

Abstract
We have already demonstrated that inactivated, replication-defective Sendai virus particles (HVJ-E) have a powerful antitumor effect by both the generation of tumor-specific cytotoxic T cells and inhibition of regulatory T cell activity. Here, we report that HVJ-E also has an antitumor effect through non-T cell immunity. Microarray analysis revealed that direct injection of HVJ-E induced the expression of CXCL10 in established Renca tumors. CXCL10 was secreted by dendritic cells in the tumors after HVJ-E injection. Quantitative real-time RT-PCR and immunohistochemistry revealed that CXCR3+ cells (predominantly NK cells) infiltrated the HVJ-E-injected tumors. Moreover, HVJ-E injection caused systemic activation of NK cells and enhanced their cytotoxity against tumor cells. In an in vivo experiment, approximately 50% of tumors were eradicated by HVJ-E injection, and this activity of HVJ-E against Renca tumors was largely abolished by NK cell depletion using anti-asialo GM1 antibody. Since HVJ-E injection induced systemic antitumor immunity by enhancing or correcting the chemokine-chemokine receptor axis, it might be a potential new therapy for cancer.
Keywords: HVJ-E, Antitumor immunity, CXCL10, NK cell, Dendritic cell
Introduction
Despite extensive progress in therapeutic and diagnostic methods, many cancers are still hard to control. In particular, the treatment of advanced or metastatic cancer and the prevention of recurrence are the most difficult problems in the field of cancer therapy.
Viruses have attracted considerable attention as anticancer agents [7], and viral vectors have been developed for anticancer gene therapy [20, 43, 58, 62]. For example, an adenovirus vector containing the p53 gene has been clinically tested against various cancers in many countries [12, 15, 44]. Various live viruses, such as mumps virus [1], Newcastle disease virus [5, 40], measles virus [4], reovirus [15, 35], and vesicular stomatitis virus [3], have also been administered into tumors in order to kill cancer cells by infection and viral replication. Based on this concept, a more elegant method has also been developed to minimize side effects [45]. Cancer-specific oncolytic viruses that chiefly replicate in tumor cells have been discovered among viral mutants [14, 30] or have been produced by genetic engineering [24, 59]. These oncolytic viruses work very well in animal tumor models, but unfortunately have been less successful in humans [33]. Moreover, tumor-selective replication of these viruses is not strict enough and replication also occurs in non-cancerous tissues [13], although its efficiency is much lower than in tumor cells.
Apart from oncolytic activity, the immune reaction to viruses has been adapted to anticancer immunotherapy. It is known that vaccinia virus envelope protein can activate both CD4+ and CD8+ T cells [49]. Vesicular stomatitis virus G protein with fusion activity has also been employed to enhance antitumor imunity [9].
Sendai virus (hemagglutinating virus of Japan: HVJ) has well-known membrane fusion activity [37]. Live Sendai virus is also used for the production of type I interferon [27, 28]. A recombinant Sendai virus vector has been developed and used for gene therapy because it allows high levels of transgene expression [61]. Furthermore, new drug delivery vectors such as HVJ-liposomes [22] and HVJ envelope vector (HVJ-E) [21] have been developed using inactivated Sendai virus particles because of the robust fusion activity of the viral envelope. Recently, we found that HVJ-E itself had a powerful antitumor effect that is mediated through enhancement of cytokine production by dendritic cells (DCs). HVJ-E has been shown to eliminate murine colon cancer by both the generation of tumor-specific cytotoxic T cells (CTL) and inhibition of regulatory T cell activity [25]. However, the influence of HVJ-E on non-T cell immunity has yet not been investigated.
In the present study, we demonstrated that intratumoral injection of HVJ-E could induce the local production of the interferon (IFN)-inducible chemokine CXCL10, which seemed to promote NK cell invasion and led to effective tumor eradication.
Materials and methods
Mice and cell lines
Female BALB/cA mice and C.B-17/IcrCrj-SCID mice aged 6–8 weeks were purchased from Oriental Yeast Co. (Kyoto, Japan) and were maintained in a temperature-controlled, pathogen-free room. All animals were handled according to the approved protocols and guidelines of the Animal Committee of Osaka University. Renca renal cell carcinoma (RCC) was purchased from the American Type Culture Collection (Manassas, VA) and was cultured in RPMI 1640 medium (Nakarai Tesque, Kyoto, Japan) with 10% fetal bovine serum (FBS) (Bio West, Miami, FL) and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin, Nakarai Tesque, Kyoto, Japan). CT26 murine colon cancer and B16 melanoma were purchased from the American Type Culture Collection and cultured in DMEM (Nakarai Tesque, Kyoto, Japan) with 10% FBS and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin).
Generation of DCs
Murine bone marrow-derived DCs were generated as described previously [17]. Briefly, after flushing bone marrow from the tibia and femur with RPMI 1640 medium, it was passed through a 40-μm mesh and erythrocytes were lysed with ammonium chloride. After washing, 1 × 106 cells were plated into 24-well plates (Costar, Corning, NY, USA) in 1 ml of RPMI 1640 medium supplemented with 10% heat-inactivated FBS (Equitech-Bio, Kerrville, TX, USA), antibiotics, 50 μM 2-mercaptoethanol (Nakarai Tesque, Kyoto, Japan), and 10 ng/ml of recombinant murine GM-CSF (R&D Systems, Minneapolis, MN, USA). The cultures were maintained by gentle pipetting to aspirate all medium every second day, after which fresh medium was added. On day 6 of culture, nonadherent and loosely adherent clusters of proliferating DCs were collected and used for the subsequent experiments. More than 90% of these DCs were positive for CD11c by flow cytometry.
Preparation of HVJ-E
HVJ (Z strain) was purified from the chorioallantoic fluid of hens’ eggs by centrifugation, and the titer was calculated as described previously [37]. The virus was inactivated by exposure to UV irradiation (99 mJ/cm2) just before use, and this procedure meant that viral replication was completely eliminated (data not shown) as described previously [21]. Then 5,000 hemagglutinating units (HAU) of HVJ-E was suspended in 100 μl of saline for injection in the in vivo experiment.
Microarray analysis
Renca cells (5 × 106) were inoculated intradermally into the backs of syngeneic BALB/c mice. When tumor nodules had grown to approximately 5–8 mm in diameter (5 days after inoculation), HVJ-E (5000 HAU in a total volume of 100 μl) or saline was injected into each tumor mass. After 24 h, the tumors were removed and RNA was isolated using an RNeasy Mini Kit (Qiagen, Tokyo, Japan) according to the manufacturer’s instructions. Microarray analysis was carried out by Hokkaido System Science Co., Ltd. (Hokkaido, Japan). In brief, RNA amplification and labeling was performed according to the LRIFLA protocol. Hybridization was done with an Agilent In Situ Hybridization Plus kit (Agilent Technologies, Palo Alto, USA) according to the manufacturer’s oligonucleotide microarray hybridization user’s manual. The arrays were scanned by an Agilent dual-laser DNA microarray scanner using SureScan technology, extracted, and analysed by Feature Extraction software (Agilent Technologies, Palo Alto, USA).
Quantitative real-time RT-PCR
Intradermal Renca tumors were produced in BALB/c mice as described above. On day 5, HVJ-E (5,000 HAU in a total volume of 100 μl) or saline was injected into each tumor mass. After 12 and 48 h, the tumors were removed and RNA was isolated using an RNeasy Mini Kit (Qiagen, Tokyo, Japan) according to the manufacturer’s instructions. A total of 1 μg of RNA was reverse-transcribed using a High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA). Primers and probes for CXCL10, CD4, CD8b, CD11c, DX5, CXCR3, IFN-α, IFN-β, IFN-γ, CD69, and GAPDH were all purchased from Applied Biosystems (Foster City, CA, USA). Real-time PCR was performed and the products were analyzed by the ABI PRISM 7900HT Sequence Detection System using SDS 2.2 software (Applied Biosystems, Foster City, CA, USA).
To quantify mRNA expression by intratumoral DCs and NK cells, we harvested the tumors of five mice from each group and minced the tissues in RPMI 1640 medium. Cells were then passed through a 40-μm mesh and a single-cell suspension was obtained. DCs and NK cells were isolated by using anti-CD11c (N418) and anti-CD49 (DX5) MicroBeads, respectively, followed by passage through MACS-positive selection columns according to the manufacturer’s instructions (Miltenyi Biotec, Gladbach, Germany). Then RNA was obtained from the isolated DCs or NK cells and real-time PCR was carried out as described above.
Measurement of CXCL10
Renca cells were seeded at 5 × 104 /well in 96-well plates (Costar, Corning, NY, USA) and were incubated overnight, after which DCs (1 × 105 cells/well) and HVJ-E were added. After 2 and 24 h of culture, CXCL10 was measured in the culture supernatant by a specific enzyme-linked immunosorbent assay (ELISA) using commercially available reagents (R&D Systems, Minneapolis, MN, USA). To assess the serum and tumor levels of CXCL10, intradermal Renca tumors were produced in BALB/c mice as described above. On day 5, HVJ-E (5,000 HAU in a total volume of 100 μl) or saline was injected into each tumor mass. After 12 h, the tumors and blood were harvested to measure CXCL10 by ELISA as described previously [39]. Briefly, tumors were homogenized and sonicated in 20 mM Tris-HCl buffer with a protease inhibitor cocktail (complete Mini, EDTA-free, Roche Diagnostics, Indianapolis, IN, USA). The specimens were centrifuged at 15,000 rpm for 20 min, followed by filtration and measurement of the total protein content using Bio-Rad Protein assay reagents (Bio-Rad Japan, Tokyo, Japan) with bovine serum albumin as the standard. A 20 μg aliquot of total protein was used for each assay. Tumor to serum gradient of CXCL10 was represented by tumor minus serum levels of CXCL10.
Immunofluorescence staining
Intradermal Renca tumors were produced in BALB/c mice as described above, and HVJ-E (5000 HAU in a total volume of 100 μl) or saline was injected into each tumor on days 5, 6, and 7. Tumor tissues were collected at 12 h after the third injection for histological examination. Tissues were embedded in Tissue-Tek OCT compound (Sakura Finetek, Tokyo, Japan), frozen in dry ice, and stored at −80°C. For immunofluorescence staining, 6-μm sections were cut with a Cryostat (Leica Microsystems AG, Wetzlar, Germany). After washing, the sections were stained with FITC-conjugated anti-mouse CD49b/Pan-NK cell monoclonal antibody (1:100, BD Biosciences, Franklin Lakes, NJ, USA) for 1.5 h at room temperature. Sections were subsequently stained with 4′,6-diamidino-2-phenylindole for 10 min at room temperature and mounted with VECTOR Shield antifade solution (Vector Laboratories, Inc., Burlingame, CA, USA).
NK cell cytotoxity assay
Intradermal Renca tumors were produced in BALB/c mice as described above. On day 5, HVJ-E (5000 HAU in a total volume of 100 μl) or saline was injected into the tumors. Twenty-four hours later, the spleen was harvested from each mouse and minced, after which NK cells were positively selected from the splenocytes by using DX5-conjugated MicroBeads according to the manufacturer’s instructions (Miltenyi Biotec, Gladbach, Germany). The isolated DX5+ NK cells had a purity >90%. The cytotoxity of the NK cells was measured by a standard 4-h 51Cr-release assay using sodium chromate-labeled Renca cells as the target.
Measurement of type I IFNs
Renca cells were seeded at 5 × 104/well in 96-well plates (Costar, Corning, NY, USA) and were incubated overnight, after which DCs (1 × 105 cells/well) and HVJ-E were added and culture was continued for 24 h. Then IFN-α and IFN-β were measured in the supernatant by a specific ELISA using commercially available reagents (PBL Biomedical Laboratories, Piscataway, NJ, USA).
Measurement of IFN-γ
After DCs (1 × 105 cells/well) and HVJ-E were cultured in 96-well plates for 24 h, the supernatant was collected. Renca cells were seeded at 5 × 104/well in 96-well plates and cultured overnight, after which NK cells (3.5 × 105/well) and the DC culture supernatant were added. After another 24 h, the IFN-γ concentration in the culture supernatant was measured by using a mouse IFN-γ DuoSet ELISA kit (R&D Systems, Minneapolis, MN, USA). For the neutralization assay of type I IFNs, NK cells were preincubated with anti-mouse IFN-α/β R2 (IFNAR2) antibody (20 μg/ml, R&D Systems, Minneapolis, MN, USA) or control goat IgG (20 μg/ml, R&D Systems) for 1 h before use.
Tumor growth in vivo
Mice were injected intradermally into the backs with 0.1 ml of a single-cell suspension containing 5 × 106 Renca cells. When the tumor nodules had grown to approximately 5–8 mm in diameter (5 days after inoculation) the mice were divided into two groups of five. On days 5, 10, and 15, HVJ-E (5000 HAU in a total volume of 100 μl) or saline was injected into each tumor then the size of the tumor masses was measured every 4 days using calipers.
Intratumoral depletion of NK cells in vivo
For intratumoral depletion of NK cells, 40 μg of an anti-asialo GM1 antibody (Wako Pure Chemical Industries, Ltd., Osaka, Japan) was injected into the tumors at the same time as HVJ-E or saline. This procedure effectively depleted NK cell subsets as shown by FACS analysis [10].
ELISPOT assay for detection of CD8+T cell responses
Spleen cells were harvested from mice at 7 days after the last intratumoral injection of HVJ-E or saline as described above. Spleen cells (5 × 107/flask) were stimulated with mitomycin C (MMC)-treated Renca cells at a ratio of 10:1 in culture medium containing 10 IU/ml of recombinant IL-2 at 37°C in 5% CO2. After 5 days, CD8+ T cells were isolated using a mouse CD8a+ isolation kit and an AutoMACS magnetic sorter (Miltenyi Biotec, Gladbach, Germany) according to the manufacturer’s instructions. Then 1 × 105 purified CD8+ T cells were cultured for 48 h with or without 1 × 105 MMC-treated Renca cells or CT26 cells. The assay was performed using a mouse IFN-γ ELISPOT kit (R&D Systems, Minneapolis, MN, USA). The number of IFN-γ-secreting CD8+ T cells was subsequently counted under a dissecting microscope (Leica, Cambridge, UK).
Statistical analysis
Statistical analysis was done with the unpaired t test and P < 0.05 was considered to indicate statistical significance.
Results
Microarray analysis of tumor gene expression after HVJ-E injection
To investigate the gene expression profile of Renca tumors after intratumoral injection of HVJ-E, microarray analysis was performed. As shown in Table 1, 44 genes were upregulated by >2.5-fold in HVJ-E-treated tumors compared with saline-treated tumors. Among 12 genes that were upregulated by >4-fold, there were nine IFN-related genes and two chemokine genes.
Table 1.
Microarray analysis of genes upregulated in Renca tumors by HVJ-E injection. Renca cells were inoculated intradermally into the backs of syngeneic BALB/c mice. HVJ-E or saline was injected into each tumor. After 24 h, the tumors were removed and microarray analysis of the isolated RNA was performed

Induction of CXCL10 by HVJ-E both in vitro and in vivo
Chemokines have been reported to play a crucial role in eliciting non-T cell immunity [32, 53] and CXCL10 was the most highly upregulated chemokine after HVJ-E injection. Therefore, we focused on CXCL10 (interferon-inducible protein 10), which is a chemokine that has been reported to display antitumor activity by inhibiting angiogenesis [46, 48] and recruiting immune cells [29, 50]. To confirm that CXCL10 expression was increased in the tumors by HVJ-E injection, quantitative real-time RT-PCR was performed, and we found that intratumoral injection of HVJ-E markedly increased the expression of CXCL10 mRNA (Fig. 1a). It was recently reported that the chemokine receptor/ligand axis plays a critical role in mediating the antitumor effect of immunotherapy [39]. To assess the influence of the intratumoral injection of HVJ-E on chemotactic gradient, we examined serum and tumor levels of CXCL10 after injection of HVJ-E or saline into Renca tumors in mice. We found that the tumor-to-serum gradient of CXCL10 was markedly increased by HVJ-E injection (Table 2). Next, we investigated the possible sources of CXCL10. It is already known that secretion of cytokines, such as type I interferon [27, 28] and IL-6 [25], by dendritic cells (DCs) increases after treatment with either live or inactivated HVJ. Furthermore, we have demonstrated that DCs have a pivotal role in the antitumor activity of HVJ-E [25]. Therefore, we focused on DCs as the probable source of CXCL10. Analysis of DCs isolated from HVJ-E-treated tumors showed that the expression of CXCL10 mRNA was increased (Fig. 1b), suggesting that DCs were one of the sources of CXCL10. Next, we tested whether HVJ-E could induce CXCL10 in vitro (Fig. 1c). We measured CXCL10 levels in the supernatant of cultures with or without Renca cells, HVJ-E, or DCs. When HVJ-E was added to cultured Renca cells, production of CXCL10 was very low. However, when HVJ-E and syngeneic mouse DCs were added to cultured Renca cells, a significant increase of CXCL10 production was observed at both 2 and 24 h. A significant increase of CXCL10 was also detected when DCs were cultured with HVJ-E, even in the absence of Renca cells, although the level was lower than in the presence of Renca cells. These results indicated that HVJ-E acted on DCs to induce CXCL10 production.
Fig. 1.

Induction of CXCL10 by HVJ-E in vivo and in vitro. CXCL10 mRNA expression in whole tumors (a) or DCs isolated from tumors (b) after injection of HVJ-E or saline was measured by quantitative real-time RT-PCR (n = 5 per group). CXCL10 mRNA expression was increased by injection of HVJ-E. Error bars indicate the SE (<5%). This experiment was repeated three times with similar results. c CXCL10 levels in the medium of cultures with (+) or without (-) Renca cells, HVJ-E, or DCs. When HVJ-E and syngeneic mouse DCs were added to cultured Renca cells, a significant increase of CXCL10 production was observed. A significant increase of CXCL10 was also detected in the medium of DCs cultured with HVJ-E even in the absence of Renca cells, although the level was lower than that obtained with Renca cells (*P < 0.05, **P < 0.01). Data points are the mean ± SE of triplicate wells. SE < 5%. This experiment was repeated four times with similar results
Table 2.
Serum and tumor levels of CXCL10 after intratumoral injection of HVJ-E or saline
| Serum CXCL10 (ng/ml) | Tumor CXCL10(ng/ml) | Chemotactic gradient (tumor-serum, ng/ml) | |
|---|---|---|---|
| Saline | 0.043 ± 0.0098 | 1.3 ± 0.45 | 1.3 |
| HVJ-E | 0.90 ± 0.10 | 10 ± 1.8 | 9.1 |
Promotion of NK cell infiltration and activation by intratumoral injection of HVJ-E
CXCL10 has been reported to recruit CXCR3-expressing cells, such as memory T cells, activated T lymphocytes, NK cells, and mononuclear cells [11, 26, 42]. Therefore, we investigated whether these cells were recruited to infiltrate tumors by HVJ-E injection. Quantitative real-time RT-PCR revealed an increase of CXCR3 mRNA expression in the tumors 12 h after HVJ-E injection, indicating that CXCR3+ cells were recruited into tumor tissue (Fig. 2a). To investigate the immune cells that were recruited in more detail, quantitative real-time RT-PCR was performed with several immune cell markers (CD8, CD4, DX5, and CD11c). A significant increase of DX5, mRNA expression (a marker for NK cells) was detected in the tumors after HVJ-E injection (Fig. 2a). To confirm the infiltration of NK cells into the tumors, we performed immunofluorescence staining using anti-pan-NK cell (DX5) antibody, which revealed that the number of infiltrating NK cells was increased by successive three times injection of HVJ-E (Fig. 2b). Even after a single injection of HVJ-E, we could detect NK cell infiltration, but the number of infiltrating cells was smaller (data not shown). Next, we investigated whether or not the infiltrating NK cells were activated. Analysis of NK cells from HVJ-E-treated tumors showed that IFN-γ mRNA expression was markedly increased, as was the expression of CD69 mRNA, another marker of NK cell activation [52] (Fig. 2c). In addition to intratumoral activation of NK cells, significant systemic NK cell activation in HVJ-E-treated mice was also revealed by the 51Cr release cytotoxicity assay using Renca cells (Fig. 2d). Moreover, we confirmed that the level of cytotoxity was much higher when we performed cytotoxic assay with YAC-1 mouse T cell lymphoma cells as targets, which have low MHC class I expression [41] and are commonly used as the prototype for an NK-sensitive tumor line [41](data not shown). These results suggested that HVJ-E promoted NK cell infiltration into tumors, and also activated the NK cells to enhance IFN-γ production.
Fig. 2.
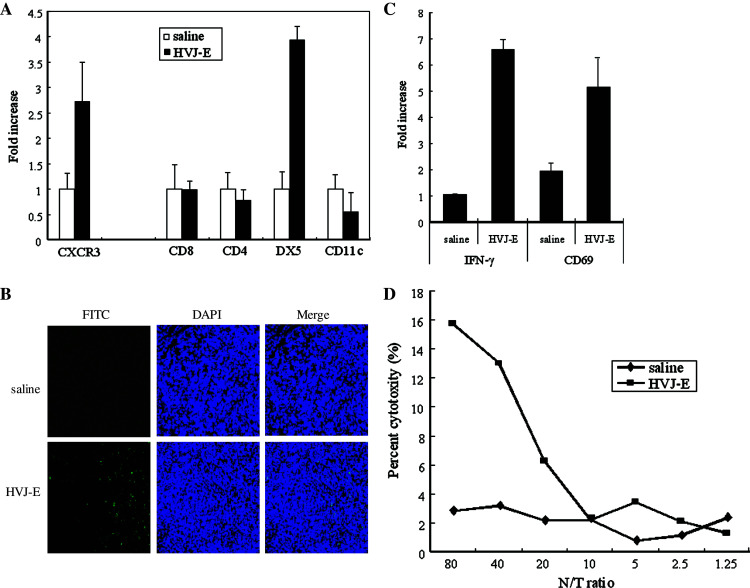
Infiltration and activation of NK cells after intratumoral injection of HVJ-E. a Intratumoral infiltration of immune cells was investigated by quantitative real-time RT-PCR (n = 5). DX5 mRNA expression showed a marked increase. SE < 5%. This experiment was repeated three times with similar results. b NK cell infiltration. Immunofluorescence staining using a FITC-conjugated anti-mouse CD49b (DX5)/Pan NK cell monoclonal antibody shows prominent infiltration of DX5-positive cells (green) into an HVJ-E-treated tumor (×200). This experiment was repeated three times with similar results. c Activation of intratumoral NK cells: NK cells were purified from tumors injected with HVJ-E or saline, and IFN-γ and CD69 mRNA expression was assayed by quantitative real-time RT-PCR. Both IFN-γ and CD69 expressions were upregulated in HVJ-E-treated tumors. SE < 5%. This experiment was repeated three times with similar results. d NK cytotoxity in vivo. Cytotoxicity assays were performed with NK cells harvested from the spleens of mice after treatment with HVJ-E (filled square) or saline (filled diamond). NK cells from HVJ-E-treated mice showed an increase of cytotoxity against Renca cells. This experiment was repeated three times with similar results
NK cell activation by type I IFNs released from HVJ-E-stimulated DCs
Next, we investigated the factor induced by HVJ-E that played an important role in NK cell activation. It is known that type I IFNs are induced by viral infection [6, 8], leading to activation of NK cells and enhanced secretion of type II IFNs [19, 57]. In this study, we found that intratumoral injection of HVJ-E markedly increased the expression of IFN-β mRNA in Renca tumors, as detected by quantitative real-time RT-PCR (Fig. 3a). This suggested that type I IFNs were being secreted in the tumor tissue. When co-culture of Renca cells and DCs was performed, a significant and dose-dependent increase of type I IFNs (predominantly IFN-β) was detected in the medium at 24 h after the addition of HVJ-E (Fig. 3b). When HVJ-E was added to Renca cells in the absence of DCs, however, secretion of type I IFNs was very low, indicating that HVJ-E acted on DCs to induce IFN secretion. We subsequently tested whether the conditioned medium of HVJ-E-stimulated DCs (H-DCCM) could activate NK cells by measuring IFN-γ as an activation marker. We cultured NK cells with or without H-DCCM and quantified the IFN-γ level in the culture supernatant. In the presence of H-DCCM, the IFN-γ level increased significantly in an HVJ-E dose-dependent manner (Fig. 3c), while there was no increase of IFN-γ secretion in the absence of H-DCCM, even when HVJ-E was added. The promotion of IFN-γ secretion by H-DCCM was abolished by the addition of anti-IFNAR2 antibody, which inhibits the signaling of type I IFNs in NK cells (Fig. 3d). These findings indicated that type I IFNs were induced by HVJ-E and subsequently promoted NK cell activation.
Fig. 3.

Activation of NK cells by type I IFNs induced from HVJ-E-stimulated DCs. a Intratumor expression of type I IFN mRNAs after in vivo injection of HVJ-E or saline injection measured by quantitative real-time RT-PCR (n = 5/group). This experiment was repeated four times with similar results. b Type I IFNs in the conditioned medium of Renca cells cultured with HVJ-E in the presence or absence of DCs. HVJ-E was added at an MOI of 0.3–3,000. Both IFN-α and IFN-β levels were increased in an HVJ-E-dose-dependent manner (*P < 0.05, **P < 0.01) only in the cultures with DCs. Data points are the mean ± SE of triplicate wells. This experiment was repeated three times with similar results. c A significant increase of IFN-γ was observed in the culture medium of Renca cells after addition of H-DCCM and NK cells (*P < 0.01), while no significant increase of IFN-γ secretion was detected with either HVJ-E or H-DCCM alone. Data points are the mean ± SE of triplicate wells. This experiment was repeated three times with similar results. d IFN-γ secretion was reduced by anti-IFNAR2 in the medium of Renca cells cultured with H-DCCM. When NK cells were preincubated with anti-IFNAR2, the increase of IFN-γ in response to HVJ-E was abolished. Data points are the mean ± SE of triplicate wells. This experiment was repeated three times with similar results. SE < 5%. Results were statistically analyzed using the unpaired t test
Intratumoral injection of HVJ-E suppresses Renca tumor growth in mice, while this effect is abolished by NK cell depletion
Next, we investigated whether direct injection of HVJ-E into established tumors could inhibit their growth. Renca cells were inoculated intradermally into the backs of syngeneic BALB/c mice, and then the growing tumors were injected three times with HVJ-E or saline. Injection of HVJ-E led to elimination of approximately 50% of the tumors and markedly inhibited the growth of the remaining lesions (Fig. 4a). We subsequently confirmed that this inhibition persisted for a longer period (data not shown). The maximum dose of HVJ-E particles that can be injected without causing side effects is 1.5 × 1010, according to the results of our previous study [31]. Three injections seemed to be necessary for tumor eradication because recurrence often occurred after one or two injections (data not shown). Survival was also significantly improved by the intratumoral injection of HVJ-E (Fig. 4b).
Fig. 4.

Suppression of the growth of Renca tumors in mice by intratumoral injection of HVJ-E. a Renca cells were inoculated intradermally into the backs of syngeneic BALB/c mice. Then HVJ-E (open circle) or saline (filled square) (n = 5 per group) was injected three times (on days 5, 10, and 15) into the resulting tumors. Tumor growth was significantly inhibited by HVJ-E injection (*P < 0.01) and approximately 50% of the mice became tumor-free. Data shown are representative of experiments that were repeated five times with similar results. b Kaplan–Meier survival curves for HVJ-E-treated and saline-treated mice. When HVJ-E (open circle) or saline (filled square) (n = 5 per group) was injected three times into the intradermal Renca tumors of BALB/c mice, the survival of HVJ-E-treated mice was significantly better than that of saline-treated mice (*P < 0.01). Data shown are representative of experiments that were repeated four times with similar results. c Loss of the antitumor effect of HVJ-E after neutralization of NK activity. Renca cells were inoculated intradermally into syngeneic BALB/c mice, and then HVJ-E was injected three times into the resulting tumors together with anti-asialo GM1 antibody (filled square) or control IgG (filled circle) (n = 5 per group). Tumor growth was inhibited in the mice treated with HVJ-E plus control IgG, whereas it was not inhibited in mice treated with HVJ-E plus anti-asialo GM1 antibody (*P < 0.05). Data shown are representative of experiments that were repeated three times with similar results. Arrows indicate the timing of injection. SE (<5%). Statistical analysis was done with the unpaired t test (a, c) and the Log-rank test (b)
To confirm that NK cells mediated the antitumor effect of HVJ-E, it was co-injected into tumors with anti-asialo GM1 antibody to cause NK cell depletion. As shown in Fig. 4c, suppression of tumor growth by HVJ-E was largely abolished after concomitant antibody injection. In addition, intraperitoneal injection of the antibody led to similar results being obtained (data not shown). These results indicated that HVJ-E predominantly acts on Renca tumors by inducing NK cell-mediated immunity.
T cell immunity was also elicited against Renca tumor
Finally, since we previously reported that HVJ-E could eliminate murine colon cancer by induction of CTL with blocking regulatory T cell activity [25], we examined the induction of T cell immunity in Renca tumors in later phase. By real-time RT-PCR analysis, we found that CD8 and CD4 mRNA expressions showed a marked increase in tumors 48h after HVJ-E injection (Fig. 5a). In addition, the ELISPOT assay showed that Renca-specific CD8+ T cell activation was induced in HVJ-E-treated mice, confirming the involvement of T cell-mediated acquired immunity also in this RCC model (Fig. 5b). Moreover, Renca tumors transplanted into severe combined immuno deficiency (SCID) mice were treated with HVJ-E or saline. We found that the growth of Renca tumors was still significantly inhibited by HVJ-E (Fig. 5c) although the inhibition was less marked than in wild-type mice. However, in CT26 tumor-bearing SCID mice, the growth inhibition by HVJ-E was completely abolished as reported previously [25]. These results suggested that the antitumor effect of HVJ-E against Renca tumors depend on both T cell- and non-T cell-mediated immunity.
Fig. 5.

Induction of T cell immunity in Renca tumors in later phase. a Intratumoral infiltration of T cells 48 h after HVJ-E treatment was investigated by quantitative real-time RT-PCR (n = 3). CD8 and CD4 mRNA expressions showed a marked increase. These experiments were repeated three times with similar results. b ELISPOT analysis of HVJ-E-treated mice (open bars) and saline-treated mice (solid bars). Spleen cells were harvested from the mice at 7 days after the last injection of HVJ-E or saline into Renca tumors. Then the spleen cells were stimulated with MMC-treated Renca cells for 5 days, after which CD8+ T cells were isolated. Subsequently, 1 × 105 purified CD8+ T cells were cultured for 48 h with or without MMC-treated Renca cells or CT26 cells and the number of IFN-γ-secreting CD8+ T cells was counted (*P < 0.05). Data points are the mean of triplicate wells. c Antitumor effect of HVJ-E in SCID mice (n = 5). Renca cells were inoculated intradermally into SCID mice, after which HVJ-E (open circle) or saline (filled square) (n = 5/group) was injected three times into the tumors that developed. Tumor growth was significantly inhibited by HVJ-E (*P < 0.05); although the inhibition was less marked compared with that in wild-type mice. Arrows indicate the timing of injection. This experiment was repeated twice with similar results. SE < 5%. Results were analyzed statistically using the unpaired t test (b, c)
Discussion
We have already shown that UV-inactivated Sendai virus particles without any specific therapeutic properties show powerful antitumor activity [25]. Recently, it has been realized that the tumor microenvironment plays a critical role in tumor progression and suppression [2, 34, 47, 54]. In this study, we performed microarray analysis to investigate how the tumor microenvironment was affected by HVJ-E injection and we found that CXCL10 mRNA was prominently upregulated in HVJ-E-treated tumors (Table 1). CXCL10 is a non-ELR (Glu-Leu-Arg) CXC chemokine that has been reported to play a critical role in regulating type 1 cytokine-induced cell-mediated immunity via the recruitment of CXCR3-expressing mononuclear cells, such as CD4 and CD8 lymphocytes, as well as NK cells [11, 26, 42, 51]. When we examined immune cell recruitment into the tumors by real-time RT-PCR and immunohistochemistry, we found that NK cells were the predominant infiltrating cells after HVJ-E injection (Fig. 2a, b). These findings suggested that HVJ-E induced NK cell-mediated immunity in our Renca tumor model. We also confirmed that HVJ-E induced systemic and intratumoral NK cell activation by measuring cytotoxity and IFN-γ secretion in vivo (Fig. 2c, d). Furthermore, we found that the secretion of type I IFNs by HVJ-E-stimulated DCs was important for NK cell activation (Fig. 3a, d). Thus, our results showed that intratumoral injection of HVJ-E induced CXCL10 production, which resulted in NK cell recruitment into the tumors and also caused systemic and intratumoral activation of NK cells through stimulation via DCs. CXCL10 is produced by several kinds of cells in response to a variety of stimuli, including IFN-α, IFN-β, IFN-γ, TNF-α, lipopolysaccharide (LPS), and viral infection [16, 36, 55, 56]. In this study, we found that HVJ-E induced CXCL10 production by DCs (Fig. 1b, c). Furthermore, we confirmed that CD11c-positive DCs produced the largest amount of CXCL10 among several kinds of immune cells (data not shown). Because the secretion of IFN-γ in Renca tumors was much enhanced after HVJ-E injection, it is likely that various other cells also produced CXCL10. However, CD11c-positive cells were obviously one of the main sources of CXCL10 production in direct response to HVJ-E stimulation. It was recently reported that CXCR3/CXCR3 ligand biological axis plays a critical role in mediating the antitumor effect of systemic IL-2 therapy [39]. During such immunotherapy, systemic levels of CXCR3 ligands (such as CXCL9 and CXCL10) increase without any marked increase in the intratumor levels of these chemokines. Accordingly, the CXCR3 ligand chemotactic gradient is attenuated and optimal recruitment of circulating immune cells into tumor tissue cannot be achieved. As shown in Table 2, intratumoral HVJ-E injection not only induced systemic activation of NK cells, but also enhanced the CXCL10 gradient by increasing the local intratumor level of this chemokine, which may be of benefit for cancer therapy.
Our previous study demonstrated that HVJ-E induced T cell-mediated immunity against colon cancer through activation of DCs and inhibition of regulatory T cells [25]. In the present study, we investigated the early response (12–24 h) to HVJ-E injection in order to focus on non-T cell immunity. Although neither by real-time PCR (Fig. 2a) nor by immunohistochemistry (data not shown) an increase of CD8 and CD4 T cells was detected during this early phase, we detected such an increase at 48 h after HVJ-E treatment (Fig. 5a) and confirmed that tumor-specific CTLs were also induced in Renca tumors (Fig. 5b). In addition, we found that antitumor activity against Renca tumors was partially retained even in SCID mice without CTLs (Fig. 5c), while activity against CT 26 tumors was completely lost [25]. Furthermore, the suppression of Renca tumor growth by HVJ-E was largely abolished after either intratumoral (Fig. 4c) or intraperitoneal (data not shown) NK cell depletion. These data suggested that NK cells could play a dominant role in the antitumor activity of HVJ-E against Renca cells. We subsequently performed cytotoxicity assays using Renca or CT26 cells and H-DCCM-stimulated NK cells. Cytotoxity of the NK cells for Renca cells was observed, while there was no significant NK activity against CT26 cells (data not shown). This observation may be explained by our subsequent finding that Renca cells showed weaker expression of MHC class I, a ligand for an NK cell inhibitory receptor [60], than CT26 cells (data not shown). Therefore, it is likely that Renca cells are more easily recognized and killed by NK cells than CT26 cells.
Thus, replication-defective HVJ-E has multiple antitumor effects. Although more detailed analysis of the mechanisms is needed, HVJ-E appears to be more advantageous for cancer treatment compared with oncolytic viruses. One advantage is its safety. Although detailed toxicity tests and pharmacodynamic studies of HVJ-E are still necessary, its safety has already been confirmed in mice, rats, and monkeys. No infective particles have been recovered from HVJ-E after inactivation by UV irradiation [21]. We observed no weight loss in the mice after intratumor administration. Second, HVJ-E can also be used for the delivery of therapeutic agents, such as plasmid DNA [38], siRNA [18] and anticancer drugs [31], to tumor cells both in vitro and in vivo. Therefore, we could expect a combined antitumor effect of such agents targeted by HVJ-E in addition to the activity of HVJ-E itself. Finally, we have established a method for producing HVJ in human cells using animal product-free medium, and also a method for purification of HVJ-E using three different columns [23]. Clinical grade HVJ-E has been produced on a pilot scale, so this vector is now ready for clinical testing.
In summary, our results suggested that intratumor injection of HVJ-E promoted NK cell infiltration into tumors by enhancing CXCL10 expression and also activated NK cells through DC stimulation. Since HVJ-E also induces systemic antitumor immunity by enhancing or correcting the chemokine-chemokine receptor axis, it may become a promising new therapeutic agent for cancer.
References
- 1.Asada T. Treatment of human cancer with mumps virus. Cancer. 1974;34:1907–1928. doi: 10.1002/1097-0142(197412)34:6<1907::AID-CNCR2820340609>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 2.Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 3.Barber GN. VSV-tumor selective replication and protein translation. Oncogene. 2005;24:7710–9. doi: 10.1038/sj.onc.1209042. [DOI] [PubMed] [Google Scholar]
- 4.Bluming AZ, Ziegler JL. Regression of Burkitt’s lymphoma in association with measles infection. Lancet. 1971;2:105–106. doi: 10.1016/S0140-6736(71)92086-1. [DOI] [PubMed] [Google Scholar]
- 5.Cassel WA, Garrett RE. Newcastle disease virus as an antineoplastic agent. Cancer. 1965;18:863–868. doi: 10.1002/1097-0142(196507)18:7<863::AID-CNCR2820180714>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 6.Colonna M, Krug A, Cella M. Interferon-producing cells: on the front line in immune responses against pathogens. Curr Opin Immunol. 2002;14:373–379. doi: 10.1016/S0952-7915(02)00349-7. [DOI] [PubMed] [Google Scholar]
- 7.Davis JJ, Fang B. Oncolytic virotherapy for cancer treatment: challenges and solutions. J Gene Med. 2005;7:1380–1389. doi: 10.1002/jgm.800. [DOI] [PubMed] [Google Scholar]
- 8.Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, Al-Shamkhani A, Flavell R, Borrow P, Reise Sousa C. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature. 2003;424:324–328. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- 9.Errington F, Bateman A, Kottke T, Thompson J, Harrington K, Merrick A, Hatfield P, Selby P, Vile R, Melcher A. Allogeneic tumor cells expressing fusogenic membrane glycoproteins as a platform for clinical cancer immunotherapy. Clin Cancer Res. 2006;12:1333–1341. doi: 10.1158/1078-0432.CCR-05-1113. [DOI] [PubMed] [Google Scholar]
- 10.Fan Z, Yu P, Wang Y, Wang Y, Fu ML, Liu W, Sun Y, Fu YX. NK-cell activation by LIGHT triggers tumor-specific CD8+ T-cell immunity to reject established tumors. Blood. 2006;107:1342–1351. doi: 10.1182/blood-2005-08-3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farber JM (1997) Mig and IP-10: CXC chemokines that target lymphocytes [PubMed]
- 12.Gabrilovich DI. INGN 201 (Advexin): adenoviral p53 gene therapy for cancer. Expert Opin Biol Ther. 2006;6:823–832. doi: 10.1517/14712598.6.8.823. [DOI] [PubMed] [Google Scholar]
- 13.Hann B, Balmain A. Replication of an E1B 55-kilodalton protein-deficient adenovirus (ONYX-015) is restored by gain-of-function rather than loss-of-function p53 mutants. J Virol. 2003;77:11588–11595. doi: 10.1128/JVI.77.21.11588-11595.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heise C, Sampson-Johannes A, Williams A, McCormick F, Von Hoff DD, Kirn DH. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med. 1997;3:639–645. doi: 10.1038/nm0697-639. [DOI] [PubMed] [Google Scholar]
- 15.Horowitz J. Adenovirus-mediated p53 gene therapy: overview of preclinical studies and potential clinical applications. Curr Opin Mol Ther. 1999;1:500–509. [PubMed] [Google Scholar]
- 16.Hoshino K, Kaisho T, Iwabe T, Takeuchi O, Akira S. Differential involvement of IFN-β in Toll-like receptor-stimulated dendritic cell activation. Int Immunol. 2002;14:1225–1231. doi: 10.1093/intimm/dxf089. [DOI] [PubMed] [Google Scholar]
- 17.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ito M, Yamamoto S, Nimura K, Hiraoka K, Tamai K, Kaneda Y. Rad51 siRNA delivered by HVJ envelope vector enhances the anti-cancer effect of cisplatin. J Gene Med. 2005;7:1044–1052. doi: 10.1002/jgm.753. [DOI] [PubMed] [Google Scholar]
- 19.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–985. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 20.Jia W, Zhou Q. Viral vectors for cancer gene therapy: viral dissemination and tumor targeting. Curr Gene Ther. 2005;5:133–142. doi: 10.2174/1566523052997460. [DOI] [PubMed] [Google Scholar]
- 21.Kaneda Y, Nakajima T, Nishikawa T, Yamamoto S, Ikegami H, Suzuki N, Nakamura H, Morishita R, Kotani H. Hemagglutinating virus of Japan (HVJ) envelope vector as a versatile gene delivery system. Mol Ther. 2002;6:219–226. doi: 10.1006/mthe.2002.0647. [DOI] [PubMed] [Google Scholar]
- 22.Kaneda Y, Saeki Y, Morishita R. Gene therapy using HVJ-liposomes: the best of both worlds? Mol Med Today. 1999;5:298–303. doi: 10.1016/S1357-4310(99)01482-3. [DOI] [PubMed] [Google Scholar]
- 23.Kaneda Y, Yamamoto S, Nakajima T. Development of HVJ envelope vector and its application to gene therapy. Adv Genet. 2005;53:307–332. [PubMed] [Google Scholar]
- 24.Kurihara T, Brough DE, Kovesdi I, Kufe DW. Selectivity of a replication-competent adenovirus for human breast carcinoma cells expressing the MUC1 antigen. J Clin Invest. 2000;106:763–771. doi: 10.1172/JCI9180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurooka M, Kaneda Y. Inactivated Sendai virus particles eradicate tumors by inducing immune responses through blocking regulatory T cells. Cancer Res. 2007;67:227–236. doi: 10.1158/0008-5472.CAN-06-1615. [DOI] [PubMed] [Google Scholar]
- 26.Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark-Lewis I, Baggiolini M, Moser B. Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. J Exp Med. 1996;184:963–969. doi: 10.1084/jem.184.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lopez CB, Garcia-Sastre A, Williams BR, Moran TM. Type I interferon induction pathway, but not released interferon, participates in the maturation of dendritic cells induced by negative-strand RNA viruses. J Infect Dis. 2003;187:1126–1136. doi: 10.1086/368381. [DOI] [PubMed] [Google Scholar]
- 28.Lopez CB, Moltedo B, Alexopoulou L, Bonifaz L, Flavell RA, Moran TM. TLR-independent induction of dendritic cell maturation and adaptive immunity by negative-strand RNA viruses. J Immunol. 2004;173:6882–6889. doi: 10.4049/jimmunol.173.11.6882. [DOI] [PubMed] [Google Scholar]
- 29.Luster AD, Leder P. IP-10, a -C-X-C- chemokine, elicits a potent thymus-dependent antitumor response in vivo. J Exp Med. 1993;178:1057–1065. doi: 10.1084/jem.178.3.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCormick F. Cancer-specific viruses and the development of ONYX-015. Cancer Biol Ther. 2003;2:S157–S160. [PubMed] [Google Scholar]
- 31.Mima H, Yamamoto S, Ito M, Tomoshige R, Tabata Y, Tamai K, Kaneda Y. Targeted chemotherapy against intraperitoneally disseminated colon carcinoma using a cationized gelatin-conjugated HVJ envelope vector. Mol Cancer Ther. 2006;5:1021–1028. doi: 10.1158/1535-7163.MCT-05-0352. [DOI] [PubMed] [Google Scholar]
- 32.Monti P, Leone BE, Marchesi F, Balzano G, Zerbi A, Scaltrini F, Pasquali C, Calori G, Pessi F, Sperti C (2003) The CC Chemokine MCP-1/CCL2 in Pancreatic Cancer progression regulation of expression and potential mechanisms of antimalignant activity 1. AACR [PubMed]
- 33.Nemunaitis J, Cunningham C, Tong AW, Post L, Netto G, Paulson AS, Rich D, Blackburn A, Sands B, Gibson B, Randlev B, Freeman S. Pilot trial of intravenous infusion of a replication-selective adenovirus (ONYX-015) in combination with chemotherapy or IL-2 treatment in refractory cancer patients. Cancer Gene Ther. 2003;10:341–52. doi: 10.1038/sj.cgt.7700585. [DOI] [PubMed] [Google Scholar]
- 34.Nishimura F, Dusak JE, Eguchi J, Zhu X, Gambotto A, Storkus WJ, Okada H (2006) Adoptive transfer of type 1 CTL mediates effective anti-central nervous system tumor response: critical roles of IFN-inducible protein-10. AACR [DOI] [PubMed]
- 35.Norman KL, Hirasawa K, Yang AD, Shields MA, Lee PW. Reovirus oncolysis: the Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection. Proc Natl Acad Sci USA. 2004;101:11099–11104. doi: 10.1073/pnas.0404310101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohmori Y. The interferon-stimulated response element and a kappa B site mediate synergistic induction of murine IP-10 gene transcription by IFN-gamma and TNF-alpha. J Immunol. 1995;154:5235–5244. [PubMed] [Google Scholar]
- 37.Okada Y. Sendai virus-induced cell fusion. Methods Enzymol. 1993;221:18–41. doi: 10.1016/0076-6879(93)21005-s. [DOI] [PubMed] [Google Scholar]
- 38.Oshima K, Shimamura M, Mizuno S, Tamai K, Doi K, Morishita R, Nakamura T, Kubo T, Kaneda Y. Intrathecal injection of HVJ-E containing HGF gene to cerebrospinal fluid can prevent and ameliorate hearing impairment in rats. Faseb J. 2004;18:212–4. doi: 10.1096/fj.03-0567fje. [DOI] [PubMed] [Google Scholar]
- 39.Pan J, Burdick MD, Belperio JA, Xue YY, Gerard C, Sharma S, Dubinett SM, Strieter RM. CXCR3/CXCR3 ligand biological axis impairs RENCA tumor growth by a mechanism of immunoangiostasis. J Immunol. 2006;176:1456–1464. doi: 10.4049/jimmunol.176.3.1456. [DOI] [PubMed] [Google Scholar]
- 40.Pecora AL, Rizvi N, Cohen GI, Meropol NJ, Sterman D, Marshall JL, Goldberg S, Gross P, O’Neil JD, Groene WS, Roberts MS, Rabin H, Bamat MK, Lorence RM. Phase I trial of intravenous administration of PV701, an oncolytic virus, in patients with advanced solid cancers. J Clin Oncol. 2002;20:2251–2266. doi: 10.1200/JCO.2002.08.042. [DOI] [PubMed] [Google Scholar]
- 41.Petersson M, Charo J, Salazar-Onfray F, Noffz G, Mohaupt M, Qin Z, Klein G, Blankenstein T, Kiessling R. Constitutive IL-10 production accounts for the high NK sensitivity, low MHC class I expression, and poor transporter associated with antigen processing (TAP)-1/2 function in the prototype NK target YAC-1 1. J Immunol. 1998;161:2099–2105. [PubMed] [Google Scholar]
- 42.Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, Koch AE, Moser B, Mackay CR. The Chemokine Receptors CXCR3 and CCR5 Mark Subsets of T Cells Associated with Certain Inflammatory Reactions. Am Soc Clin Investig. 1998;104(4):746–754. doi: 10.1172/JCI1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robbins PD, Tahara H, Ghivizzani SC. Viral vectors for gene therapy. Trends Biotechnol. 1998;16:35–40. doi: 10.1016/S0167-7799(97)01137-2. [DOI] [PubMed] [Google Scholar]
- 44.Roth JA. Adenovirus p53 gene therapy. Expert Opin Biol Ther. 2006;6:55–61. doi: 10.1517/14712598.6.1.55. [DOI] [PubMed] [Google Scholar]
- 45.Russell SJ. RNA viruses as virotherapy agents. Cancer Gene Ther. 2002;9:961–966. doi: 10.1038/sj.cgt.7700535. [DOI] [PubMed] [Google Scholar]
- 46.Sgadari C, Angiolillo AL, Cherney BW, Pike SE, Farber JM, Koniaris LG, Vanguri P, Burd PR, Sheikh N, Gupta G, Teruya-Feldstein J, Tosato G. Interferon-inducible protein-10 identified as a mediator of tumor necrosis in vivo. Proc Natl Acad Sci USA. 1996;93:13791–13796. doi: 10.1073/pnas.93.24.13791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shurin MR, Shurin GV, Lokshin A, Yurkovetsky ZR, Gutkin DW, Chatta G, Zhong H, Han B, Ferris RL. Intratumoral cytokines/chemokines/growth factors and tumor infiltrating dendritic cells: friends or enemies? Cancer Metastasis Rev. 2006;25(3):333–356. doi: 10.1007/s10555-006-9010-6. [DOI] [PubMed] [Google Scholar]
- 48.Sun Y, Finger C, Alvarez-Vallina L, Cichutek K, Buchholz CJ. Chronic gene delivery of interferon-inducible protein 10 through replication-competent retrovirus vectors suppresses tumor growth. Cancer Gene Ther. 2005;12:900–912. doi: 10.1038/sj.cgt.7700854. [DOI] [PubMed] [Google Scholar]
- 49.Tang J, Murtadha M, Schnell M, Eisenlohr LC, Hooper J, Flomenberg P. Human T-cell responses to vaccinia virus envelope proteins. J Virol. 2006;80:10010–10020. doi: 10.1128/JVI.00601-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tannenbaum CS, Tubbs R, Armstrong D, Finke JH, Bukowski RM, Hamilton TA. The CXC chemokines IP-10 and Mig are necessary for IL-12-mediated regression of the mouse RENCA tumor. J Immunol. 1998;161:927–932. [PubMed] [Google Scholar]
- 51.Taub DD. Recombinant human interferon-inducible protein 10 is a chemoattractant for human monocytes and T lymphocytes and promotes T cell adhesion to endothelial cells. J Exp Med. 1993;177:1809–1814. doi: 10.1084/jem.177.6.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Terme M, Tomasello E, Maruyama K, Crepineau F, Chaput N, Flament C, Marolleau JP, Angevin E, Wagner EF, Salomon B, Lemonnier FA, Wakasugi H, Colonna M, Vivier E, Zitvogel L. IL-4 confers NK stimulatory capacity to murine dendritic cells: a signaling pathway involving KARAP/DAP12-triggering receptor expressed on myeloid cell 2 molecules. J Immunol. 2004;172:5957–5966. doi: 10.4049/jimmunol.172.10.5957. [DOI] [PubMed] [Google Scholar]
- 53.Trifilo MJ, Montalto-Morrison C, Stiles LN, Hurst KR, Hardison JL, Manning JE, Masters PS, Lane TE. CXC Chemokine ligand 10 controls viral infection in the central nervous system: evidence for a role in innate immune response through recruitment and activation of natural killer cells. J Virol. 2004;78:585. doi: 10.1128/JVI.78.2.585-594.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van de Broek I, Leleu X, Schots R, Facon T, Vanderkerken K, Van Camp B, Van Riet I. Clinical significance of chemokine receptor(CCR 1, CCR 2 and CXCR 4) expression in human myeloma cells: the association with disease activity and survival. Haematologica(Roma) 2006;91:200–206. [PubMed] [Google Scholar]
- 55.Vanguri P. IFN and virus-inducible expression of an immediate early gene, crg-2/IP-10, and a delayed gene, IA alpha in astrocytes and microglia. J Immunol. 1994;152:1411–8. [PubMed] [Google Scholar]
- 56.Vanguri P, Farber JM. Identification of CRG-2. An interferon-inducible mRNA predicted to encode a murine monokine. J Biol Chem. 1990;265:15049–15057. [PubMed] [Google Scholar]
- 57.Walzer T, Dalod M, Robbins SH, Zitvogel L, Vivier E. Natural-killer cells and dendritic cells: “l’union fait la force”. Blood. 2005;106:2252–2258. doi: 10.1182/blood-2005-03-1154. [DOI] [PubMed] [Google Scholar]
- 58.Wilson DR. Viral-mediated gene transfer for cancer treatment. Curr Pharm Biotechnol. 2002;3:151–164. doi: 10.2174/1389201023378445. [DOI] [PubMed] [Google Scholar]
- 59.Wirth T, Zender L, Schulte B, Mundt B, Plentz R, Rudolph KL, Manns M, Kubicka S, Kuhnel F. A telomerase-dependent conditionally replicating adenovirus for selective treatment of cancer. Cancer Res. 2003;63:3181–3188. [PubMed] [Google Scholar]
- 60.Yokoyama WM, Seaman WE. THE Ly-49 AND NKR-P1 gene families encoding Lectin-like receptors on natural killer cells: the NK gene complex. Annu Rev lmmunol. 1993;11:613–635. doi: 10.1146/annurev.iy.11.040193.003145. [DOI] [PubMed] [Google Scholar]
- 61.Yonemitsu Y, Kitson C, Ferrari S, Farley R, Griesenbach U, Judd D, Steel R, Scheid P, Zhu J, Jeffery PK, Kato A, Hasan MK, Nagai Y, Masaki I, Fukumura M, Hasegawa M, Geddes DM, Alton EW. Efficient gene transfer to airway epithelium using recombinant Sendai virus. Nat Biotechnol. 2000;18:970–973. doi: 10.1038/79463. [DOI] [PubMed] [Google Scholar]
- 62.Young LS, Searle PF, Onion D, Mautner V. Viral gene therapy strategies: from basic science to clinical application. J Pathol. 2006;208:299–318. doi: 10.1002/path.1896. [DOI] [PubMed] [Google Scholar]