

Abstract
The aim of this study is to identify candidate factors which may be responsible for the functional inactivation and depletion of NK cells by tumor cells. Inhibition of NFκB activity by an IκB super-repressor in HEp2 cells, a cell line commonly used as an oral tumor model, blocked tumor-induced NK cell death, and increased the function of NK cells significantly. Increased expression of CD69 early activation antigen on NK cells as well as augmented proliferation and secretion of IFN-γ by NK cells were observed when these cells were co-incubated with IκB super-repressor transfected HEp2 cells (HEp2-IκB(S32AS36A)). More importantly, the secretion of IL-6 was significantly inhibited when NK cells were co-cultured with HEp2-IκB(S32AS36A) cells. In addition, the survival and function of cytotoxic effector cells remained significantly elevated in the presence of IFN-γ-treated HEp2-IκB(S32AS36A) cells when compared to either untreated or IFN-γ-treated vector-alone transfected HEp2 cells. Similar findings to those obtained using purified peripheral blood NK cells were also observed when non-fractionated peripheral blood mononuclear cells were used in the co-cultures of immune effectors with HEp2 cell transfectants. Addition of recombinant human IL-6 to the co-cultures of immune effectors with the NFκB knockdown HEp2 tumor cells substantially decreased the levels of secreted IFN-γ. Thus, the results presented in this paper suggest that the inhibition of NFκB function in oral tumors may serve to activate and expand the function and numbers of NK cells. Moreover, NFκB-mediated increase in IL-6 secretion by oral tumors may in part be responsible for the observed inactivation and death of the immune effectors.
Keywords: Apoptosis, NFκB, TNF-α, IFN-γ, NK, IL-6, Natural killer, NK
Introduction
Depressed natural killer (NK) and cytotoxic T lymphocyte (CTL) proliferation and function are evident in the early stages of the squamous cell carcinomas of the oral cavity [1–3]. Functional deficiency of cytotoxic cells have also been reported in a variety of other cancers notably breast [4–7], renal [8], and colon [9]. It has been hypothesized that the absence and/or paralysis of cytotoxic cells residing within the inflammatory infiltrate of advanced cancer patients contributes to poor prognosis [10]. Furthermore, freshly isolated tumor-infiltrating lymphocytes are not cytotoxic to autologous tumor cells and show significantly reduced clonogenicity [11–14].
The reason for the functional paralysis of lymphocytes in patients with clinically detectable tumors is still a matter of controversy. It has been suggested that the lack of tumor recognition by lymphocytes and/or the immunosuppressive nature of various tumors obtained from tumor-bearing individuals represent some of the reasons for the failure of the generation of effective immunity in cancer patients. We have previously demonstrated that co-incubation of NK cells with a variety of tumor cell lines mediated significant inactivation and cell death of NK cells [15].
It appears that a net balance between activating and inhibitory signals received from tumor cells dictates whether cytotoxic NK or T cells will become functionally activated or inhibited in the presence of tumor target cells. The list for both the activating and inhibitory signals delivered by different tumor cell ligands to NK cells has been increasing steadily [16]. Thus, the identification and characterization of factors that regulate and aid in altering the balance toward functional activation of cytotoxic lymphocytes are necessary for designing strategies to eliminate multi-resistant tumor cells. In this paper we present NFκB as one such factor.
An important role for NFκB/Rel proteins has been attributed to the malignant transformation and increased hyperplasia previously [17]. Factors contributing to tumor metastasis in lung cancer are clearly shown to be regulated by NFκB [18]. Constitutive activation of NFκB in several cancers including lung [19], breast [20], pancreatic [21], gastric [22], melanoma [23], and head and neck cancers [24] is shown to be an important contributor to the progression and metastasis of these cancers. NFκB is also known to protect tumor cells from undergoing cell death [25]. In most cell types, NFκB is present as a heterodimer consisting of 50 kDa (p50) and 65 kDa (p65) subunits. NFκB exists in a complex with IκB in resting cells [26, 27]. Phosphorylation of IκB proteins on specific serine residues, such as serines 32 and 36 of IκB-α and serines 19 and 23 of IκB-β, by I kappa kinases (IκKs), and their subsequent ubiquitination results in the degradation of IκB and translocation of NFκB to the nucleus where it regulates various genes carrying the NFκB response elements.
It is not fully clear how NFκB nuclear function in oral tumors modulates and shapes the function of interacting immune effectors. We have shown previously that inhibition of NFκB function by the IκB super-repressor in the HEp2 oral laryngeal cell line leads to significant activation of human NK cell cytotoxic function and increase in IFN-γ secretion. Similarly, inhibition of NFκB by Sulindac, a non-steroidal anti-inflammatory drug (NSAID), was able to increase the functional activation of NK and dendritic cells (DCs) [28]. However, many important questions still remain unanswered with regard to the role of NFκB nuclear function in tumor-induced NK cell death and inactivation. Since our objective is to enhance the function of cytotoxic immune effectors in oral tumors, dissection of the underlying mechanisms of immune activation and expansion when NFκB function is inhibited in HEp2 cells may help design strategies to block the well-recognized inactivation and death of the cytotoxic effectors in patients with aggressive oral tumors. Furthermore, targeted inhibition of NFκB function in both the intestinal epithelial cells and the myeloid cells was recently shown to result in a significant decrease in the size and the numbers of tumor cells [29]. Thus, establishing the underlying mechanisms of NFκB function in tumors in recruitment and activation of tumor enhancing versus inhibitory factors elaborated by different immune subsets in the inflammatory infiltrate is important for designing strategies to prevent tumor progression.
The induction of many important chemo- and cytokine genes is positively regulated by the function of nuclear NFκB in immune cells. However, accumulating evidence indicates that this factor may exhibit an opposite effect on cytokine secretion by skin epithelial cells [30, 31]. Therefore, the functional consequences of NFκB activation may differ from cell to cell and from tissue to tissue. In order to understand how the decreased expression of nuclear NFκB in HEp2 cells can further contribute to the functional activation of tumor-interacting NK cells, we asked the following questions; (1) does the decrease in NFκB nuclear function in HEp2 tumor cells increase survival and expansion of tumor-interacting NK cells? (2) does the decrease in NFκB nuclear function in HEp2 tumor cells mediate an increase in activating cytokines while decreasing the secretion of inhibitory cytokines in the co-cultures of NK-HEp2 cells? and finally, (3) how does the treatment of NFκB-depleted HEp2 cells with IFN-γ modulate the function and survival of NK cells since IFN-γ treatment of tumor cells has previously been shown to result in their resistance to cytotoxic immune effector function [32].
We demonstrate in the present study that the interaction of immune effectors with NFκB depleted HEp2 cells results in the elevation of a number of important immune-modulating cytokines. We found that the inhibition of NFκB in HEp2 tumor cells was capable of providing signals for the survival, proliferation, and expansion of NK cells. Furthermore, inverse modulation of IFN-γ and IL-6 cytokine secretion was clearly seen in the cultures of NK cells with IκB(S32AS36A) transfected HEp2 cells indicating that blocking NFκB in HEp2 tumor cells serves to switch the balance from an immune inhibitory type response to an immune-activating type response. Thus, targeting the NFκB-signaling module in tumors is likely to provide the added benefit of blocking tumor progression at multiple stages of cancer development. Targeted therapies may prevent the selection and progression of aggressive oral tumors by partly blocking tumor cell survival and proliferation as well as by restoring robust anti-tumor immune responses.
Material and methods
Cell lines, plasmids, and reagents
HEp2 tumor cell lines were obtained from ATCC and maintained in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% FCS. RPMI 1640 supplemented with 10% FCS was used for the cultures of NK and T cells. Recombinant IL-2 was obtained from Hoffman La Roche, New Jersey. IFN-γ was a generous gift from Dr. Yoichi Mizutani. TNF-α was purchased from Peprotech. The NK cell purification kit was obtained from Miltenyi Biotech (Auburn, CA, USA). Polyclonal anti-IFN-γ antibodies were prepared in rabbits in our laboratory. The anti-IFN-γ and anti-GMCSF monoclonal antibodies were purchased from R&D. Control IgG1 was derived from a control hybridoma (ATCC) and purified IgG1 and PE-conjugated CD69 mAbs were purchased from Coulter/Immunotech (Miami, FL, USA). pRcCMV empty vector and pRcCMV-IκB (S32AS36A) constructs were generous gifts from Dr. G. Cheng and they were described previously [33].
Transfections and the generation of HEp2 cell transfectants
The generation of HEp2 cell transfectants was described previously [28]. The stability of IκB(S32AS36A) super-repressor transfected HEp2 cells were regularly checked by western blot analysis and EMSA using nuclear extracts prepared from the HEp2 cell transfectants. For further description of HEp2 tumor line please refer to Jewett et al. [28].
Purification of NK cells
PBMCs from healthy donors were isolated as described before [15]. Briefly, peripheral blood lymphocytes were obtained after Ficoll-hypaque centrifugation and purified NK cells were negatively selected by using an NK cell isolation kit (Miltenyi Biotech). The purity of NK cell population was found to be greater than 90% based on flow cytometric analysis of anti-CD16 antibody-stained cells. The levels of contaminating CD3+ T cells remained low, at 2.4±1%, throughout the experimental procedures.
3H thymidine incorporation assay
After incubation of NK cells with tumor cell transfectants for the number of days indicated in the result section, 3H thymidine at 1 μCi/well was added to each well, and the incubation was continued for another 16–18 h. The NK cell samples were then harvested by a PhD harvester and the levels of incorporated 3H thymidine were determined by counting the amount of radioactivity in the samples by a liquid scintillation counter (LSC). Tumor cell transfectants were irradiated with 10–20 Gy for 1 h using γ irradiator prior to their co-culture with the cytotoxic effector cells.
Surface staining
The cells were washed twice with ice-cold PBS containing 1% BSA and 0.01% sodium azide. PE- and FITC-conjugated antibodies against CD16, CD69, CD3, CD8, CD4, and their corresponding isotype control antibodies were each added separately or in combination with 2×105 to 5×105 cells in 50 μl of cold PBS-BSA followed by incubation on ice for 30 min. The cells were then washed twice and fixed in 1% paraformaldehyde solution. Isotype control antibody staining was used to set the cursor for measuring specific staining. The Epics Elite (Coulter Electronics, FL, USA) flow cytometer was used for the analysis.
NK cell cytotoxicity assay
The 51Cr release assay was performed as described previously [15, 34]. Briefly, different numbers of purified NK cells were incubated with 51Cr-labeled tumor target cells. After a 4 h incubation period the supernatants were harvested from each sample and counted for released radioactivity using the gamma counter. The percentage-specific cytotoxicity was calculated as follows;
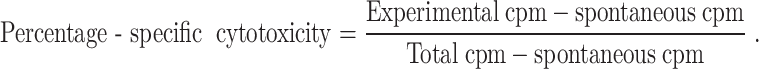 |
LU 30/107 is calculated by using the inverse of the number of effector cells needed to lyse 30% of tumor target cells × 100.
Multiplex cyto- and chemokine protein arrays
The fluorokine MAP cytokine multiplex array kits for measuring a number of Th1 and Th2 type cytokines (please see Results section) were purchased from R&D Systems and the procedures were conducted as suggested by the manufacturer. To analyze and obtain the cyto- and chemokine concentrations, a standard curve was generated by a threefold dilution of recombinant cytokines provided by the manufacturer. Analysis was performed using the software provided by Luminex.
Results
Stable expression of IκB(S32AS36A) super-repressor in HEp2 cells was previously shown to inhibit NFκB DNA-binding activity in HEp2 cells, and significant induction of NK cell cytotoxicity against these cells [28]. Both untreated and IL-2-treated NK cells mediated greater lysis of IκB(S32AS36A) transfected HEp2 cells when compared to vector-alone transfected HEp2 cells [28].
Increased expression of CD69 early activation antigen on NK cells after their interaction with NFκB knockdown HEp2 cells
Increased CD69 surface expression on the NK cells is a measure of NK cell functional activation. Thus, untreated and IL-2-treated purified NK cells were co-incubated with untreated and IFN-γ-treated HEp2-IκB(S32AS36A) cells at an effector to target (E:T) ratio of 1:1 and the levels of CD69 activation antigen were determined after an overnight incubation at 37°C on NK cells (Table 1). It is important to note that in every experiment which IL-2-treated immune cells were used; IL-2 was present in the entire period of co-culture of immune cells with the tumor cells. Inhibition of NFκB activity in HEp2 cells elevated the levels of CD69 activation antigen on NK cells after their interaction with HEp2-IκB(S32AS36A) cells in the presence and absence of IL-2 treatment (Table 1). CD69 expression on NK cells was also increased when co-incubated in the presence of vector-alone transfected HEp2 cells. However, this increase was much lower than that observed on NK cells co-incubated with HEp2-IκB(S32AS36A) cells (Table 1). Treatment of vector-alone transfected HEp2 cells with IFN-γ prior to their co-culture with the NK cells decreased the IL-2-mediated upregulation of CD69 expression on NK cells. In contrast, treatment of IκB(S32AS36A) transfected HEp2 cells with IFN-γ was unable to decrease CD69 expression on NK cells (Table 1). Therefore, these results further corroborated the findings that increased lysis of NFκB knockdown HEp2 tumor cells by the NK cells is not due to the heightened sensitivity of tumor cells to the apoptotic factors elaborated by the NK cells in the co-cultures of NK and tumor cells.
Table 1.
Increased induction of CD69 expression on NK cells by HEp2-IκB(S32AS36A) cells
| NK Treatment | % CD69 expressing cells | ||||
|---|---|---|---|---|---|
| No tumors | Hep2-vec | HEp2-IκB(S32AS36A) cells | |||
| IL-2 (−/+) | −IFN-γ | +IFN-γ | −IFN-γ | +IFN-γ | |
| − | 0.7±0.0 | 17.8±3.9 | 14±2 | 96±1 | 95.3±1.53 |
| + | 16.3±1 | 42.5±4.6 | 19.3±1.15 | 95.7±0.6 | 95.7±0.6 |
NK cells at a concentration of 1×106/ml were treated with and without IL-2 (500 units/ml) overnight before their co-culture with untreated and IFN-γ (200 units/ml) treated vector-alone and IκB(S32AS36A) transfected HEp2 cells (E:T ratio 1:1). After an overnight incubation the expression of CD69 early activation antigen were determined on NK cells using surface staining with PE conjugated anti-CD69 antibody. IL-2 was present at all times in the co-cultures of NK cells with the tumor cells. Tumor cells were left untreated or treated with IFN-γ for 12–18 h prior to their co-culture with the NK cells. IFN-γ-treated HEp2 cell transfectants were washed three times before they were added to the NK cells. The P values for the difference between the expression of CD69 on untreated or IL-2-treated NK cells co-cultured in the presence of vector-alone versus IκB(S32AS36A) transfected HEp2 cells were at less than 0.05 for all the samples tested. One of 4 representative experiments is shown in this table
Increased survival and function of NK cells co-cultured with NFκB knockdown HEp2 cells
Untreated and IL-2-treated NK cells were co-cultured in the presence of vector-alone and IκB(S32AS36A) transfected HEp2 cells and the levels of NK cell expansion were determined both by counting the numbers of the live NK cells and by measuring the incorporation of 3H thymidine in the proliferating NK cells. As demonstrated in Table 2, the numbers of IL-2-treated NK cells were significantly decreased in the presence of vector-alone transfected HEp2 cells when compared to the IL-2-treated NK cells which were cultured in the absence of tumor cells. Similarly, the numbers of NK cells were decreased in untreated NK cells co-cultured in the presence of vector-alone transfected HEp2 cells when compared to those cultured in the absence of tumor cells (Table 2). However, the overall numbers of untreated NK cells were also decreased in the absence of tumor cells when compared to the input numbers of NK cells at the initiation of the cultures. In contrast, the numbers of NK cells were increased when untreated and IL-2-treated NK cells were co-cultured in the presence of HEp2-IκB(S32AS36A) cells when compared to either vector-alone transfected HEp2 cells or NK cells in the absence of tumor cells (Table 2). Likewise, the levels of 3H thymidine incorporation in IL-2-treated NK cells in the presence of vector-alone transfected HEp2 cells was significantly decreased when compared to either HEp2-IκB(S32AS36A) cells or IL-2-treated NK cells in the absence of tumor cells (Tables 2, 3).
Table 2.
Survival of NK cells co-cultured in the presence of HEp2-IκB(S32AS36A) cells
| NK Treatment | No tumors | HEp2-vec | Hep2-IκB(S32AS36A) cells | ||
|---|---|---|---|---|---|
| IL-2 (−/+) | −IFN-γ | +IFN-γ | −IFN-γ | +IFN-γ | |
| − | 4.1×105±0.2×105 | 2.1×105±0.6×105 | 5.5×104±0.7×104 | 1.27×106±1×105 | 7.6×105±0.5×105 |
| + | 1.21×106±0.1×105 | 2.15×105±0.2×105 | 1.4×105±0.7×105 | 3.66×106±9.3×105 | 3.24×106±1.8×105 |
NK cells (1×106/ml in 1 or 2 ml of culture medium) were treated with and without IL-2 (500 units/ml) overnight before their co-culture with untreated and IFN-γ (200 units/ml) treated vector-alone and IκB(S32AS36A) transfected HEp2 cells (E:T ratio 1:1). After 5 days of incubation the concentration of viable NK cells per milliliter of culture media as indicated in the table was determined in each sample by microscopic evaluation obtained by two independent observers using trypan blue exclusion assay. HEp2 cell transfectants were treated with IFN-γ overnight after which they were washed three times before they were added to the NK cells. The P values for the difference between the co-cultures of untreated and IL-2-treated NK cells with vector-alone transfected HEp2 cells versus IκB(S32AS36A) transfected HEp2 cells were significant at less than 0.05. The P value between untreated and IL-2-treated NK cells which did not receive any tumors and those that were co-cultured in the presence of vector-alone transfected HEp2 cells were at less than 0.05. One of four representative experiments is shown in this table
Table 3.
Inhibition of NK cell Thymidine incorporation after interaction of NK cells with vector-alone transfected HEp2 cells but not with HEp2-IκB(S32AS36A) cells
| Donor | NK Treatment | No tumors | HEp2-vec | HEp2-IκB(S32AS36A) cells | ||
|---|---|---|---|---|---|---|
| IL-2 (−/+) | −IFN-γ | +IFN-γ | −IFN-γ | +IFN-γ | ||
| #1 | − | 4053±41 | 7243±164 | 4227.5±916 | 6226±100 | 6444±254 |
| + | 30494±1000 | 18557±933 | 6444±127 | 52353±4998 | 47116±5528 | |
| #2 | − | 3185±441 | 6311±534 | 5168±712 | 7754±873 | 2690±723 |
| + | 39615±509 | 27186±913 | 10756±366 | 58684±2131 | 52775±1460 | |
| #3 | − | 2007±558 | 3848±426 | 2923±385 | 3551±1372 | 2009±697 |
| + | 43836±3613 | 39075±2368 | 34396±3536 | 74834±5699 | 74575±8491 | |
NK cells (1×106/ml in 1 or 2 ml of culture medium) were treated with and without IL-2 (500 units/ml) overnight before their co-culture with untreated and IFN-γ (200 units/ml) treated vector-alone and IκB(S32AS36A) transfected HEp2 cells (E:T ratio 1:1). 100 μl of the NK tumor cell co-cultures were transferred immediately to the 96 well plates in triplicates and the plates were incubated for 5 days. Tumor cells were irradiated for 1 h before they were used in the co-cultures of NK and HEp2 cells. After 5 days of incubation 3H thymidine at 1 μCi/well was added to each well, and the incubation was continued for another 16–18 h. The levels of 3H thymidine incorporation were determined as described in Materials and methods section. The P values for the difference between the co-cultures of NK cells with vector-alone transfected HEp2 cells versus IκB(S32AS36A) transfected HEp2 cells were significant at less than 0.05 for IL-2-treated samples. The P value between IL-2-treated NK cells which did not receive any tumors and those that were co-cultured in the presence of vector-alone transfected HEp2 cells were at less than 0.05. Three of several representative experiments are shown in this table
IFN-γ treatment of tumor cells has previously been shown to decrease cytotoxic activity of immune effector cells [32]. Thus, we were interested to know what effect if any IFN-γ treatment may have on the ability of HEp2 tumor cell transfectants in mediating NK cell inactivation and depletion. We found that the loss of IL-2-treated NK cell numbers and proliferation was more dramatic when NK cells were co-cultured in the presence of IFN-γ-treated vector-alone transfected HEp2 cells (Table 3). In contrast, no significant decreases in the numbers and proliferation of NK cells could be observed when IL-2-treated NK cells were co-cultured in the presence of IFN-γ-treated HEp2-IκB(S32AS36A) cells (Table 3). Since NK cells do not proliferate in the absence of IL-2 treatment, no clear differences could be seen between the samples when NK cells were left untreated.
Since vector-alone transfected HEp2 cells were capable of decreasing the proliferation as well as the numbers of viable IL-2-treated NK cells, we were interested to know whether this correlated with a decreased IL-2-treated NK cell cytotoxic function. In agreement with the above-mentioned experiments when IL-2-treated NK cells were co-cultured with cold vector-alone transfected HEp2 cells prior to incubation with 51Cr-labeled HEp2 tumor target cells in a cytotoxicity assay, they were unable to lyse either 51Cr-labeled vector-alone transfected HEp2 cells or 51Cr-labeled HEp2-IκB(S32AS36A) cells when compared to IL-2-treated NK cells either cultured in the absence of tumor cells or in the presence of HEp2-IκB(S32AS36A) cells (Table 4). In contrast, IL-2-treated NK cells cultured in the presence of HEp2-IκB(S32AS36A) cells when added to either 51Cr-labeled vector-alone transfected HEp2 cells or 51Cr-labeled HEp2-IκB(S32AS36A) cells were able to mediate lysis of both lines, and indeed their levels of cytotoxicity against HEp2 cell transfectants exceeded those obtained by the IL-2-treated NK cells cultured in the absence of tumor cells (Table 4). Similar findings were obtained when NK cell activity was assessed against K562 cells (data not shown). Collectively, these results indicated that the loss of function of NK cells in long-term cultures (5 days) is likely due to decrease in the numbers of viable NK cells in the co-culture of NK cells with vector-alone transfected HEp2 cells. Therefore, we chose to limit the time of co-cultures to 4–18 h for functional assessment since minimal loss of NK cell viability could be observed at those time points.
Table 4.
Inhibition of HEp2 tumor cell cytotoxicity against 51Cr-labeled HEp2 tumor cells transfectants after exposure of NK cells to cold HEp2-vec but not cold HEp2-IκB(S32AS36A) cells
| HEp2-vec (51Cr labeled) | Cytotoxicity (LU30/107 cells) | |
|---|---|---|
| −IL-2 | +IL-2 | |
| NK cells pre-incubated with | ||
| No targets | 5.3±0.14 | 13.1±0.0 |
| HEp2-vec (cold) | 4.1±0.56 | 7.6±0.98 |
| HEp2-IκB(S32AS36A) (cold) | 7.7±0.0 | 22.78±1.1 |
| HEp2-IκB(S32AS36A) (51Cr labeled) | ||
| NK cells pre-incubated with | ||
| No targets | 14.3±0.0 | 22.35±0.07 |
| Hep2-vec (cold) | 4.9±0.0 | 7.99±0.0 |
| HEP2-IκB(S32AS36A)(cold) | 8.2±0.0 | 44.5±9.2 |
NK cells were left untreated or treated with IL-2 (500 units/ml) and cultured in the absence (no targets) and presence of each of cold vector-alone and IκB(S32AS36A) transfected HEp2 cells (E:T ratio 1:1). After an overnight treatment NK-tumor cell co-cultures were washed twice and added to 51Cr-labeled HEp2-vec and HEp2-IκB(S32AS36A) cells (E:T ratio 1:1). After 4–6 h of incubation at 37°C, supernatants were harvested and the levels of released 51Cr radioactivity were determined by a γ counter. LU 30/107 is calculated by using the inverse of the number of effector cells needed to lyse 30% of tumor target cells × 100. The P values for the difference between the co-cultures of NK cells with vector-alone transfected HEp2 cells and with IκB(S32AS36A) transfected HEp2 cells are less than 0.05 for all the samples tested. One of 6 representative experiments is shown in this table
Decreased IL-6 secretion and increased secretion of IFN-γ in co-cultures of NK cells and NFκB knockdown HEp2 tumor cells
In order to determine how NFκB depletion in tumor cells affects the type of cytokines produced in the tumor cell/immune cell microenvironment, we cultured untreated and IL-2-treated NK cells and PBMCs in the presence of HEp2 cell transfectants. After an overnight incubation, the levels of a panel of Th1 and Th2 cytokines were determined in the co-cultures of immune cells with HEp2 cell transfectants (Table 5). We determined the levels of secreted cytokines in the supernatants obtained from both the PBMCs and NK cells in order to assess whether the profiles of secreted cytokines or the magnitude of secreted cytokines remained similar or differed between PBMCs and NK cells after their interaction with vector-alone and IκB(S32AS36A) transfected HEp2 cells. HEp2-IκB(S32AS36A) cells alone in the absence of PBMCs released significantly lower amounts of IL-6 when compared to vector-alone transfected HEp2 cells (Table 5). More importantly, supernatants from the co-cultures of HEp2-IκB(S32AS36A) cells and PBMCs/NK cells contained significantly lower levels of IL-6 when compared to those obtained from the co-cultures of vector-alone transfected HEp2 cells with the immune effectors (Tables 5, 6). In contrast, HEp2-IκB(S32AS36A) cells triggered significantly higher levels of IFN-γ secretion from PBMCs and NK cells when compared to vector-alone transfected HEp2 cells (Tables 5, 6). When the levels of Th1 and Th2 type cytokines were considered on a multiplex cytokine array system, we observed an inverse relationship between the secreted IL-6 and those of IFN-γ, GM-CSF, and TNF-α in both PBMCs and NK cells (Tables 5, 6). The levels of secreted IL-4, IL-10, and IL-12 were, in general, too low to be able to ascribe any differences between IκB(S32AS36A) and vector-alone transfected HEp2 cells. Even though the ratios of IL-6 to IFN-γ differ from donor to donor and the type of immune cells, in general the data obtained indicated that significantly lower ratios of IL-6 to IFN-γ secretion is obtained when immune cells were activated by HEp2-IκB(S32AS36A) cells as compared to vector-alone transfected HEp2 cells (Tables 5, 6). Indeed, for the most part, there was a direct relationship between increased immune cell cytotoxicity and cell survival and decreased ratios of IL-6 to IFN-γ in the co-cultures of PBMCs and NK cells with HEp2 cell transfectants (Tables 5, 6). Therefore, the ratios of IL-6 to IFN-γ may be better indicators of immune cell survival and function than each cytokine alone after interaction with oral tumors (see below).
Table 5.
Decreased IL6 and increased IFN-γ secretion in supernatants obtained from the co-cultures of PBMCs and HEp2-IκB(S32AS36A) cells
| No tumors | Concentration (pg/ml) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| GMCSF | IFN-γ | IL-10 | IL-12 | IL-4 | IL-6 | IL-8 | TNF-α | Ratio of IL-6/IFN-γ | |
| +PBMC (−IL-2) | 123.3 | 24.01 | 8.48 | <3.2 | 1.26 | 205.35 | >10,000 | 82.84 | 8.55 |
| +PBMC (+IL-2) | 112.85 | 343.03 | 5.02 | <3.2 | 0.42 | 321.69 | – | 24.45 | 0.94 |
| Hep2-pRcCMV | |||||||||
| −PBMC | 0.36 | 0.53 | 0.18 | <3.2 | <3.2 | 365.79 | 17.55 | 2.89 | – |
| +PBMC (−IL-2) | 139.98 | 20.85 | 14.73 | <3.2 | <3.2 | >10,000 | 5,957.08 | 47.14 | 479.6 |
| +PBMC (+IL-2) | 599.98 | 3,098.13 | 9.47 | <3.2 | <3.2 | >10,000 | 8,040.46 | 151.92 | 3.23 |
| HEp2-IκB(S32AS36A) | |||||||||
| −PBMC | 0.1 | 0.14 | 0.07 | <3.2 | 0.7 | 13.72 | 6.38 | 3.05 | – |
| +PBMC (−IL-2) | 49.96 | 273.96 | – | <3.2 | <3.2 | 625.96 | >10,000 | 287.12 | 2.28 |
| +PBMC (+IL-2) | 1,058.82 | >10,000 | 10.75 | <3.2 | 5.11 | 611.41 | – | 385.08 | 0.06 |
PBMCs (3×106 cells/ml) obtained from a healthy donor were treated in the presence and absence of IL-2 (500 units/ml) overnight before their co-culture with vector-alone and IκB(S32AS36A) transfected HEp2 cells (E:T ratio 10:1). Tumor cell transfectants were each cultured alone or in combination with PBMCs as indicated in the table and the supernatants were removed from the cultures after an overnight incubation. The levels of Th1 and Th2 type cytokine secretion were determined using antibody-coated multiplex microbead immunoassay. For simplification of the tables standard deviations are not presented and they ranged from 0 to 10% of the amount obtained for each cytokine. The P values for the difference in the secretion of IL-6, IFN-γ, TNF-α, and GM-CSF between untreated or IL-2-treated PBMCs co-cultured in the presence of vector-alone versus IκB(S32AS36A) transfected HEp2 cells were at less than 0.05. One of six representative experiments is shown in this table
Table 6.
NK cells co-cultured in the presence of IFN-γ-treated HEp2-IκB(S32AS36A) cells retained their ability to secrete increased levels of Th1 type cytokines
| No tumors | Concentration (pg/ml) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| GMCSF | IFN-γ | IL-10 | IL-12 | IL-4 | IL-6 | IL-8 | TNF-α | Ratio of IL-6/IFN-γ | |
| +NK (−IL-2) | 0.21 | 2.22 | 0.59 | 0.31 | 2.62 | 9.49 | 5.37 | 0.97 | 4.3 |
| +NK (+IL-2) | 7.21 | 71.91 | 1.8 | 0.45 | 3.8 | 24.41 | 8.44 | 14.77 | 0.34 |
| HEp2-pRcCMV | |||||||||
| +NK (−IL-2) | 1.05 | 3.25 | 0.74 | 0.42 | 4.65 | 97.99 | 9.48 | 1.16 | 30.15 |
| +NK (+IL-2) | 413.26 | 1967.7 | 2.07 | 0.72 | 6.22 | 922.73 | 37.72 | 49.54 | 0.47 |
| HEp2-IκB(S32AS36A) | |||||||||
| +NK (−IL-2) | 7.68 | 8.46 | 0.82 | 0.38 | 5.17 | 46.88 | 10.74 | 1.93 | 5.54 |
| +NK (+IL-2) | 1276.8 | 5519.7 | 3.04 | 0.85 | 7.38 | 465.9 | 57.79 | 180.01 | 0.08 |
| HEp2-pRcCMV (+IFN-γ) | |||||||||
| +NK (−IL-2) | 0.68 | 2.7 | 0.74 | 0.37 | 6 | 128.12 | 9 | 1.1 | 47.45 |
| +NK (+IL-2) | 201.57 | 666.5 | 2.11 | 0.63 | 7 | 1069.1 | 31.44 | 36.3 | 1.6 |
| HEp2-IκB(S32AS36A) (+IFN-γ) | |||||||||
| +NK (−IL-2) | 6.31 | 28.6 | 0.89 | 0.42 | 6.31 | 57.24 | 11.3 | 2.3 | 2.0 |
| +NK (+IL-2) | 1084 | 5872.3 | 3.44 | 0.97 | 8.19 | 556.91 | 47.71 | 158.4 | 0.095 |
NK cells (1×106/ml) were treated with and without IL-2 (500 units/ml) overnight before their co-culture with untreated and IFN-γ (200 units/ml) treated vector-alone and IκB(S32AS36A) transfected HEp2 cells (E:T ratio 1:1). After an overnight incubation the levels of Th1 and Th2 type cytokine release were determined using antibody-coated multiplex microbead immunoassay. For simplification of the table standard deviations are not presented and they ranged from 0 to 10% of the amount obtained for each cytokine. The P values for the decrease in secretion of IFN-γ and GM-CSF between the untreated and IFN-γ-treated NK cells co-cultured in the presence of vector-alone transfected HEp2 cells were at less than 0.05. One of four representative experiments is shown in this table
Treatment of vector-alone but not NFκB knockdown HEp2 tumor cells with IFN-γ decreased the levels of IFN-γ but not IL-6 secretion in the co-cultures of NK and HEp2 cells
We have previously shown that the treatment of vector-alone transfected HEp2 cells with IFN-γ results in a decrease in both the cytotoxicity and IFN-γ secretion by the NK cells [28]. Thus, we wanted to determine whether treatment of HEp2 cells with IFN-γ will also negatively affect the ability of NK cells to secrete both activating and inhibitory cytokines. We therefore treated the tumor cell transfectants with IFN-γ overnight and washed them extensively to remove excess IFN-γ before they were co-cultured with purified NK cells in an overnight assay. Supernatants were then removed from the co-cultures of NK-HEp2 cells and assayed for the levels of Th1 and Th2 type cytokines. Addition of IFN-γ to vector-alone transfected HEp2 cells decreased IFN-γ, GM-CSF, and TNF-α secretion in the co-cultures of NK and vector-alone transfected HEp2 cells, whereas it had no effect on IL-6 secretion (Table 6). In contrast, treatment of HEp2-IκB(S32AS36A) cells with IFN-γ did not decrease the levels of IFN-γ secretion, and levels of GM-CSF and TNF-α were decreased less in the co-cultures of NK and HEp2-IκB(S32AS36A) cells when compared to those obtained from the co-cultures of NK cells with vector-alone transfected HEp2 cells. The co-cultures of NK and HEp2-IκB(S32AS36A) cells contained lower levels of IL-6 when compared to those obtained from the co-cultures of NK and vector-alone transfected HEp2 cells, and IFN-γ treatment of HEp2-IκB(S32AS36A) cells did not modify this effect (Table 6). However, the magnitude of cytokines secreted (with the exception of IL-6) in the co-cultures of NK cells and IFN-γ-treated HEp2-IκB(S32AS36A) cells remained substantially higher than those obtained in the co-cultures of NK cells with untreated and IFN-γ-treated vector-alone transfected HEp2 cells. The levels of IL-4, IL-10, and IL-12 were too low to attribute any significant differences. Thus, IκB(S32AS36A) transfected HEp2 cells still retained their ability to trigger more of the Th1 type cytokines by the NK cells when treated with IFN-γ.
Inhibition of IFN-γ secretion by the addition of IL-6 to the co-cultures of immune cells with NFκB knockdown HEp2 tumor cells
Untreated PBMCs were co-cultured in the presence and the absence of vector-alone and IκB(S32AS36A) transfected HEp2 cells at an effector to target ratio of 10:1. Exogenous recombinant IL-6 was then added to each culture condition indicated in the Fig. 1 and the levels of secreted IFN-γ were determined after an overnight incubation. Addition of IL-6 to the co-cultures of PBMCs and IκB(S32AS36A) transfected HEp2 cells inhibited greater than 50% of secreted IFN-γ in the supernatants (Fig. 1). In contrast, as shown above, the addition of vector-alone transfected HEp2 cells to PBMCs was unable to trigger appreciable levels of IFN-γ secretion. Likewise, no induction of IFN-γ secretion could be observed either by PBMCs or by HEp2 tumor cells alone. The results indicated that IL-6 is a potent inhibitor of IFN-γ secretion in the co-cultures of PBMCs and HEp2-IκB(S32AS36A) tumor cells.
Fig. 1.

Untreated PBMCs at a concentration of 3×106/ml were co-cultured in the presence and the absence of vector-alone and IκB(S32AS36A) transfected HEp2 cells at an effector to target ratio of 10:1. Exogenous recombinant IL-6 at a concentration of 10 ng/ml was then added to each culture condition indicated in the figure and the levels of secreted IFN-γ were determined after an overnight incubation
Discussion
Many aggressive and metastatic tumor cells exhibit constitutively elevated NFκB activity [35]. Increased NFκB activity is shown to have a causal relationship to neoplastic transformation and uncontrolled cell growth in many cell types [35]. Human leukemias and lymphomas, as well as human solid tumors exhibit constitutively activated nuclear NFκB [35]. However, although previous studies have attributed a significant role for NFκB in survival, oncogenesis, and tumor progression, no studies have been conducted thus far to demonstrate the significance of elevated NFκB function in tumor cells in the modulation of cytotoxic immune effector function. We have previously shown that inhibition of NFκB in HEp2 cells resulted in an activation of NK cell cytotoxic function and in an increase in the levels of IFN-γ secretion by the NK cells [28]. Furthermore, inhibition of NFκB nuclear activity in primary oral tumors also resulted in an increase in NK cell cytotoxic function as well as in the elevation of IFN-γ secretion in supernatants removed from the co-cultures of NK cells with the primary oral tumors (manuscript in preparation). In this paper we demonstrate that the increase in the function of NK cells is paralleled by an increase in CD69 expression, survival, and expansion of NK cells, and in an inverse modulation of IFN-γ and IL-6 secretion when NK cells come in contact with NFκB-depleted HEp2 tumor cells. In addition, HEp2 tumor cells depleted of NFκB nuclear function demonstrate significant ability to secrete chemokines (manuscript in preparation). Finally, the addition of IL-6 to the co-cultures of lymphocytes with NFκB knockdown HEp2 tumor cells resulted in the inhibition of IFN-γ secretion substantially. Collectively, the results obtained from these studies suggested that blocking NFκB activity in tumors of oral origin may recruit, activate, and expand the cytotoxic immune effectors in the tumor microenvironment.
The role of the NFκB family of transcription factors as tumor promoters has been firmly established previously [34–38]. However, other studies suggest a tumor suppressor role for NFκB function [36–40]. It appears that the duality of NFκB function as either a tumor promoter or a tumor suppressor largely depends on cell type, stage of maturation, and the nature and extent of mutations sustained by the cells prior to the modulation of NFκB function. Thus, at the earlier stages of tumorigenesis NFκB in certain cell types may behave as a tumor promoter, whereas, at the later stages when cells have accumulated one or more mutations at critical sites, it may act as a tumor suppressor. However, in either case, changes in NFκB function in tumor cells, as shown in this paper, may profoundly affect the function and survival of cytotoxic immune effectors. Indeed, decrease in cellular NFκB function may well be one of the important “danger signals” required for the homing and expansion of cytotoxic immune effectors at the site of pathology in order to eliminate transformed tumor cells.
There are conflicting results with regard to the role of NFκB in skin disease. Inhibition of NFκB function in dermal keratinocytes resulted in an increased proliferation and hyperplasia [36] and eventual development of cutaneous squamous cell carcinomas of skin if mice were allowed to survive and reach adulthood [38, 41]. However, deletion of IKKβ was shown in a separate study to significantly block NFκB function in epidermis without the induction of impaired differentiation and hyper-proliferation of keratinocytes [31]. Indeed, keratinoctyes lacking IKKβ exhibited decreased rather than an increased proliferation in this model system when compared to control animals. Furthermore, these animals developed a TNF-α-dependent inflammatory skin disease [31]. Blocking of NFκB in HEp2 cells neither change nor decreased the rate of tumor cell growth when compared to vector-alone transfected HEp2 cells (unpublished observation). In addition, even though NFκB knockdown HEp2 cells were capable of forming tumors in nude mice, the size of the tumors were smaller than those obtained by the injection of vector-alone transfected HEp2 cells (manuscript in preparation).
To the best of our knowledge the study reported in this paper is the first to characterize the interaction and crosstalk between NFκB knockdown laryngeal tumor cells with the cytotoxic immune effectors obtained from human donors. Furthermore, previous studies using animal model system did not address the specific contribution of either NFκB knockdown cells or the interacting immune effector cells or both in the activation of immune cell function, whereas we were able to measure the effect of each of the cell subpopulations separately and in combination in our studies. It is clear that NFκB is a broadly acting transcription factor, capable of regulating many critical processes within the cells. However, by identifying key cell surface or secreted molecules and/or factors involved in expansion and activation of cytotoxic immune effectors when NFκB is inhibited in HEp2 tumor cells, we may be able to design strategies to prevent the well-documented inactivation and death of cytotoxic immune effectors by tumors.
Inhibition of NFκB activity in HEp-2 cells resulted in a significant enhancement of NK cell cytotoxicity [28]. The increased lysis of IκB super-repressor transfected HEp-2 cells by NK cells was not due to an increased sensitivity of these cells to NK cell elaborated apoptotic factors since supernatants obtained from the co-cultures of NK cells and HEp2-IκB(S32AS36A) cells were unable to lyse HEp2-IκB(S32AS36A) cells in 4–6 h (data not shown). More importantly, the increased NK cell cytotoxicity observed against IκB(S32AS36A) transfected HEp-2 cells was blocked by antibodies directed against CD54 adhesion molecule [28]. Thus, induction of increased lysis of HEp2-IκB(S32AS36A) cells by NK cells is due to the increased activation of NK cells and subsequent killing of the IκB(S32AS36A) transfected HEp-2 cells by direct cell–cell contact in a 4 h 51Cr release assay.
We have previously shown that NK cells lost cytotoxic activity and underwent cell death when co-cultured in the presence of a number of tumor target cells [15]. NK cells which bound and killed their respective tumors (killer cells) were inactivated to a lesser extent than the NK cells which bound but did not kill their tumor target cells (binder cells) [15]. Likewise, NK cells co-cultured with IκB(S32AS36A) transfected HEp2 cells were not inactivated and their numbers increased substantially over time, whereas the numbers of NK cells decreased after their interaction with vector-alone transfected HEp2 cells. Therefore, blocking of NFκB function in HEp2 cells was found to decrease substantially the ability of HEp2 cells to induce cell death and inactivation of NK cells.
Decreased binding of inhibitory MHC class I antigens to their respective receptors on NK cells might be one potential mechanism for the survival and increased function of NK cells in the co-cultures of NK and HEp2-IκB(S32AS36A) cells. Indeed, decreased expression of MHC class I antigens were observed on the surface of untreated and IFN-γ-treated HEp2-IκB(S32AS36A) cells when compared to vector-alone transfected HEp2 cells [28]. Since the functions of inhibitory MHC Class I antigens are dominant, slight down-modulation of inhibitory MHC Class I antigen expression might relieve the protection of targets from the NK cell-mediated cytolysis and result in significant activation of the NK cell function. Therefore, an overall increase in the activation of NK cells should ensue under the conditions when target cells express lower expression of MHC class I antigen on the surface.
When NK cells were exposed to cold IκB(S32AS36A) transfected HEp2 cells before they were used in the cytotoxicity assay, results indicated that they were capable of mediating increased lysis of hot vector-alone transfected HEp2 cells. However, if the NK cells were first exposed to cold vector-alone transfected HEp2 cells, they were unable to lyse hot vector-alone transfected HEp2 cells in a second round of killing. This observation is significant since these results indicated that modification of NFκB function in certain tumors can be exploited as a potential source for the increase in the function of killer cells. Such strategy could be effective for the expansion and generation of lymphocytes which would later target NFκB intact tumor cells. We are in the process of establishing whether HLA-A28 peptide-specific lysis of tumor cells by CD8+ T cells can also be augmented in the presence of IκB(S32AS36A) transfected HEp2 cells. Indeed, increased survival and function of CD8+ T cells were also observed when they were co-cultured in the presence of HEp2-IκB(S32AS36A) cells but not vector-alone transfected HEp2 cells (manuscript submitted). Furthermore, our preliminary experiments indicated that dendritic cells (DC) loaded with IκB(S32AS36A) but not vector-alone transfected HEp2 cells were capable of activating peripheral blood lymphocytes (PBL) to secrete significantly higher levels of TNF-α, IFN-γ, and IL-12 when compared to those obtained either by the DCs alone or by PBLs alone [28]. Indeed, comparing the function of PBMCs with the purified NK cells after their exposure to HEp2 cell transfectants suggests that results obtained using PBMCs are likely due to more than just NK cell activation. Therefore, generation of an effective in vitro tumor vaccine by targeted inhibition of NFκB in tumor cells may be one potential strategy to augment the overall anti-tumor immunity against tumors.
Inhibition of NFκB in HEp2 cells increased secretion of IFN-γ, TNF-α, and GM-CSF by PBMCs and purified NK cells, whereas secretion of IL-6 was significantly inhibited. PBMCs/NK cells and vector-alone transfected HEp2 cells each secreted moderate levels of IL-6; however, co-incubation of these cells together increased substantially secreted levels of IL-6. Therefore, as suggested by the findings, it is possible that cross signaling and activation of effector cells and the tumor cells in the co-cultures of effectors and targets may establish an amplification loop, resulting in synergistic augmentation of secreted cytokines elaborated by both the effector cells and the target cells. These possibilities are under investigation at present in our laboratory.
Several fold higher release of IL-6 was seen in untreated and IL-2-treated PBMCs co-cultured in the presence of vector-alone transfected HEp2 cells when compared to PBMCs cultured with HEp2-IκB(S32AS36A) cells (Tables 5, 6). In contrast, the reverse was observed for the secretion of IFN-γ in the co-cultures of PBMCs/NK cells and HEp2-IκB(S32AS36A) cells. Much higher induction of IFN-γ was observed in the co-cultures of immune effectors and HEp2-IκB(S32AS36A) cells than in vector-alone transfected HEp2 cells. It is also important to note that the ratios of IL-6 to IFN-γ was significantly lower for the IL-2-treated PBMCs and NK cells when compared to untreated cells co-cultured with the HEp2 cell transfectants. Indeed, when comparing the ratios of IL-6 with IFN-γ, the values obtained for untreated PBMC or NK cell co-cultures with HEp2-IκB(S32AS36A) cells were close to those obtained by the IL-2-treated PBMCs and NK cells co-cultured with vector-alone transfected HEp2 cells (Tables 5, 6). Thus, it appears that NFκB deletion may serve to switch cytokine profiles from an immune inhibitory type profile to more of an immune-enhancing type profile.
IL-6 is secreted constitutively by oral squamous cell carcinomas [42] and it is found to be elevated in oral cancer patients [43]. IL-6 is known to interfere with IFN-γ signaling by the induction of Th2 differentiation via activation of NFAT and secretion of IL-4, which subsequently inhibits Th1 polarization by the induction of suppressor of cytokine signaling (SOCS)-1 expression in CD4+ T cells [44]. Furthermore, the induction of Stat3 activation by IL-6 is shown to be the mediator of SOCS-1 activation [44]. Since blocking of Stat3 function in tumor cells is known to inhibit tumor-mediated inactivation of adaptive immunity [45], it is likely that IL-6 induced Stat3 in the co-cultures of NK cells and vector-alone transfected HEp2 cells may in part be responsible for the induction of NK cell inactivation and cell death. Indeed, the addition of IL-6 to the co-cultures of immune cells with the NFκB knockdown tumor cells inhibited secreted levels of IFN-γ in the supernatants. Furthermore, the addition of antibody to either IL-6 or to IL-6 receptors increased the levels of IFN-γ secretion in the supernatants obtained from the co-cultures of immune cells with vector-alone transfected HEp2 cells (manuscript in preparation). Therefore, it appears that NFκB in HEp2 tumor cells serves as the master molecular switch between IL-6 and IFN-γ secretion in the co-cultures of NK cells and vector-alone transfected HEp2 cells.
Finally, IFN-γ treatment significantly augmented the secretion of MCP-1 and Rantes in NFκB knockdown HEp2 tumor cells and subsequently increased the overall secretion of both chemokines in the co-cultures of HEp2-IκB(S32AS36A) cells and PBMCs or NK cells (manuscript in preparation). Both MCP-1 and Rantes were shown to play significant roles in the recruitment of NK cells to the site of infection and malignancy [46, 47]. Thus, based on the findings obtained in this study, it is tempting to speculate that NFκB knockdown tumor cells may be able to recruit greater numbers of immune cells to the site of the tumors due to an increased production of key chemokines (manuscript submitted). Next, because of a decrease in inhibitory signals provided, for instance, by inhibitory MHC Class I antigens and IL-6, NK cells may be able to remain viable and in turn increase their functional activation at the site of pathology. Enhancement in survival, in addition to increased signals for proliferation, may then provide the means for the expansion of the number of NK cells at the tumor site.
The NSAIDs are known to inhibit NFκB activity and are clinically shown to have significant anti-carcinogenic effects [48–54]. Therefore, the protective effect of NSAIDs against a variety of cancers could relate to their ability to block NFκB and increase the cytotoxic function of immune effectors [28]. Indeed the treatment of HEp2 cells in the presence of Sulindac down-regulated nuclear NFκB expression, decreased IL-6 secretion and increased the survival and activation of immune effector cells in the co-cultures of Sulindac-treated HEp2 cells and NK cells (manuscript in preparation). Thus, the local use of NSAIDs as chemopreventive agents in oral cancer patients is well warranted not only for their inhibitory effect on tumor survival, but also for their potential promoting effect on the survival and expansion of cytotoxic immune effectors. Furthermore, specific targeting of IL-6 cytokine and NFκB transcription factor by genetic or pharmacological means should be beneficial for the treatment of oral tumors.
In conclusion, we have presented evidence for the function of NFκB in HEp2 tumors in inactivation and depletion of NK cell function. Strategies to inhibit NFκB activation in oral tumors may result in the increased recruitment of the NK cells to the site of malignancy, and in increased survival and expansion as well as in functional activation of the NK cells which will eventually lead to an increased susceptibility of these tumors to NK cell-mediated cytotoxicity.
Footnotes
This work was supported by RO1-DE12880 from NIDCR-NIH.
References
- 1.Castiglione F, Destefanis M, Marenco D, Gosso P, Manera G, Porcile G. The mammographic screening project in 2 local health units of Piemonte. Epidemiol Prev. 1996;20:157. [PubMed] [Google Scholar]
- 2.Veltri RW, Rodman SM, Maxim PE, Baseler MW, Sprinkle PM. Immune complexes, serum proteins, cell-mediated immunity, and immune regulation in patients with squamous cell carcinoma of the head and neck. Cancer. 1986;57:2295. doi: 10.1002/1097-0142(19860615)57:12<2295::AID-CNCR2820571211>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 3.Schantz SP, Shillitoe EJ, Brown B, Campbell B. Natural killer cell activity and head and neck cancer: a clinical assessment. J Natl Cancer Inst. 1986;77:869. [PubMed] [Google Scholar]
- 4.Marrogi AJ, Munshi A, Merogi AJ, Ohadike Y, El-Habashi A, Marrogi OL, Freeman SM. Study of tumor infiltrating lymphocytes and transforming growth factor-beta as prognostic factors in breast carcinoma. Int J Cancer. 1997;74:492. doi: 10.1002/(SICI)1097-0215(19971021)74:5<492::AID-IJC3>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 5.Camp BJ, Dyhrman ST, Memoli VA, Mott LA, Barth RJ., Jr In situ cytokine production by breast cancer tumor-infiltrating lymphocytes. Ann Surg Oncol. 1996;3:176. doi: 10.1007/BF02305798. [DOI] [PubMed] [Google Scholar]
- 6.Hartveit F. Breast cancer: poor short-term prognosis in cases with moderate lymphocyte infiltration at the tumour edge: a preliminary report. Oncol Rep. 1998;5:423. [PubMed] [Google Scholar]
- 7.Gimmi CD, Morrison BW, Mainprice BA, Gribben JG, Boussiotis VA, Freeman GJ, Park SY, Watanabe M, Gong J, Hayes DF, Kufe DW, Nadler LM. Breast cancer-associated antigen, DF3/MUC1, induces apoptosis of activated human T cells. Nat Med. 1996;2:1367. doi: 10.1038/nm1296-1367. [DOI] [PubMed] [Google Scholar]
- 8.Kolenko V, Wang Q, Riedy MC, O’Shea J, Ritz J, Cathcart MK, Rayman P, Tubbs R, Edinger M, Novick A, Bukowski R, Finke J. Tumor-induced suppression of T lymphocyte proliferation coincides with inhibition of Jak3 expression and IL-2 receptor signaling: role of soluble products from human renal cell carcinomas. J Immunol. 1997;159:3057. [PubMed] [Google Scholar]
- 9.Mulder WM, Bloemena E, Stukart MJ, Kummer JA, Wagstaff J, Scheper RJ. T cell receptor-zeta and granzyme B expression in mononuclear cell infiltrates in normal colon mucosa and colon carcinoma. Gut. 1997;40:113. doi: 10.1136/gut.40.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayry P, Totterman. TH. Cytological and functional analysis of inflammatory infiltrates in human malignant tumors I Composition of the inflammatory infiltrates. Eur J Immunol. 1978;8:866. doi: 10.1002/eji.1830081208. [DOI] [PubMed] [Google Scholar]
- 11.Miescher S, Stoeck M, Qiao L, Barras C, Barrelet L, von Fliedner V. Preferential clonogenic deficit of CD8-positive T-lymphocytes infiltrating human solid tumors. Cancer Res. 1988;48:6992. [PubMed] [Google Scholar]
- 12.Qin J, Han B, Pang J. The relationship between TIL from human primary hepatic carcinoma and prognosis. Zhonghua Yi Xue Za Zhi. 1997;77:167. [PubMed] [Google Scholar]
- 13.Han X, Papadopoulos AJ, Ruparelia V, Devaja O, Raju KS. Tumor lymphocytes in patients with advanced ovarian cancer: changes during in vitro culture and implications for immunotherapy. Gynecol Oncol. 1997;65:391. doi: 10.1006/gyno.1997.4668. [DOI] [PubMed] [Google Scholar]
- 14.Itoh K, Tilden AB, Balch CM. Interleukin 2 activation of cytotoxic T-lymphocytes infiltrating into human metastatic melanomas. Cancer Res. 1986;46:3011. [PubMed] [Google Scholar]
- 15.Jewett A, Bonavida B. Target-induced inactivation and cell death by apoptosis in a subset of human NK cells. J Immunol. 1996;156:907. [PubMed] [Google Scholar]
- 16.Lanier LL. Turning on natural killer cells. J Exp Med. 2000;191:1259. doi: 10.1084/jem.191.8.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell. 1996;87:13. doi: 10.1016/S0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 18.Andela VB, Schwarz EM, Puzas JE, O’Keefe RJ, Rosier RN. Tumor metastasis and the reciprocal regulation of prometastatic and antimetastatic factors by nuclear factor kappaB. Cancer Res. 2000;60:6557. [PubMed] [Google Scholar]
- 19.Mukhopadhyay T, Roth JA, Maxwell SA. Altered expression of the p50 subunit of the NF-kappa B transcription factor complex in non-small cell lung carcinoma. Oncogene. 1995;11:999. [PubMed] [Google Scholar]
- 20.Nakshatri H, Bhat-Nakshatri P, Martin DA, Goulet RJ, Jr, Sledge GW., Jr Constitutive activation of NF-kappaB during progression of breast cancer to hormone-independent growth. Mol Cell Biol. 1997;17:3629. doi: 10.1128/mcb.17.7.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang W, Abbruzzese JL, Evans DB, Larry L, Cleary KR, Chiao PJ. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin Cancer Res. 1999;5:119. [PubMed] [Google Scholar]
- 22.Sasaki N, Morisaki T, Hashizume K, Yao T, Tsuneyoshi M, Noshiro H, Nakamura K, Yamanaka T, Uchiyama A, Tanaka M, Katano M. Nuclear factor-kappaB p65 (RelA) transcription factor is constitutively activated in human gastric carcinoma tissue. Clin Cancer Res. 2001;7:4136. [PubMed] [Google Scholar]
- 23.McNulty SE, Tohidian NB, Meyskens FL., Jr RelA, p50 and inhibitor of kappa B alpha are elevated in human metastatic melanoma cells and respond aberrantly to ultraviolet light B. Pigment Cell Res. 2001;14:456. doi: 10.1034/j.1600-0749.2001.140606.x. [DOI] [PubMed] [Google Scholar]
- 24.Nakayama H, Ikebe T, Beppu M, Shirasuna K. High expression levels of nuclear factor kappaB, IkappaB kinase alpha and Akt kinase in squamous cell carcinoma of the oral cavity. Cancer. 2001;92:3037. doi: 10.1002/1097-0142(20011215)92:12<3037::AID-CNCR10171>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 25.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 26.Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 27.Thanos D, Maniatis T. NF-kappa B: a lesson in family values. Cell. 1995;80:529. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- 28.Jewett A, Wang MY, Teruel A, Poupak Z, Bostanian Z, Park NH. Cytokine dependent inverse regulation of CD54 (ICAM1) and major histocompatibility complex class I antigens by nuclear factor kappaB in HEp2 tumor cell line: effect on the function of natural killer cells. Hum Immunol. 2003;64:505. doi: 10.1016/S0198-8859(03)00039-9. [DOI] [PubMed] [Google Scholar]
- 29.Greten FR, Karin M. The IKK/NF-kappaB activation pathway-a target for prevention and treatment of cancer. Cancer Lett. 2004;206:193. doi: 10.1016/j.canlet.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 30.Santos M, Perez P, Segrelles C, Ruiz S, Jorcano JL, Paramio JM. Impaired NF-kappa B activation and increased production of tumor necrosis factor alpha in transgenic mice expressing keratin K10 in the basal layer of the epidermis. J Biol Chem. 2003;278:13422. doi: 10.1074/jbc.M208170200. [DOI] [PubMed] [Google Scholar]
- 31.Pasparakis M, Courtois G, Hafner M, Schmidt-Supprian M, Nenci A, Toksoy A, Krampert M, Goebeler M, Gillitzer R, Israel A, Krieg T, Rajewsky K, Haase I. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature. 2002;417:861. doi: 10.1038/nature00820. [DOI] [PubMed] [Google Scholar]
- 32.Lukacher AE. IFN-gamma suspends the killing license of anti-tumor CTLs. J Clin Invest. 2002;110:1407. doi: 10.1172/JCI200217209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DiDonato J, Mercurio F, Rosette C, Wu-Li J, Suyang H, Ghosh S, Karin M. Mapping of the inducible IkappaB phosphorylation sites that signal its ubiquitination and degradation. Mol Cell Biol. 1996;16:1295. doi: 10.1128/mcb.16.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jewett A, Cavalcanti M, Bonavida B. Pivotal role of endogenous TNF-alpha in the induction of functional inactivation and apoptosis in NK cells. J Immunol. 1997;159:4815. [PubMed] [Google Scholar]
- 35.Rayet B, Gelinas C. Aberrant rel/nfkb genes and activity in human cancer. Oncogene. 1999;18:6938. doi: 10.1038/sj.onc.1203221. [DOI] [PubMed] [Google Scholar]
- 36.Seitz CS, Lin Q, Deng H, Khavari PA. Alterations in NF-kappaB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-kappaB. Proc Natl Acad Sci USA. 1998;95:2307. doi: 10.1073/pnas.95.5.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dajee M, Lazarov M, Zhang JY, Cai T, Green CL, Russell AJ, Marinkovich MP, Tao S, Lin Q, Kubo Y, Khavari PA. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421:639. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- 38.van Hogerlinden M, Rozell BL, Toftgard R, Sundberg JP. Characterization of the progressive skin disease and inflammatory cell infiltrate in mice with inhibited NF-kappaB signaling. J Invest Dermatol. 2004;123:101. doi: 10.1111/j.0022-202X.2004.22706.x. [DOI] [PubMed] [Google Scholar]
- 39.Perkins ND. NF-kappaB: tumor promoter or suppressor? Trends Cell Biol. 2004;14:64. doi: 10.1016/j.tcb.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 40.Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell. 2004;6:203. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 41.van Hogerlinden M, Rozell BL, Ahrlund-Richter L, Toftgard R. Squamous cell carcinomas and increased apoptosis in skin with inhibited Rel/nuclear factor-kappaB signaling. Cancer Res. 1999;59:3299. [PubMed] [Google Scholar]
- 42.Thomas GR, Chen Z, Leukinova E, Van Waes C, Wen J. Cytokines IL-1 alpha, IL-6, and GM-CSF constitutively secreted by oral squamous carcinoma induce down-regulation of CD80 costimulatory molecule expression: restoration by interferon gamma. Cancer Immunol Immunother. 2004;53:33. doi: 10.1007/s00262-003-0433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.St John MA, Li Y, Zhou X, Denny P, Ho CM, Montemagno C, Shi W, Qi F, Wu B, Sinha U, Jordan R, Wolinsky L, Park NH, Liu H, Abemayor E, Wong DT. Interleukin 6 and interleukin 8 as potential biomarkers for oral cavity and oropharyngeal squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 2004;130:929. doi: 10.1001/archotol.130.8.929. [DOI] [PubMed] [Google Scholar]
- 44.Diehl S, Rincon M. The two faces of IL-6 on Th1/Th2 differentiation. Mol Immunol. 2002;39:531. doi: 10.1016/S0161-5890(02)00210-9. [DOI] [PubMed] [Google Scholar]
- 45.Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola D, Dalton W, Jove R, Pardoll D, Yu H. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. 2004;10:48. doi: 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- 46.Morrison BE, Park SJ, Mooney JM, Mehrad B. Chemokine-mediated recruitment of NK cells is a critical host defense mechanism in invasive aspergillosis. J Clin Invest. 2003;112:1862. doi: 10.1172/JCI200318125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dorner BG, Scheffold A, Rolph MS, Huser MB, Kaufmann SH, Radbruch A, Flesch IE, Kroczek RA. MIP-1alpha, MIP-1beta, RANTES, and ATAC/lymphotactin function together with IFN-gamma as type 1 cytokines. Proc Natl Acad Sci U S A. 2002;99:6181. doi: 10.1073/pnas.092141999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Giovannucci E, Egan KM, Hunter DJ, Stampfer MJ, Colditz GA, Willett WC, Speizer FE. Aspirin and the risk of colorectal cancer in women. N Engl J Med. 1995;333:609. doi: 10.1056/NEJM199509073331001. [DOI] [PubMed] [Google Scholar]
- 49.Kopp E, Ghosh S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science. 1994;265:956. doi: 10.1126/science.8052854. [DOI] [PubMed] [Google Scholar]
- 50.Grilli M, Pizzi M, Memo M, Spano P. Neuroprotection by aspirin and sodium salicylate through blockade of NF-kappaB activation. Science. 1996;274:1383. doi: 10.1126/science.274.5291.1383. [DOI] [PubMed] [Google Scholar]
- 51.Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. 1998;396:77. doi: 10.1038/23948. [DOI] [PubMed] [Google Scholar]
- 52.Thun MJ, Namboodiri MM, Heath CW., Jr Aspirin use and reduced risk of fatal colon cancer. N Engl J Med. 1991;325:1593. doi: 10.1056/NEJM199112053252301. [DOI] [PubMed] [Google Scholar]
- 53.Suh O, Mettlin C, Petrelli NJ. Aspirin use, cancer, and polyps of the large bowel. Cancer. 1993;72:1171. doi: 10.1002/1097-0142(19930815)72:4<1171::AID-CNCR2820720407>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 54.Rosenberg L, Palmer JR, Zauber AG, Warshauer ME, Stolley PD, Shapiro S. A hypothesis: nonsteroidal anti-inflammatory drugs reduce the incidence of large-bowel cancer. J Natl Cancer Inst. 1991;83:355. doi: 10.1093/jnci/83.5.355. [DOI] [PubMed] [Google Scholar]