micro isolator units and provided with sterile food and water ad libitum throughout the studies. All procedures involving mice were approved by the Wyeth Animal Care and Use Committee according to established guidelines.
Conjugation of anti-CD20 mAb with calicheamicin
Chimeric human IgG1 anti-CD20 mAb, rituximab (Rituxan, Biogen-Idec Pharmaceuticals, San Diego, CA and Genentech, South San Francisco, CA), was purchased from Med World Pharmacy, Chestnut Ridge, NY. Rituximab was covalently conjugated via amide bonds between its lysine ε–NH2 groups and N-hydroxysuccinimide activated derivatives of N-acetyl gamma calicheamicin with either an acid stable dimethyl amide or acid labile dimethyl hydrazide AcBut linker as follows. The antibody was buffer exchanged to a pH 8–8.5 non-nucleophilic buffer (10 mM HEPES/100 mM NaCl) and concentrated to approximately 10 mg/ml. For the AcBut derivative, an excipient (sodium octanoate) that prevents protein aggregation was added to a final concentration of 150 mM. Finally, 5% of the protein mass of the N-hydroxysuccinimide activated AcBut calicheamicin derivative was added as a concentrated solution (10 mg/ml) in ethanol. This reaction mixture was then incubated at 25°C for 2 h. Similarly, the N-hydroxysuccinimide activated DMA (dimethyl acid) calicheamicin derivative was conjugated to the buffer exchanged antibody, but using 20% dimethylformamide as a co-solvent in place of the excipient used in the AcBut reaction. Progress of each reaction was monitored by SEC–HPLC, and after completion the conjugates were separated from aggregated antibody and free calicheamicin on a preparative SEC column. Only the monomeric IgG conjugates as defined by analytical size-exclusion profiling were used in various investigations. The amount of calicheamicin on each of the monomeric conjugates prepared was between 20 and 25 μg/mg protein (2–3 moles/mole).
Human CD33-targeted CMA-676 is comprised of a humanized IgG4 anti-CD33 mAb, hP67.6, linked to CalichDMH via the AcBut linker with an average drug loading of 35 μg of CalichDMH per mg of hP67.6 protein (3.2 moles/mole). Human CD22-targeted CMC-544 is comprised of a humanized IgG4 anti-CD22 mAb, G5/44, linked to CalichDMH via the AcBut linker with an average drug loading of 65–73 μg of CalichDMH per mg of G5/44 protein (approximately 6 moles/mole). Additionally, an acid-stable Amide conjugate of anti-CD22 mAb was made by conjugating G5/44 or its murine counterpart, m5/44, to N-acetyl gamma calicheamicin dimethyl acid (CalichDMA). The average drug loading of the amide-linked calicheamicin conjugates was 25 μg of CalichDMA per mg of antibody (approximately 2 moles/mole). All conjugates were confirmed as having low endotoxin (<5.0 EU/ml) as determined by the limulus amebocyte lysate test (BioWhittaker, Walkersville, MD). Doses of calicheamicin conjugates are expressed as equivalents of CalichDMH and that of unconjugated antibody are expressed as antibody protein.
Flow cytometry
Binding of the anti-CD20 antibody or anti-CD20 calicheamicin conjugates was confirmed with Ramos cells and compared with that of G5/44 and its AcBut and amide-linked calichDM conjugates. Briefly, 0.3–10 μg of antibody or antibody conjugate was incubated with 100,000 cells for 30 min at 4°C. Cells were washed twice with PBS containing 1% BSA to remove unbound antibody and then a FITC labeled goat anti-human (H + L) secondary antibody (100 fold diluted) was added for 30 min at 4°C. The cells were again washed twice to remove unbound secondary antibody and then resuspended in 1% formaldehyde (in PBS) with 1% BSA. Fluorescent intensities were measured on a Becton Dickinson FACSort. Results are plotted in arbitrary units of mean fluorescent intensity (MFI). Expression of CD32 on BCL was also determined by flow cytometry.
Tumor digest for primary culture
Ramos subcutaneous tumors were excised aseptically and minced with 2 ml of collagenase (Type 4, 2 mg/ml stock, 203 Units/mg, Worthington, Lakewood, NJ) and incubated for 4 h at 37°C with 5% CO2. The suspension was thoroughly mixed until the tumor was completely dissociated. The tumor suspension was pelleted by centrifugation and then resuspended in 25 ml of fresh culture media. After an overnight incubation in culture media, the expression of CD20, CD22, and CD33 was analyzed by flow cytometry.
Cytotoxicity assays
A vital dye (MTS, Promega, Madison, WI) staining was used to determine the number of surviving cells following exposure to various drug treatments. For each cell line a calibration curve (cell number vs. optical density after 2 h) was established to estimate an appropriate initial seeding density. Cells were then seeded in 96-well microtiter plates at a density of 5,000–10,000 cells per well. After seeding, the cells were exposed to various concentrations of unconjugated calicheamicin (CalichDMH), anti-CD20 mAb, NAc-gamma calicheamicin derivatives conjugated to anti-CD20 or CMA-676. Calicheamicin conjugate concentrations were based on the quantity of calicheamicin equivalents. Concentrations of unconjugated mAb were based on protein content. Following determination of the number of cells surviving 96 h of drug exposure, the IC50 was calculated by logistic non-linear regression from the dose–response curves and reported as the calicheamicin concentration from each treatment group that causes 50% loss of cell viability.
Assessment of ADCC and CDC
Human whole blood was collected and allowed to clot for 60 min at room temperature after which the tubes were centrifuged to collect serum for CDC analysis. Either 10,000 or 50,000 Ramos B cells were mixed with increasing concentrations of rituximab or its conjugates in the presence or absence of human serum (1:60 final dilution). Samples were incubated at 37°C for 4 h after which the activity of lactate dehydrogenase (LDH) in the cell-free supernatants was assessed using the Cytotox-1 homogeneous membrane integrity kit (Promega). Ramos cells were lysed to assess the maximum LDH releasable from these cells. LDH activity in the absence of antibody or conjugate, and complement represented the background release.
The percent cytotoxicity was calculated by the following equation:
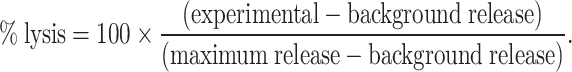 |
Peripheral blood mononuclear cells (PBMCs) were used as effector cells to assess the ADCC activity of rituximab and its conjugates. Peripheral blood was collected in heparinized collection tubes and PBMCs were separated from the heparinized blood using Ficoll–Hypaque gradient centrifugation. PBMCs were washed in PBS and resuspended in RPMI 1640 supplemented with 10% FBS and used as effector cells in ADCC assays.
For ADCC activity, 5,000 Ramos B cells were mixed with 250,000 PBMCs [effector cell to target cell ratio (E:T) of 50:1] with rituximab or its conjugates and incubated at 37°C for 4 h. The release of LDH in the cell-free culture medium was assessed using the Cytotox-1 homogeneous membrane integrity kit as described above. The negative controls included Ramos cells alone, PBMCs alone, a mixture of Ramos cells and PBMCs in the absence of antibodies, Ramos cells and antibodies without PBMCs, and PBMCs and antibodies without Ramos cells. Maximum release of LDH was derived from cells treated with the lysis buffer. Percent lysis was calculated as described above.
Subcutaneous BCL xenografts
Female, athymic (nude) mice were exposed to total body irradiation (400 rads) to further suppress their residual immune system and facilitate the establishment of BCL xenografts. An additional group of mice were not irradiated before tumor cell implantation. After 3 days, irradiated or non-irradiated mice were injected s.c. with 1 × 107 Ramos cells suspended in Matrigel (Collaborative Biomedical Products, Belford, MA, diluted 1:1 in RPMI 1640 medium) in the dorsal, right flank. When the tumors reached a mass of approximately 0.5 g for the moderate tumor size study or 2 g for the large tumor size study, they were staged to maximize uniformity of the tumor mass prior to the administration of therapy (n = 5–7 mice/treatment group). Compounds were administered i.p. in sterile saline (0.2 ml/mouse) on day 1 and the same treatment was repeated twice, 4 days apart (Q4Dx3). Length and width (in cm) of the tumors were measured at least once a week and their mass was calculated as tumor mass (g) = [0.5 × (tumor width)2 (tumor length)]. Mean (±SEM) tumor mass for each treatment group was calculated and compared to the vehicle-treated group for statistical significance using analysis of variance and subsequent pairwise comparisons to the vehicle-treated group by a one-tailed t test with the error term for the t test based on the pooled variance across all treatment groups. Tumor mass values for each treatment group were recorded up to 60 days after the initiation of treatment or until the tumors grew to 15% of the body weight at which time these mice were euthanized according to institutional regulations.
Assessment of anti-tumor efficacy against disseminated BCL
Male scid mice were injected intravenously (iv) with 1 × 106 Ramos BCL in a volume of 0.2 ml in the tail vein. Dissemination and growth of BCL was allowed to occur for 9 days prior to the initiation of drug therapy. Mice with disseminated BCL (8–9 mice/treatment group) were administered vehicle (PBS), rituximab (10 mg/kg), rituximab conjugates (AcBut or amide linked, 80 μg CalichDM/kg), or CMC-544 (80 μg CalichDM/kg) for a maximum of three doses administered intraperitoneally (i.p.) on days 9, 13, and 17 (Q4Dx3). Mice with disseminated BCL were monitored daily for the presence of hind-limb paralysis or death. Mice exhibiting hind-limb paralysis were euthanized by CO2 asphyxiation according to institutional regulations. The average survival time (days ± SD) was calculated for each group.
Results
Influence of conjugation on binding of calicheamicin conjugates to BCL
Binding of the anti-CD20 mAb rituximab and its calicheamicin conjugates and that of the anti-CD22 mAb G5/44 and its conjugates to Ramos B lymphoma cells was examined by flow cytometry. As shown in Fig. 1, conjugation to calicheamicin caused a modest decrease in the binding of rituximab. In spite of this, rituximab conjugated to calicheamicin still exhibited strong binding to CD20+ Ramos B cells. In contrast, the binding of calicheamicin conjugates of the anti-CD22 mAb to Ramos B cells was approximately tenfold lower than that of calicheamicin conjugates of rituximab, consistent with the higher expression of CD20 over that of CD22 on Ramos B cells.
Fig. 1.

Binding of calicheamicin-conjugated rituximab to Ramos BCL. Ramos BCL were exposed for 30 min at 4°C to increasing concentrations of chimeric anti-CD20 rituximab, humanized anti-CD22 mAb G5/44 or their calicheamicin conjugates. Binding of the above antibody conjugates to Ramos BCL was detected with FITC-conjugated goat anti-human IgG Fc antibody by flow cytometry
In vitro growth inhibition by conjugates of rituximab
The effect of the anti-CD20 mAb conjugated to CalichDM with the acid-labile AcBut linker or the acid-stable Amide linker on the growth of various CD20+ CD33− cell lines (Ramos, Raji, Daudi, ARH-77), CD20+ CD33+ cell line (RL) or CD20− CD33+ cell line (RPMI-8226) was examined in vitro. As shown in Fig. 2, a CD20-specific conjugate of CalichDMH with an acid-labile AcBut linker exhibited more potent growth inhibition on CD20+ Ramos BCL than unconjugated CalichDMH or the nonbinding CD33-specific acid-labile conjugate, CMA-676. In contrast, the CD20-specific conjugates were weaker than unconjugated CalichDMH in potency against CD20− CD33+ RPMI-8226 myeloma cells. The CD20-specific acid-stable conjugate of CalichDM was less efficacious than unconjugated CalichDMH against either cell line but was more active than CD33-specific nonbinding conjugate, CMA-676, against CD20+ Ramos BCL. Table 1 summarizes results of a similar evaluation of CalichDM conjugates against five additional B cell lines. Of the cell lines tested, Daudi and Raji were the only lines among those evaluated that expressed CD32/FcγRII (MFI = 30.4 and 8, respectively). The results suggest that the CD20-specific, acid-labile conjugate of CalichDMH provides superior growth inhibition against CD20+ BCL compared to that of the CD20-specific, acid-stable conjugate and that of unconjugated CalichDMH. Unconjugated rituximab, when used up to a concentration of 10 μg/ml, did not significantly inhibit the growth of BCL. These results suggest that rituximab conjugated to calicheamicin can be an effective inhibitor of the growth of CD20+ BCL.
Fig. 2.

Effect of calicheamicin conjugated rituximab on the in vitro growth of CD20+ CD33− Ramos BCL or CD20− CD33+ RPMI-8226 myeloma cells. Ramos or RPMI-8226 cells were cultured with increasing concentrations of unconjugated CalichDMH or calicheamicin-conjugated to CD20-specific chimeric rituximab, or CD33-specific humanized hP67.6. Calicheamicin was conjugated to rituximab via either the acid-labile AcBut or acid-stable Amide linker. After a period of 96 h, the viable cell number in each culture was assessed as described in Materials and methods. When used at a concentration as high as 10 μg/ml, Rituximab did not have any effect on the growth of either cell type
Table 1.
Effect of CD20-specific calicheamicin immunoconjugates on the growth of human B cell lines in vitro
| Human B cell line | CD20 expression (mean fluorescence intensity) | IC50 (pM CalichDM equivalents) | Unconjugated CalichDMH | ||
|---|---|---|---|---|---|
| Conjugated CalichDM | |||||
| CD20-targeted AcBut-linked | CD20-targeted Amide-linked | CD33-targeted AcBut-linked | |||
| CD20+ CD33− Ramos | 339 | 270 | 3,700 | 8,300 | 2,200 |
| CD20+ CD33− Daudi | 33 | 130 | 400 | 2,300 | 2,700 |
| CD20+ CD33− Raji | 572 | 2,500 | 3,900 | 5,200 | 7,600 |
| CD20+ CD33+ RL | 473 | 1,200 | 5,400 | 600 | 600 |
| CD20+ CD33 − ARH-77 | 504 | 600 | 4,700 | 1,620 | 1,500 |
| CD20− CD33+ RPMI-8226 | 10 | 1,700 | 12,500 | 200 | 400 |
Rituximab (10 μg/ml) had no significant growth inhibitory effect on any of the above cell lines
ADCC and CDC Activity
The impact of the conjugation of rituximab to calicheamicin on its ability to mediate ADCC and CDC was examined. We have previously demonstrated that rituximab can facilitate both these activities against CD20+ target cells using mononuclear effector cells (MNC) or complement (C′) of both human and murine origin [17]. Results shown in Fig. 3 indicate that rituximab as well as its calicheamicin conjugates exerted ADCC and CDC activities against CD20+ Ramos BCL using human peripheral blood-derived MNCs and C′, respectively. Similar results were obtained using two other CD20+ target cells, Raji and RL BCL (data not shown). Thus, conjugation of rituximab to calicheamicin does not interfere with its ability to mediate either effector function.
Fig. 3.

In vitro ADCC (left panel) and CDC (right panel) of rituximab, rituximab conjugated to CalichDMH with an AcBut linker (Rituximab–AcBut–Calich) or conjugated to CalichDM with the Amide linker (Rituximab–Amide–Calich) against Ramos BCLs. Ramos BCLs were incubated at 37°C with increasing concentrations of antibody or conjugate in the presence or absence of either mononuclear cells (MNC) from human plasma as effector cells (effector to target ratio = 50) or human serum (1:60 dilution) as a source of complement (C′). Four hours later, the cell free supernatants were assayed for the released LDH activity as an indication of cell lysis
In vivo anti-tumor efficacy of immunoconjugates against subcutaneous BCL xenografts
CalichDM conjugates of rituximab were examined for their efficacy against subcutaneous Ramos BCL xenografts established in nude mice. Human CD22-targeted conjugate of CalichDM and CD33-targeted CMA-676 were used as a binding and nonbinding control conjugate, respectively. Each conjugate was dosed at 160 μg of conjugated calichDM/kg Q4Dx3, a dose and a regimen previously shown to be efficacious in xenograft models [15]. The expression of CD20, CD22 and CD33 in Ramos subcutaneous xenografts was determined by flow cytometric analysis of tumor digests that demonstrated that CD20 was expressed four-fold higher than CD22 while CD33 was minimally expressed (MFI for CD20, CD22, and CD33 was 46.7, 11.1, and 3.3, respectively).
Tumors were allowed to grow to an initial mass of 500 mg (moderate size, Fig. 4 left panel) or 2 g (large size, Fig. 4 right panel) before the initiation of therapy. The rituximab–AcBut–Calich. conjugate significantly inhibited (P < 0.05 vs. vehicle-treated mice) tumor growth in each of the studies, regardless of the initial tumor mass. The rituximab–Amide–CalichDM conjugate also significantly inhibited tumor growth though generally much less potently than the AcBut conjugate. The dose of rituximab used in the study (6.8 mg/kg) approximated the amount of rituximab protein present in each dose of the rituximab–CalichDM conjugates (7.0 mg/kg for the AcBut-linked conjugate and 6.6 mg/kg for the amide-linked conjugate). Rituximab produced a modest, though statistically significant (P < 0.05) inhibition in tumor growth relative to the vehicle-treated mice (Fig. 4). Increasing the dose of Rituximab to 20 mg/kg did not increase its anti-tumor activity [17]. The non-binding control conjugate, CMA-676, was far less effective against the BCL xenografts (Fig. 4a). We have previously demonstrated that unconjugated CalichDMH, at doses similar to those used for conjugated CalichDMH, fail to inhibit growth of established BCL xenografts in nude mice [15]. For the non-irradiated mice, the relative tumor suppression for rituximab, rituximab–Amide–CalichDM, and rituximab–AcBut–Calich-treated mice was not appreciable changed (data not reported).
Fig. 4.

Effect of unconjugated rituximab, rituximab conjugated to CalichDMH with an AcBut linker (Rituximab–AcBut–Calich), rituximab conjugated to CalichDM with an Amide linker (Rituximab–Amide–Calich), CMA-676, or anti-CD22 mAb conjugated to Calich DM with the Amide linker (Anti-CD22–Amide–Calich.) on the growth of moderate (left panel) or large (right panel) Ramos BCL xenografts. Each conjugate was administered ip at a dose of 160 μg/kg CalichDM, Q4Dx3. Unconjugated rituximab was administered i.p. at 6.8 mg/kg Q4Dx3. CMA-676 was used as a nonbinding control conjugate. Mean tumor mass (±SEM) is plotted for each treatment group. *P < 0.05 versus vehicle-treated mice
When the BCL xenografts were allowed to grow to a large initial mass of 2 g prior to treatment (Fig. 4b), the rituximab–AcBut–Calich. conjugate was still able to significantly retard tumor growth. The Amide-linked conjugate was not as effective. A CD22-specific Amide conjugate of CalichDM was as active as the rituximab–AcBut–Calich. conjugate against large established BCL xenografts. However, neither conjugate caused complete regression of these large BCL xenografts. In contrast, we have previously shown that CD22-specific AcBut conjugate of CalichDMH (CMC-544) causes almost complete regression of such large BCL xenografts [15]. These results suggest that CD20-targeted conjugates of CalichDMH can be used in the treatment of established BCL.
Anti-tumor efficacy against disseminated BCL
Rituximab and its calicheamicin conjugates were evaluated with CMC-544 in a systemically disseminated BCL tumor model (Fig. 5). BCL, disseminated via systemic circulation, infiltrate the CNS and cause hind limb paralysis and death of the afflicted scid mice [18–20]. Rituximab, administered at 10 mg/kg, produced a modest but significant delay in hind limb paralysis compared with vehicle-treated mice (average survival time of 26.7 ± 2.6 days for vehicle-treated mice vs. 40.2 ± 10.4 days for rituximab-treated mice, P < 0.05). The rituximab–Amide–Calich. conjugate produced similar results as unconjugated rituximab (average survival time of 40.7 days, P < 0.05 vs. vehicle-treated mice) while the rituximab–AcBut–Calich. conjugate (average survival time 50.4 ± 10.0 days, P < 0.05 vs. rituximab) conferred longer protection than rituximab. The same dose of CalichDMH delivered by the CD22-targeted conjugate, CMC-544, produced complete (100%) protection against the disseminated BCL over the course of the study.
Fig. 5.

Effect of rituximab, its calicheamicin conjugates and CMC-544 on survival of SCID mice with disseminated Ramos BCL. SCID mice were injected with Ramos BCL into the tail vein and 9 days later were administered ip with rituximab (10 mg/kg), rituximab conjugated to CalichDMH with an AcBut linker (Rituximab–AcBut–Calich) or conjugated to CalichDM with an Amide linker (Rituximab–Amide–Calich), or CMC-544. Each conjugate was administered at 80 μg/kg CalichDM, Q4Dx3. Mice were monitored up to 70 days for the presence of hind-limb paralysis or death due to the disseminated disease
Discussion
In this study, we explored the potential therapeutic use of a CD20-specific antibody-targeted chemotherapy using rituximab as a calicheamicin-targeting agent. Our in vitro and in vivo results described here demonstrate that rituximab-conjugated to calicheamicin can provide stronger and more durable anti-tumor activity against BCL than rituximab alone. In this study, calicheamicin was conjugated to rituximab via both acid-labile (AcBut) and acid-stable (Amide) linkers. The acid-labile AcBut linker is the same linker used in CD33-targeted gemtuzumab ozogamicin (CMA-676 or Mylotarg) and CD22-targeted inotuzumab ozogamicin (CMC-544). This linker may allow rapid hydrolysis of the hydrazone functionality within the AcBut linker facilitating the release of CalichDMH from its conjugated state under acidic conditions such as those that exists in lysosomal vesicles without extensive intracellular processing of the targeting antibody. In contrast, the Amide linker does not allow the release of CalichDM from its conjugated state under acidic conditions and, thus, calicheamicin immunoconjugates bearing the Amide linker require internalization and intracellular processing of the immunoconjugate in order to liberate the active drug [21, 22]. Both acid-labile and acid-stable calicheamicin conjugates of murine anti-CD22 mAb m5/44 and its humanized counterpart, G5/44, have been described and shown to be efficacious in preclinical models of BCL [23].
Binding of calicheamicin-conjugated rituximab was quantitatively greater than that of CD22-targeted CMC-544, consistent with the higher expression of CD20 over that of CD22 on BCL. Conjugation to calicheamicin via either linker maintained the ability of conjugated rituximab to mediate both ADCC and CDC activities. Thus, the conjugation to calicheamicin does not alter the effector functional capabilities of the targeting antibody. Our in vitro studies with calicheamicin conjugates of rituximab showed that the conjugate with an acid-labile linker was more effective than its acid-stable counterpart and unconjugated CalichDMH in inhibiting growth of CD20+ BCL. Daudi BCL was the only exception where the acid-stable conjugate was more effective than unconjugated CalichDMH.
Antibodies bound to CD20 on the surface of normal or malignant B cells are usually not known to internalize [24, 25]. However, Law et al. [26] demonstrated that even though unconjugated rituximab did not internalize in BCL, its conjugation to a derivative of a microtubule-disrupting drug, auristatin, allowed the conjugate to be internalized resulting in a strong inhibition of BCL growth. Irrespective of their stage of maturation, malignant B cells express CD32/FcγRII in varying quantities and as shown by Vervoordeldonk et al. [27] this Fcγ receptor was shown to facilitate anti-CD19 mAb-mediated modulation of CD19 on malignant B cells. In addition, CD32 was shown to internalize upon interaction with aggregated human IgG [28]. Since Daudi cells express CD32 (this report and [28]), it is conceivable that the human IgG1Fc portion of CD20-bound rituximab engages CD32 on the cell membrane of the same or apposing BCL facilitating its internalization. Such an event may explain the enhanced growth-inhibitory effect of the acid-stable immunoconjugate of rituximab over that of unconjugated calicheamicin against CD20+ CD32+ BCL like Daudi. It would be interesting to establish a correlation between the degree of expression of CD32 by BCL as a biomarker and their subsequent responsiveness to antibody therapeutics like rituximab [29, 30].
Rituximab conjugated to CalichDMH via the acid-labile AcBut linker caused a strong inhibition of growth of small established subcutaneous BCL xenografts. In contrast, rituximab conjugated to CalichDM via the acid-stable Amide linker was far less effective in inhibiting subcutaneous BCL growth than its acid-labile counterpart. The anti-tumor activity of acid-stable conjugate of rituximab was similar to that of unconjugated rituximab. This functional difference in the anti-tumor efficacy of acid-labile and acid-stable calicheamicin conjugates of rituximab was also apparent in their activity against large BCL xenografts. A similar acid-stable conjugate targeted to CD22, an internalizable molecule, was able to exhibit efficacy similar to that of acid-labile calicheamicin conjugate of rituximab. The lower efficacy of acid-stable conjugate of rituximab may indicate its poor internalization in Ramos BCL. Due to the lack of internalization of antibody-bound CD20 [24, 25], the Amide-linked conjugate of rituximab may not deliver calicheamicin in the intracellular compartments to bring about its cytotoxic activity. However, its surface binding and retention may still allow interaction with FcR-expressing effector cells and complement and thus is able to derive anti-tumor activity similar to that of unconjugated rituximab.
The superior efficacy of the acid-labile conjugate of rituximab can also be explained in part on the prolonged retention (due to the lack of its internalization) of the bound conjugate on the surface of BCL. Most metabolically active cycling tumor cells acidify their extracellular environment due to their increased rate of glycolysis [31]. Acidification of the pericellular microenvironment of metabolically active BCL may facilitate hydroysis of the hydrazone bond in the linkage of conjugated CalichDMH to BCL bound rituximab conjugate freeing CalichDMH in the extracellular proximity [32] that may then attack the same BCL. Such a focused pericellular release of CalichDMH may explain the superior anti-BCL efficacy of rituximab conjugate with the acid-labile linkage. Such a release is not possible with an acid-stable conjugate unless the conjugate-antigen complex is internalized and processed intracellularly as observed with a CD22-targeted acid-stable conjugate. Thus the use of an acid-labile linker such as the AcBut linker allows anti-tumor efficacy to be achieved even with noninternalizable targeted antigens such as CD20.
Studies from a number of investigators have established that the binding of rituximab to CD20 on BCL causes its accumulation into detergent-insoluble cell membrane functional domains, enriched in glycosphingolipids and cholesterol, known as lipid rafts [33, 34]. Molecules that move into lipid rafts are poorly internalized resulting in their sustained display on the cell surface [33–35]. A number of key signaling receptors partition into lipid rafts which presumably allows them to resist internalization, and thereby, continue to transduce signals that effect cellular growth and other functions [33]. In fact, the very ability of rituximab to induce translocation of CD20 into lipid rafts on the surface of B cells is considered crucial to its ability to fix complement and bring about complement-mediated lysis [36–38]. Prolonged surface retention of rituximab-bound CD20 in lipid rafts may also facilitate its interaction with FcR-bearing effector cells resulting in the effective mediation of ADCC. Consistent with this notion is the behavior of the acid-stable conjugate of rituximab that provided anti-BCL activity similar to that of unconjugated rituximab.
In contrast with CD20, antibody binding to CD22 results in its rapid internalization making CD22 an ideal candidate for efficient intracellular delivery of cytotoxic agents. CMC-544 is a CD22-targeted immunoconjugate of calicheamicin with an acid-labile linker that has been shown to cause regression of both developing and established small and large human BCL xenografts and to protect mice with disseminated BCL [15, 18]. The present study demonstrates that CD22-targeted calicheamicin and CD20-targeted calicheamicin with the acid-labile linker cause similar growth inhibition of established small and large BCL xenografts. However, CD22-targeted CMC-544 provided superior protection against disseminated BCL. The expression of CD20 on BCL is often greater than that of CD22. Hence, the pericellular accumulation of conjugated calicheamicin is likely to be higher with the CD20-targeted conjugate than that with CD22-targeted conjugate. However, superior internalization of CD22 may compensate for its quantitatively lower surface expression. Ultimately, it is the quantitative intracellular delivery of the cytotoxic agent such as calicheamicin that will always be a critical determinant of the overall anti-tumor activity of its immunoconjugates. This study demonstrates that the acid-labile, CD20-specific immunoconjugate of calicheamicin may cause meaningful anti-tumor activity against established BCL and further provides the necessary preclinical basis in support of clinical evaluation of the CD20-targeted calicheamicin therapeutic strategy in B-NHL.
Acknowledgments
We thank Fred Immermann of Wyeth Biometrics Research for statistical analysis of the data.
Abbreviations
- BCL
B-cell lymphoma
- CalichDM
N-Acetyl gamma calicheamicin dimethyl derivative(s)
- CalichDMH
CalichDM hydrazide
Footnotes
All authors are employed by Wyeth Research.
References
- 1.Damle NK. Tumor-targeted chemotherapy with immunoconjugates of calicheamicin. Expert Opin Biol Ther. 2004;4:1445–1452. doi: 10.1517/14712598.4.9.1445. [DOI] [PubMed] [Google Scholar]
- 2.Bross PF, Beitz J, Chen G, et al. Gemtuzumab ozogamicin: approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. 2001;7:1490–1496. [PubMed] [Google Scholar]
- 3.Sievers EL, Appelbaum FR, Spielberger RT, et al. Selective ablation of acute myeloid leukemia using antibody-targeted chemotherapy: a phase I study of an anti-CD33 calicheamicin immunoconjugate. Blood. 1999;93:3678–3684. [PubMed] [Google Scholar]
- 4.Sievers E, Larson R, Stadmauer E, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol. 2001;19:3244–3254. doi: 10.1200/JCO.2001.19.13.3244. [DOI] [PubMed] [Google Scholar]
- 5.Hamann PR, Hinman LM, Hollander I, et al. A potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconj Chem. 2002;13:47–58. doi: 10.1021/bc010021y. [DOI] [PubMed] [Google Scholar]
- 6.Lee M, Dunne T, Chang C, et al. Calicheamicins, a novel family of antibiotics. 4. Structural elucidations of calicheamicins. J Am Chem Soc. 1992;114:985–987. doi: 10.1021/ja00029a030. [DOI] [Google Scholar]
- 7.Zein N, Sinha A, McGahren W, Ellestad G. Calicheamicin γI: an antitumor antibiotic that cleaves double-stranded DNA site specifically. Science. 1988;240:1198–1201. doi: 10.1126/science.3240341. [DOI] [PubMed] [Google Scholar]
- 8.Grillo-Lopez A. Rituximab (Rituxan/MabThera): the first decade (1993–2003) Expert Rev Anticancer Ther. 2003;3:767–779. doi: 10.1586/14737140.3.6.767. [DOI] [PubMed] [Google Scholar]
- 9.Ghobrial I, Witzig T. Radioimmunotherapy: a new treatment modality for B-cell non-Hodgkin’s lymphoma. Oncology. 2004;18:623–630. [PubMed] [Google Scholar]
- 10.Uchida J, Hamaguchi Y, Oliver JA, et al. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J Exp Med. 2004;199:1659–1669. doi: 10.1084/jem.20040119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaetano ND, Cittera E, Nota R, et al. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol. 2003;171:1581–1587. doi: 10.4049/jimmunol.171.3.1581. [DOI] [PubMed] [Google Scholar]
- 12.Manches O, Lui G, Chaperot L, et al. In vitro mechanisms of action of rituximab on primary non-Hodgkin lymphomas. Blood. 2003;101:949–954. doi: 10.1182/blood-2002-02-0469. [DOI] [PubMed] [Google Scholar]
- 13.Hainsworth JD, Litchy S, Burris HA, et al. Rituximab as first-line and maintenance therapy for patients with indolent non-Hodgkin’s lymphoma. J Clin Oncology. 2002;20:4261–4267. doi: 10.1200/JCO.2002.08.674. [DOI] [PubMed] [Google Scholar]
- 14.Edwards JC, Szczepanski L, Szechinski J, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 15.DiJoseph JF, Armellino DC, Boghaert E, et al. Antibody-targeted chemotherapy with CMC-544: a CD22-targeted immunoconjugate of calicheamicin for the treatment of B lymphoid malignancies. Blood. 2004;103:1807–1814. doi: 10.1182/blood-2003-07-2466. [DOI] [PubMed] [Google Scholar]
- 16.Advani A, Giné E, Gisselbrecht C et al (2005) Preliminary report of a phase 1 study of cmc-544, an antibody-targeted chemotherapy agent, in patients with b-cell non-Hodgkin’s lymphoma (NHL). Blood 106(11): abstract No. 230
- 17.DiJoseph JF, Dougher MM, Kalyandrug LB, et al. Antitumor efficacy of a combination of CMC-544 (inotuzumab ozogamicin), a CD22-targeted cytotoxic immunoconjugate of calicheamicin, and rituximab against non-Hodgkin’s B-cell lymphoma. Clin Cancer Res. 2006;12:242–249. doi: 10.1158/1078-0432.CCR-05-1905. [DOI] [PubMed] [Google Scholar]
- 18.DiJoseph JF, Goad ME, Dougher MM, et al. Potent and specific anti-tumor efficacy of CMC-544, a CD22-targeted immunoconjugate of calicheamicin, against systemically disseminated B-cell lymphoma. Clin Cancer Res. 2004;10:8620–8629. doi: 10.1158/1078-0432.CCR-04-1134. [DOI] [PubMed] [Google Scholar]
- 19.Flavell DJ, Noss A, Pulford KAF, Ling N, Flavell SU. Systemic therapy with 3BIT, a triple combination cocktail of anti-CD19, -CD22, and -CD38-saporin immunotoxins, is curative of human B-cell lymphoma in severe combined immunodeficient mice. Cancer Res. 1997;57:4824–4829. [PubMed] [Google Scholar]
- 20.Flavell DJ, Boehm DA, Emery L, Noss A, Ramsay A, Flavell SU. Therapy of human B-cell lymphoma bearing SCID mice is more effective with anti-CD19- and anti-CD38-saporin immunotoxins used alone in combination than with either immunotoxin alone. Int J Cancer. 1995;62:337–344. doi: 10.1002/ijc.2910620318. [DOI] [PubMed] [Google Scholar]
- 21.Hinman LM, Hamann PR, Wallace R, Menendez AT, Dur FE, Upeslacis J. Preparation and characterization of monoclonal antibody conjugates of the calicheamicins: a novel and potent family of antitumor antibiotics. Cancer Res. 2003;53:3336–3342. [PubMed] [Google Scholar]
- 22.Hamann P, Hinman L, Beyer C, et al. An anti-CD33 antibody–calicheamicin conjugate for treatment of acute myeloid leukemia. Choice of linker. Bioconj. Chem. 2002;13:40–46. doi: 10.1021/bc0100206. [DOI] [PubMed] [Google Scholar]
- 23.DiJoseph JF, Popplewell A, Tickle S, et al. Antibody-targeted chemotherapy of B-cell lymphoma using calicheamicin conjugated to murine or humanized antibody against CD22. Cancer Immunol Immunother. 2005;54:11–24. doi: 10.1007/s00262-004-0572-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Press OW, Farr AG, Borroz KI, Anderson SK, Martin PJ. Endocytosis and degradation of monoclonal antibodies targeting human B-cell malignancies. Cancer Res. 1989;49:4906–4912. [PubMed] [Google Scholar]
- 25.Vangeepuram N, Ong GL, Mattes MJ. Processing of antibodies bound to B-cell lymphomas and lymphoblastoid cell lines. Cancer. 1997;80(Suppl):2425–2430. doi: 10.1002/(SICI)1097-0142(19971215)80:12+<2425::AID-CNCR14>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 26.Law CL, Cerveny CG, Gordon KA, et al. Efficient elimination of B-lineage lymphomas by anti-CD20–Auristatin conjugates. Clin Cancer Res. 2004;10:7842–7851. doi: 10.1158/1078-0432.CCR-04-1028. [DOI] [PubMed] [Google Scholar]
- 27.Vervoordeldonk SF, Merle PA, van Leeuwen EF, van der Schoot CE, von dem Borne AE, Slaper-Cortenbach IC. Fc gamma receptor II (CD32) on malignant B cells influences modulation induced by anti-CD19 monoclonal antibody. Blood. 1994;83:1632–1639. [PubMed] [Google Scholar]
- 28.Van Den Herik-Oudijk IE, Westerdaal NA, Henriquez NV, Capel PJ, Van De Winkel JG. Functional analysis of human Fc gamma RII (CD32) isoforms expressed in B lymphocytes. J Immunol. 1994;152:574–585. [PubMed] [Google Scholar]
- 29.Flieger D, Renoth S, Beier I, Sauerbruch T, Schmidt-Wolf I. Mechanism of cytotoxicity induced by chimeric mouse human monoclonal antibody IDEC-C2B8 in CD20-expressing lymphoma cell lines. Cell Immunol. 2000;205:55–63. doi: 10.1006/cimm.2000.1693. [DOI] [PubMed] [Google Scholar]
- 30.Miettinen HM, Matter K, Hunzinker W, et al. Fc receptor endocytosis is controlled by a cytoplasmic domain determinant that actively prevents coated pit localization. J Cell Biol. 1992;116:875–888. doi: 10.1083/jcb.116.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Costello LC, Franklin RB. ‘Why do tumour cells glycolyse?’: from glycolysis through citrate to lipogenesis. Mol Cell Biochem. 2005;280:1–8. doi: 10.1007/s11010-005-8841-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boghaert ER, Khanke K, Sridharan L, et al. Tumoricidal effect of calicheamicin immuno-conjugates using a passive targeting strategy. Int J Oncol. 2006;28:675–684. [PubMed] [Google Scholar]
- 33.Harder T, Engelhardt (2004) Membrane domains in lymphocytes-from lipid rafts to protein scaffolds. Traffic 5:265–275 [DOI] [PubMed]
- 34.Polyak MJ, Tailor SH, Deans JP. Identification of a cytoplasmic region of CD20 required for its redistribution to a detergent-insoluble membrane compartment. J Immunol. 1998;161:3242–3248. [PubMed] [Google Scholar]
- 35.Fujimoto M, Kuwano Y, Wananabe R, et al. B cell antigen receptor and CD40 differentially regulate CD22 tyrosine phosphorylation. J Immunol. 2006;176:873–879. doi: 10.4049/jimmunol.176.2.873. [DOI] [PubMed] [Google Scholar]
- 36.Gaetano ND, Cittera E, Nota R, et al. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol. 2003;171:1581–1587. doi: 10.4049/jimmunol.171.3.1581. [DOI] [PubMed] [Google Scholar]
- 37.Cragg MS, Glennie MJ. Antibody specificity controls effector mechanisms of anti-CD20 reagents. Blood. 2004;103:2738–2743. doi: 10.1182/blood-2003-06-2031. [DOI] [PubMed] [Google Scholar]
- 38.Cragg MS, Morgan SM, Claude Chan HT, et al. Complement-mediated lysis by anti-CD20 mAb correlated with segregation into lipid rafts. Blood. 2003;101:1045–1052. doi: 10.1182/blood-2002-06-1761. [DOI] [PubMed] [Google Scholar]