

Abstract
Background: Data from a phase II trial combining chemoradiotherapy with interferon-alpha (IFN-α) (CapRI scheme) for adjuvant treatment of pancreatic carcinoma are very encouraging. Methods: Eight human ductal pancreatic carcinoma cell lines were treated with the CapRI scheme [5-fluorouracil (5-FU), Cisplatin, IFN-α and radiation]. Natural killer (NK) and T cells preincubated with IFN-α were tested in cytotoxicity assays against these cell lines and the mechanism of cell lysis was investigated. The induction of the immunoproteasome in tumour cells after IFN-α stimulation was analysed by immunoblot and RT-PCR. Results: IFN-α activated NK cells and increased their cytotoxicity. This cytotoxicity was mediated as well by Fas-induced apoptosis as by perforin release. Pre-treatment of tumour cells with 5-FU and combinations showed a significant increase in the susceptibility of tumour cells against NK cells. Treatment of tumour cells with IFN-α induced a switch to the immunoproteasome and enhanced their vulnerability to T cells. This is the first description of this phenomenon in pancreatic carcinoma cells with implications for their immunogenicity. Discussion: IFN-α activates NK cells against pancreatic carcinoma cells and 5-FU treatment makes tumour cells more susceptible. Furthermore, IFN-α induces the immunoproteasome with impact on the immunogenicity of pancreatic carcinoma cells. These mechanisms may be responsible for the improved clinical outcome of CapRI.
Keywords: Combination therapy, Immunoproteasome, NK cells, Fas
Introduction
Carcinoma of the exocrine pancreas has an especially poor prognosis. The 5-year survival rate is <1% with a median survival of 4–6 months. Even after surgical intervention with a curative intention, the 2-year survival rate is in specialized centres at the best 25% [23].
Investigators from the Virginia Mason Clinic have recently published data from a phase II trial where postoperative cisplatin (CDDP), 5-fluorouracil (5-FU), interferon-alpha-2b (IFN-α), and external-beam radiation (RT) was administered following pancreaticoduodenectomy. We termed this regimen CapRI for adjuvant treatment of pancreatic adenocarcinoma with chemoradioimmunotherapy. They have treated 43 patients with mainly stage III tumours. 84% had positive lymph nodes and 19% had positive resection cut margins. After a mean follow-up of 31.9 months, 67% of the patients were still alive. Actuarial overall survival for the 1-, 2-, and 5-year survival rate were 95, 64, and 55%, respectively. The treatment was quite toxic and 42% patients were hospitalized during treatment, but there were no treatment-related deaths [22].
Various chemo- and/or radiation regimens have been tested in small studies for the treatment of adjuvant resected pancreatic carcinoma. Most of them use 5-FU or gemcitabine as a chemotherapeutic agent, sometimes in combination with other agents such as CDDP. These protocols are often combined with radiation [25]. The data from the ESPAC-1 showed that radiochemotherapy has only limited success [19].
Comparing the data from CapRI and ESPAC-1, we hypothesize that IFN-α is the agent that turns a slightly effective treatment (chemoradiotherapy) into a potent therapy. Several mechanisms are described which might explain why IFN-α plays a potential role in combination therapies.
IFN-α belongs to the group of type I interferons, which are already used in cancer therapy (e.g. malignant melanoma, renal cell carcinoma, hairy cell leukaemia, and chronic myeloid leukaemia) [1, 12]. IFN-α is produced by monocytes/macrophages, lymphoblastoid cells, fibroblasts, and plasmacytoid dendritic cells [7]. Several other cell types are known to produce type I interferons after viral infections. IFN-α binds to the IFN-α/β receptor CD118; binding to the EBV-receptor CD21 is also described [4]. Binding to the IFN receptor activates the transcription of several different genes inducing the synthesis of host cell proteins that contribute to the inhibition of viral replication [5, 17].
In addition to its anti-viral properties, IFN-α exhibits several other features that might be of interest especially for the use in combination treatments of cancer, as there are (a) direct inhibitory effects on tumour cell growth [9, 21], (b) radio- and chemosensitizing effects [8, 13], (c) anti-angiogenic properties [3, 24, 28], (d) enhancement of immunogenicity of tumours (increase of MHC class I expression) [21], and (e) modulation of the immune system: IFN-α plays an essential role in the differentiation, maturation and function of dendritic cells (DC), enhances the survival of T cells by the expression of anti-apoptotic genes, induces the generation of CD8+ memory cells, enhances macrophage activities, and activates natural killer (NK) cells thus releasing cytokines [15, 16, 20, 21]. Previously, we have tested the impact of direct effects, radio- and chemosensitizing properties and anti-angiogenic features of IFN-α in the CapRI-regimen and found only moderate influence by these mechanisms [14]. Therefore, we focused in this work on the immunomodulatory aspect of IFN-α and tested the influence of IFN-α on various immunological effector cells regarding their cytotoxic properties.
Intracellular proteins are degraded to peptides by the proteasome: a large proteolytic complex whose primary function is the controlled elimination of proteins marked for degradation by ubiquitin, and then presented to cytolytic T lymphocytes (CTLs) by MHC class I molecules. Stimulation with IFN-γ induces the replacement of the three catalytic subunits of the proteasome by their homologous subunits LMP-2, MECL-1 and LMP-7, to form the so-called ‘immunoproteasomes’ (iPS). The efficiency of the modified proteasome to eliminate proteins remains unchanged, but its preferences for cleavage are modified. Several antigenic peptides, including tumour epitopes, were found to be processed differentially by the two proteasome types, thus implying a different ability to be recognized by CTLs [26]. Here, we investigated whether the incubation with IFN-α induces the switch to the immunoproteasome and its implications for immunogenicity.
Materials and methods
Preparation of T cells and NK cells
Peripheral blood mononuclear cells (PBMC) were isolated from buffy coats by Ficoll density gradient centrifugation. The cells were allowed to adhere in six-well plates at a density of 5×106 cells/ml for 1 h at 37°C RPMI 1640 medium supplemented with 10% foetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. They were maintained at 37°C under 5% CO2 in a humidified atmosphere. Non-adherent cells (PBL) were further purified:
T cells were isolated using sheep red blood cells (SRBC from the animal farm of the Heidelberg University) for rosetting. In brief, 106 PBL/ml were mixed 10:1 with neuraminidase-treated SRBC and centrifuged at 400g for 5 min. After 1 h incubation on ice to form E rosettes, the cells were gently resuspended and a Ficoll density gradient centrifugation was performed. The erythrocytes in the pelleted fraction were lysed with NH4Cl solution.
NK cells were enriched using the MACS technique according to the manufacturer’s instructions. In brief, cells were labelled with anti-CD56 beads and enriched on an LS column. The purity was between 90 and 95%. Effector cells were stimulated with 1,000 U/ml IFN-α over night when indicated.
Treatment of pancreatic cancer cell lines
Human pancreatic cancer cell lines ASPC-1, CAPAN-2, DAN-G, KciMoh-1, MiaPaCa-1, PANC-1, PatScl, and PK 9 were cultured in RPMI 1640 medium supplemented with 10% foetal bovine serum. Tumour cells were treated when indicated either with 5-FU 65 μg/ml continuously, days 1–5; CDDP 3 μg/ml on day 1, for 60 min; radiation 1.8 Gy/day, day 1–5; IFN-α 1,000 U/ml, days 1, 3, and 5; CR consisting of 5-FU, CDDP and radiation or the entire combination therapy (CapRI).
MTT assay for cell proliferation
The non-radioactive proliferation assay “EZ4U” kit (Biomedica, Vienna, Austria) was used according to the manufacturer’s instructions. Tumour cells were seeded at a density of 2,000 cells per well in triplicate in a 96-well plate. The proliferation rate was determined after 5 h of incubation; the absorbance of a medium blank was subtracted. The proliferation index was calculated by setting the proliferation of untreated cells to 1.
Determination of apoptosis, cell death, and cell numbers
Cells were investigated on day five of treatment. Apoptotic cells were detected on an Epics®XL. MCL flow cytometer (Beckman-Coulter, Krefeld, Germany) by AnnexinV staining using AnnexinV/PI staining. Cells were analysed immediately after staining. Late apoptosis was defined as Annexin V/PI double positive cells. Unstained cells were used to set up controls. Cell numbers of viable cells were determined by Trypan blue exclusion.
Flow cytometric analysis
Human pancreatic cancer cell lines and lymphocytes were stained using monoclonal antibodies directed against human HLA-ABC, HLA-DR, Fas, FasL, CD3, CD16 and CD56, CD19, CD21 and CD118 (IFN-receptors). For each flow cytometry measurement, gates for negative control were set to less than 2.0%. Data from at least 10,000 cells were collected and analysed. Negative controls consisted of the cells labelled with a corresponding isotype control. Mean fluorescence was normalized to mean fluorescence of the control antibodies.
Cytotoxicity assay
A standard chromium release assay was used to determine the cytotoxic activity as described elsewhere [21].
Mechanism of induction of apoptosis
Effector cells (2.5×106 in 2.5 ml of RPMI-1640) were pre-treated for 2 h at 37°C with either 100 nM of CMA, which inhibits perforin-based cytotoxic activity due to accelerated degradation of perforin by an increase in the pH of lytic granules, or 10 μM of BFA which is an inhibitor of intracellular glycoprotein transport which selectively inhibits Fas-mediated cytotoxicity [10]. The cells were washed three times and analysed in a cytotoxicity assay.
Cytotoxicity assay for pre-treated tumour cells
Tumour cells pre-treated with chemotherapy could not be investigated in a standard chromium release assay because they showed a too high spontaneous release of chromium or were not suitable for labelling. Therefore, we developed a flow cytometric based cytotoxicity assay. Treated and untreated tumour cells were stained either with PKH-2 or PKH-26 mixed 1:1 and incubated over 4 h with effector cells at an E:T of 80:1. The percentage of red and green tumour cells was then analysed by flow cytometry. Since the tumour cell lysis of untreated tumour cells at this given effector to target cell ratio is known, the approximately cell lysis of pre-treated tumour cells could be calculated using the following formula:
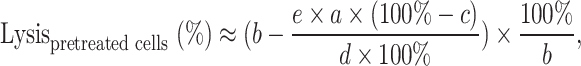 |
where a is the percentage of untreated cells in control mix (without PBL) (%), b, the percentage of treated cells in control mix (without PBL) (%), c, the known lysis of untreated cells at given E:T ratio (from 51Cr-release assay) (%), d, the percentage of remaining untreated cells after 4-h incubation with PBL (%), e, the percentage of remaining treated cells after 4-h incubation with PBL (%); in which
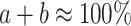 and
and
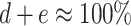 .
.
Granzyme B ELISpot
The Human Granzyme B ELISpot Set was used according to the manufacturer’s instructions. NK cells or PBL with and without IFN-α stimulation were seeded at a density of 45,000 cells per well in quadruplicates in the provided 96 well plates. Tumour cells were seeded at a density of 560 cells per well and co-cultured with the effector cells for 4 h. Spots were counted using the KS ELISPOT System, release 4.1 (Carl Zeiss Light Microscopy, Göttingen, Germany). Wells without cells, effector cells alone and tumour cells alone served as controls and were subtracted from the results.
RNA extraction and RT-PCR
Tumour cells numbering 3×106 were incubated with RNAlater and stored at −20°C until RNA preparation. RT-PCR was performed using QIAGENT® one-step RT-PCR kit according to the manufacturer’s protocol. Sense and antisense primers used for PCR amplifications were as follows: hß-actin: 5′-ACT CCA TCA TGA AGT GTG ACG-3′ and 5′-CAT ACT CCT GCT TGC TGA TCC-3(249 bp); hLMP-2: 5′-CGT TGT GAT GGG TTC TGA TTC C-3′ and 5′-GTT CAT TGC CCA AGA TGA CTC G-3′ (547 bp); hLMP-7: 5′-AAT GCA GGC TGT ACT ATC TGC G-3′ and 5′-TGC AGC AGG TCA CTG ACA TCT G-3′ (406 bp) and h-MECL-1: 5′-CCT TCG AGA ACT GCC AAA GAA ATG C-3′ and 5′-CAA GCT CTA AGC CTC AGC TTA CTC C-3′ (802 bp). PCR was performed under the following conditions: 94°C for 30 s (denaturation); 55 (hß-actin), 60 (hLMP-2 and hLMP-7) or 62°C (hMECL-1) for 30 s (annealing) and 72°C for 30 s (elongation) followed by 7 min at 72°C for 30 cycles. The PCR products were separated on a 1.5% agarose gel containing 0.5 μg/ml ethidium bromide and photographed under UV light.
Immunoblot analysis of PS subunits
Tumour cells numbering 3×106 were resuspended in 200 μl ice-cold lysis buffer (10 mM imidazole pH 7.3, 0.5 M NaCl, 1% Triton X-100, 0.2 mM sodium ortho-vanadate, 0.2 mM PMSF, 2 mM sodium azide) and incubated for 30 min on ice. The solution was then centrifuged with 13,000 rcf for 30 min at 4°C. The amount of protein in the supernatant was determined using the Bicinchoninic Acid Kit for Protein Determination according to the manufacturer’s instructions.
Ten micrograms of each total cell lysate was mixed with electrophoresis sample buffer and separated by SDS-PAGE followed by transfer onto a nitrocellulose membrane (0.2 μm). A Ponceau stain was performed to confirm that equal quantities of proteins were loaded. Membranes were soaked overnight in PBS containing 5% milk and 0.1% Tween 20 at 4°C, and were then probed for 2 h by gentle shaking at 4°C with the specific antibody in PBS containing 1% milk and 0.1% Tween 20 at the dilution indicated by the manufacturer. Rabbit polyclonal antisera, PW8350, PW8345 and PW8355, directed against the iPS subunits MECL-1, LMP-2, and LMP-7 were used (Affiniti Research Products, Mamhead, UK). After extensive washing with PBS/1% Tween 20, membranes were incubated for 1 h in the dark at room temperature with horseradish peroxidase-conjugated monoclonal anti-rabbit (1:10,000) antibody. Following extensive washing with PBS/1% Tween 20, the enzyme substrate was added and allowed to react in the dark for 5 min. Chemiluminescence was detected with an autoradiographic film.
Statistical analysis
Non-parametrical analysis (Wilcoxon test) and paired t test on SPSS 11.5 were used to analyse statistical significance where appropriate. A P-value <0.05 was considered as significant.
Results
Influence of IFN-α on lymphocytes
As known, IFN-α showed a proliferative effect on peripheral blood lymphocytes (PBL) from healthy donors. After an over-night incubation with IFN-α the cell number increased about 13.3±8.8% compared to a decrease for untreated cells of 17.9±5.9% (P<0.05). Stimulated PBL showed a significant increase in FasL expression (42.0±6.9% vs. 64.0±20.1%; P<0.01; data not shown).
Next, we tested the cytotoxic activity of IFN-α-stimulated PBL against eight human pancreatic carcinoma cell lines. PBL from healthy donor showed no remarkable cytotoxicity (12.5±6.3% at an effector to target ratio of 80:1) whereas PBL preincubated with IFN-α showed an enhanced cytotoxicity (34.3±5.5%; P<0.02, Fig. 1).
Fig. 1.

Cytotoxicity of PBL after IFN-α stimulation PBL from healthy donors were stimulated over night with IFN-α and tested for their cytotoxic activity against pancreatic carcinoma cell lines at different E:T ratios in a standard chromium release assay. Data is shown as mean±standard error from eight cell lines each with at least three separate experiments. An asterisk indicates statistical significance (P<0.05)
We purified T cells from PBL to investigate which subpopulation was mainly involved in killing. 83.5±5.3% of the enriched T cell fraction was positive for CD3, whereas the negative fraction consisted of 62.1±8.1% CD16+ cells; 59.7±2.2% CD56+ cells; 35.4±8.1% NKG2D+ cells; 44.2±6.2% CD161+ cells; 24.2±6.9% CD19+ cells and 3.7±0.7% CD3+ cells. The cells in the negative fraction expressed to 45.9±8.9% the IFN-receptor CD118 (data not shown).
T cells with or without IFN-α stimulation failed completely to lyse pancreatic tumour cells (lysis <5%) while T cell-depleted lymphocytes (i.e. mostly NK cells) killed without any stimulation 23.0±25.9% of the tumour cells (E:T ratio 80:1; Fig. 2). After preincubation with IFN-α, the killing rate was 42.7±31.6% of the tumour cells.
Fig. 2.

Cytotoxicity of immunological subpopulations against tumour cells PBL from healthy donors were separated into a T cell and a non-T cell fraction by a rosetting procedure and stimulated over night with IFN-α. Cytotoxic activity was determined in a standard chromium release assay. Data is shown as mean±standard error from eight cell lines each with at least three separate experiments. An asterisk indicates statistical significance (P<0.05)
Analysis of killing mechanism
NK cells and PBLs were investigated in a Granzyme B assay. NK cells were enriched via CD56 expression by MACS-technique (purity 87.4±3.6%, data not shown) and tested together with PBL against six untreated pancreatic carcinoma cell lines (ratio 80:1). PBL with or without IFN-α prestimulation showed no real detectable increase in Granzyme B release (0.9 vs. 1.8 spots/45,000 cells). NK cells even without stimulation showed after co-culture with tumour cells, a significant increase in Granzyme B release compared to PBL (2.8±1.0 spot, P<0.03; Fig. 3a). A strong increase in numbers of spots could be achieved with IFN-α stimulated NK cells (41.2±6.0 spots/45,000 cells; P<0.001).
Fig. 3.

a, b Analysis of the killing mechanism: a NK cells and PBL with and without IFN-α stimulation were seeded out in quadruplicates. Human untreated pancreatic cancer cells were added and incubated for 4 h. Data are shown as mean±standard error from six cell lines. An asterisk indicates statistical significance (P<0.05) three asterisks indicate a P<0.001. b The pathway of apoptosis induced by effector cells was investigated by the degradation of perforin-containing granules or selectively inhibition of Fas-mediated cytotoxicity. Effector cells were incubated either with Concanamycin A (CMA) or Brefeldin A (BMA) before they were used in a 51Cr-release assay. Panc 1 cells were used as targets. Data is shown as mean±standard error from three separate experiments. An asterisk indicates statistical significance (P<0.05)
Furthermore, the mechanism of apoptosis induced by effector cells was investigated by use of Concanamycin A (CMA) and Brefeldin A (BFA). CMA is an inhibitor of vacuolar type H+-ATPase which inhibits perforin-based cytotoxic activity, mostly due to accelerated degradation of perforin by an increase in the pH of lytic granules which does not affect Fas-dependent cytototoxicity. BFA is an inhibitor of intracellular glycoprotein transport which selectively inhibits Fas-mediated cytotoxicity. Inhibition of Fas-mediated cytotoxicity in T cell-depleted lymphocytes resulted in a statistically significant decrease of cytotoxicity. The cell lysis at an effector to target ratio of 80:1 dropped from 51.3±8.1 to 8.8±1.3%, P<0.02. Degradation of perforin release showed as well an influence on the cytotoxic activity of T cell-depleted PBL (21.2±3.9% lysis at an E:T ratio of 80:1); however, this was not significant (P=0.06; Fig. 3b).
Cytotoxic activity of IFN-α pre-treated lymphocytes against chemotreated tumour cells
Next, we analysed if pre-treatment with chemo- and/or radiotherapy enhances the susceptibility of tumour cells against immunological effector cells. Pre-treated tumour cells were incubated with IFN-α stimulated PBL and cytolysis was determined in a flow-cytometry-based cytotoxicity assay. Treatment with IFN-α, CDDP and/or radiation did not influence the susceptibility of tumour cells, whereas pre-treatment with 5-FU or the whole CapRI-scheme led to an increase of cytolysis from 34% up to approximately 70% (P<0.001; Fig. 4a).
Fig. 4.

a/b Cytotoxicity against chemo-radiotherapy-pre-treated tumour cells: a Tumour cells were pre-treated as indicated over 5 days. PBL from healthy donors were stimulated over night with IFN-α and tested for their cytotoxic activity against pre-treated pancreatic carcinoma cell lines at an effector to target ratio of 80:1. Cytolysis was investigated as described in Materials and methods. Data are shown as mean±standard error from eight cell lines each with at least three separate experiments. Three asterisks indicate statistical significance of P<0.001. b Tumour cells were pre-treated over 5 days with 5-FU. NK cells and T cells from healthy donors were stimulated over night with IFN-α and tested for their cytotoxic activity at an effector to target ratio of 80:1. Cytolysis was investigated as described in Materials and methods. Data are shown as mean±standard error from two cell lines (Capan-2, DAN-G) each with three separate experiments. Three asterisks indicate statistical significance of P<0.001
Subsequently, we investigated in Capan-2 and DAN-G cells if 5-FU treatment makes them more susceptible to any specific subpopulation. Again, 5-FU treatment makes them more vulnerable against IFN-α stimulated PBL (30% vs. 63% at an effector to target ratio of 80:1). Interestingly, we found a highly significant increase in cytotoxicity after the 5-FU treatment for NK cells (from 37 to 69%) as well as for T cells. However, the increase in T cell-mediated cytolysis was much more impressive (from 3 to 48%; Fig. 5b).
Fig. 5.

Impact of IFN-α on the immunogenicity of tumour cells. Mean fluorescence of HLA expression on tumour cells after 5 days of IFN-α stimulation with a representative overlay of flow cytometric analysis (DAN-G cells). b Fas and FasL expression on tumour cells after 5 days of IFN-α stimulation or cytostatic/radiation treatment. Data are shown as mean±standard error from eight cell lines each with at least three separate experiments. An asterisk indicates statistical significance (P<0.05)
After the addition of CMA, the cell lysis dropped from 63 to 36% (P<0.01). Inhibition of Fas-based cytotoxicity resulted in the reduction of cytolysis down to 44% (P<0.01; data not shown), cell lysis of untreated cells was 40%. Since we had shown that Fas-mediated cytotoxicity is mainly responsible for lysis of untreated tumour cells, we conclude that the gain in cytolysis after treatment with 5-FU is induced by an increased vulnerability against perforin.
Influence of IFN-α on the immunogenicity of pancreatic carcinoma cells
Seven out of eight investigated tumour cells were even in their untreated status nearly to 100% positive for MHC class I expression. Therefore, it was not possible to investigate if IFN-α was able to affect the percentage of positive cells. However, tumour cells showed a significant increase in mean fluorescence of MHC class I (4.9±4.8 vs. 13.30±7.0; P<0.03; Fig. 5) after IFN-α treatment. Mean fluorescence of MHC class II was not significantly enhanced.
No treatment showed any significant effect on Fas expression on tumour cells, although there was a tendency towards down regulation after 5-FU treatment (30±26 vs. 12±16). FasL expression was down regulated after 5-FU treatment; this was significant for treatment with the complete CapRI-scheme (62±26% vs. 28±21%; P<0.05; Fig. 5b).
Induction of the immunoproteasome
Treatment of tumour cell lines with IFN-γ induces the switch from proteasome to immunoproteasome, which has profound implications for the immunogenicity of tumour cells. Here, we investigated whether the iPS is induced in pancreatic tumour cells after IFN-α treatment. Eight pancreatic carcinoma cells were treated with IFN-α and additional chemo- and/or radiotherapy and the iPS were detected in a non-quantitative way on mRNA- and protein level (Fig. 6a, b).
Fig. 6.

a/b Detection of subunits of the immunoproteasome: Tumour cells were treated as described. a The subunits LMP-2, LMP-7, MECL-1 of the immunoproteasome were detected by immunoblot and b also detected by RT-PCR. 1 untreated cells, 2 IFN-α treated cells, 3 after 5-FU and IFN-α-treatment, 4 treatment with CDDP + IFN-α, 5 treatment with RT + IFN-α, 6 CapRI-treatment, 7 incubation with IFN-γ (positive control), and 8 T2 cells (negative control). The figure shows representative plots from ASPC cells. Eight cell lines each with at least two separate experiments were investigated
The subunit LMP-7 could be detected in nearly all cell lines both on mRNA level and on protein level, even in the untreated situation. As all three subunits are needed to form the iPS, we looked for co-expression of LMP-2, LMP-7, and MECL-1. This was found on mRNA level for all cell lines after IFN-γ treatment (positive control) but not on T2 cells which are genetically defective for iPS (negative control). Six out of eight cell lines showed a switch to the iPS after IFN-α; CDDP + IFN-α; radiation + IFN-α and the whole CapRI-treatment. For combined treatment with 5-FU and IFN-α, the results were not equally pronounced, the bands were weaker and only two out of eight cell lines were positive for all three subunits (Fig. 6b). The weaker expression after treatment with IFN-α and 5-FU containing regimens has to be analysed carefully as 5-FU showed a strong cytotoxic effect. After 5 days of treatment, the RNA synthesis has already stopped (Fig. 6b) but protein is still detectable at a lower level (Fig. 6b).
On protein level, the situation was similar. Interestingly, the number of positive cell lines was higher compared to the results from RT-PCR. All cell lines were positive after IFN-α; CDDP + IFN-α; radiation + IFN-α and whole CapRI-treatment and six out of eight were positive after 5-FU + IFN-α treatment (Fig. 6a).
Cytotoxic activity of T cells against IFN-α treated tumour cells
Since we saw a switch to the immunoproteasome in tumour cells after IFN-α stimulation, we analysed if IFN-α pre-treatment enhances the susceptibility of tumour cells against HLA-matched T cells. IFN-α pre-treated tumour cells were incubated with IFN-α stimulated T cells and cytolysis was determined in a standard chromium-release assay. T cells failed completely to lyse untreated pancreatic carcinoma cells (0.1±4.2%) while IFN-α pre-treated pancreatic carcinoma cells were moderately susceptible (14.3±2.5% at an E:T ratio of 80:1; P<0.001; Fig. 7).
Fig. 7.

Cytotoxicity of stimulated T cells against IFN-α treated tumour cells. T cells from a healthy donor (HLA A2+) were separated and stimulated over night with IFN-α. HLA-matched tumour cells (Panc1, DAN-G) were stimulated with IFN-α as described. T cells were tested for their cytotoxic activity against stimulated and unstimulated pancreatic carcinoma cell lines. Data are shown as mean±standard error from two cell lines each with at least three separate experiments. Three asterisks indicate statistical significance of P<0.001
Discussion
IFN-α is well known to stimulate the immune system by several strategies. Aside from direct activating effects on immunological effector cells, such as cytotoxic T cells and NK cells, IFN-α induces the differentiation and maturation of DC and enhances the immunogenicity of tumour cells by up regulation of MHC molecules. IFN-α also enhances the survival of T cells, and it is important for the proliferation and long-term survival of antigen-specific T cells [1]. According to these references, we saw an increased proliferation rate of PBL after IFN-α stimulation. More interestingly, we saw a significant increase in cytotoxic activity against pancreatic carcinoma cell lines after one single over-night IFN-α stimulation. Tumour cells which are normally resistant against PBL could be lysed by IFN-α stimulated PBL. The cytotoxic activity came mainly from NK cells and was mediated as well by Fas-induced apoptosis (Fig. 3b) as by perforin release (Fig. 3a). This activity of NK cells was surprising to us as most of our target cells are strong MHC class I positive. We hypothesize that we have a mismatch between MHC class I and KIR resulting in no or reduced inhibition in our allo-system. Another explanation is the strong expression of NKG2D-ligands on our pancreatic carcinoma cell lines compensating the inhibition. However, metastases from primary pancreatic carcinoma are known to down regulate MHC class I. displaying classical NK cell targets.
Combination therapies as the CapRI protocol seem to be superior to mono-therapies not only through the addition of different strategies but also just by synergistic effects. Interestingly, we observed that the treatment with 5-FU makes tumour cells more vulnerable to the attacks from IFN-α stimulated immune cells. These observations underline that the whole is much more than the sum of the parts in combination therapy.
Furthermore, we saw in accordance with the published data that the IFN-α treatment enhances the immunogenicity of tumour cells. The density of MHC molecules on tumour cells increased significantly after IFN-α treatment. Moreover, immune cells expressed more Fas ligand (FasL) after IFN-α stimulation while FasL expression on tumour cells was unaffected by IFN-α and decreased after 5-FU treatment. Fas expression on tumour cells was not significantly affected, although there was a strong tendency towards down regulation of Fas after 5-FU treatment. In the counterattack model of tumorigenesis, it has been proposed that tumours develop resistance to attack from FasL expressing cytotoxic T cells by down regulating Fas (immune escape), while at the same time up regulating FasL expression to induce apoptosis in Fas-expressing T cells [11]. For these cell lines, we cannot support this model—just in contrast—we observed an increase in FasL expression on immune cells and therefore an enhanced ability of immune cells to kill via the Fas pathway. Decreasing apoptosis in the effector cells due to the down regulation of FasL on the surface of pre-treated target cells could be one possible explanation for the observed enhanced susceptibility of tumour cells after 5-FU treatment. Supporting the importance of Fas regulation, we were able to show that T cells kill IFN-α pre-treated tumour cells by using the Fas/FasL pathway.
Treatment of pancreatic carcinoma cells with IFN-α resulted in most of the investigated cell lines in a switch to the immunoproteasome. This is the first description of this phenomenon in pancreatic carcinoma cells with implications for their immunogenicity. Chen et al. [2] reported that the immunoproteasome plays an important role in determining the immunodominance hierarchy so that it has direct impact on CD8+ T cell responses by modifying the repertoire of responding CD8+ T cells. Furthermore, a number of peptides that are poorly processed by the standard proteasome are described to be more effectively produced in the presence of the immunoproteasome [26]. Nevertheless, at this moment, several known tumour epitopes which are recognized by autologous CTLs on tumour cells are described to be poorly presented after IFN-γ induced switch to the immunoproteasome [18]. Presently it is discussed that the immunoproteasome is more competent at producing class-I binding peptides but with exception to the majority of epitopes derived from self-proteins. However, there are also several reports from tumour epitopes which are better processed by cells carrying iPS [2, 17, 25]. Here, we observed an increase in T cell-mediated cytolysis after IFN-α treatment of pancreatic carcinoma cells. Therefore, we hypothesize that after the switch to the immunoproteasome, the peptides presented on MHC class I molecules on pancreatic carcinoma cells are more immunogenic than the normal one. In general, T cells seem to be able to enter pancreatic carcinoma though the infiltrating cells often show a reduced activation status [6, 27]. Here, we could show that although IFN-α stimulation of T cells alone failed to induce cytolytic activity, IFN-α pre-treated tumour cells are getting vulnerable to T cells.
In conclusion, NK cells could be activated by IFN-α against pancreatic carcinoma cells, and pancreatic carcinoma cells are more susceptible against immunological attacks after 5-FU treatment. Furthermore, IFN-α induces the immunoproteasome in pancreatic carcinoma cells which are then more immunogenic for T cells. As IFN-α is described to have direct effects on tumour cells and acts also synergistic with chemoradiation (CR), we could presume a complex network of enhancing interactions between the different agents used in the CapRI scheme.
Footnotes
Jan Schmidt, Emilia M. Patrut, and Jianhua Ma have contributed equally
References
- 1.Belardelli F, Ferrantini M, Proietti E, Kirkwood JM. Interferon-alpha in tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002;13:119. doi: 10.1016/S1359-6101(01)00022-3. [DOI] [PubMed] [Google Scholar]
- 2.Chen W, Norbury CC, Cho Y, Yewdell JW, Bennink JR. Immunoproteasomes shape immunodominance hierarchies of antiviral CD8(+) T cells at the levels of T cell repertoire and presentation of viral antigens. J Exp Med. 2001;193:1319. doi: 10.1084/jem.193.11.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Decatris M, Santhanam S, O’Byrne K. Potential of interferon-alpha in solid tumours: part 1. BioDrugs. 2002;16:261. doi: 10.2165/00063030-200216040-00003. [DOI] [PubMed] [Google Scholar]
- 4.Delcayre AX, Salas F, Mathur S, Kovats K, Lotz M, Lernhardt W. Epstein Barr virus/complement C3d receptor is an interferon alpha receptor. EMBO J. 1991;10:919. doi: 10.1002/j.1460-2075.1991.tb08025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Domanski P, Witte M, Kellum M, Rubinstein M, Hackett R, Pitha P, Colamonici OR. Cloning and expression of a long form of the beta subunit of the interferon alpha beta receptor that is required for signaling. J Biol Chem. 1995;270:21606. doi: 10.1074/jbc.270.27.15974. [DOI] [PubMed] [Google Scholar]
- 6.Emmrich J, Weber I, Nausch M, Sparmann G, Koch K, Seyfarth M, Lohr M, Liebe S. Immunohistochemical characterization of the pancreatic cellular infiltrate in normal pancreas, chronic pancreatitis and pancreatic carcinoma. Digestion. 1998;59:192. doi: 10.1159/000007488. [DOI] [PubMed] [Google Scholar]
- 7.Gutterman JU. Cytokine therapeutics: lessons from interferon alpha. Proc Natl Acad Sci USA. 1994;91:1198. doi: 10.1073/pnas.91.4.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holsti LR, Mattson K, Niiranen A, Standertskiold-Nordenstam CG, Stenman S, Sovijarvi A, Cantell K. Enhancement of radiation effects by alpha interferon in the treatment of small cell carcinoma of the lung. Int J Radiat Oncol Biol Phys. 1987;13:1161. doi: 10.1016/0360-3016(87)90189-1. [DOI] [PubMed] [Google Scholar]
- 9.Iacopino F, Ferrandina G, Scambia G, Benedetti-Panici P, Mancuso S, Sica G. Interferons inhibit EGF-stimulated cell growth and reduce EGF binding in human breast cancer cells. Anticancer Res. 1996;16:1919. [PubMed] [Google Scholar]
- 10.Kataoka T, Shinohara N, Takayama H, Takaku K, Kondo S, Yonehara S, Nagai K. Concanamycin A, a powerful tool for characterization and estimation of contribution of perforin- and Fas-based lytic pathways in cell-mediated cytotoxicity. J Immunol. 1996;156:3678. [PubMed] [Google Scholar]
- 11.Kim R, Emi M, Tanabe K, Uchida Y, Toge T. The role of Fas ligand and transforming growth factor beta in tumor progression: molecular mechanisms of immune privilege via Fas-mediated apoptosis and potential targets for cancer therapy. Cancer. 2004;100:2281. doi: 10.1002/cncr.20270. [DOI] [PubMed] [Google Scholar]
- 12.Kirkwood J. Cancer immunotherapy: the interferon-alpha experience. Semin Oncol. 2002;29:18. doi: 10.1053/sonc.2002.33078. [DOI] [PubMed] [Google Scholar]
- 13.Kurzrock R, Talpaz M, Guttermann J. Interferons: clinical applications. Philadelphia: Lippincott; 1991 . [Google Scholar]
- 14.Ma J, Patrut E, Schmidt J, Knaebel H-P, Büchler M, Märten A. Synergistic effects of Interferon-alpha in combination with Chemoradiation on human pancreatic adenocarcinoma. World J Gastroenterol. 2005;11(10):1521–1528. doi: 10.3748/wjg.v11.i10.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marrack P, Kappler J, Mitchell T. Type I interferons keep activated T cells alive. J Exp Med. 1999;189:521. doi: 10.1084/jem.189.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matikainen S, Sareneva T, Ronni T, Lehtonen A, Koskinen PJ, Julkunen I. Interferon-alpha activates multiple STAT proteins and upregulates proliferation-associated IL-2Ralpha, c-myc, and pim-1 genes in human T cells. Blood. 1999;93:1980. [PubMed] [Google Scholar]
- 17.Mogensen KE, Lewerenz M, Reboul J, Lutfalla G, Uze G. The type I interferon receptor: structure, function, and evolution of a family business. J Interferon Cytokine Res. 1999;19:1069. doi: 10.1089/107999099313019. [DOI] [PubMed] [Google Scholar]
- 18.Morel S, Levy F, Burlet-Schiltz O, Brasseur F, Probst-Kepper M, Peitrequin AL, Monsarrat B, Van Velthoven R, Cerottini JC, Boon T, Gairin JE, Van den Eynde BJ. Processing of some antigens by the standard proteasome but not by the immunoproteasome results in poor presentation by dendritic cells. Immunity. 2000;12:107. doi: 10.1016/S1074-7613(00)80163-6. [DOI] [PubMed] [Google Scholar]
- 19.Neoptolemos J, Stocken D, Friess H, Bassi C, Dunn J, Hickey H, Beger H, Fernandez-Cruz L, Dervenis C, Lacaine F, Falconi M, Pederzoli P, Pap A, Spooner D, Kerr D, Büchler M. The final results of the European Study Group for Pancreatic Cancer randomized controlled trial of adjuvant chemoradiotherapy and chemotherapy in patients with resectable pancreatic canecr. N Engl J Med. 2004;350:1200. doi: 10.1056/NEJMoa032295. [DOI] [PubMed] [Google Scholar]
- 20.Paquette RL, Hsu NC, Kiertscher SM, Park AN, Tran L, Roth MD, Glaspy JA. Interferon-alpha and granulocyte-macrophage colony-stimulating factor differentiate peripheral blood monocytes into potent antigen-presenting cells. J Leukoc Biol. 1998;64:358. doi: 10.1002/jlb.64.3.358. [DOI] [PubMed] [Google Scholar]
- 21.Pfeffer LM, Dinarello CA, Herberman RB, Williams BR, Borden EC, Bordens R, Walter MR, Nagabhushan TL, Trotta PP, Pestka S. Biological properties of recombinant alpha-interferons: 40th anniversary of the discovery of interferons. Cancer Res. 1998;58:2489. [PubMed] [Google Scholar]
- 22.Picozzi VJ, Kozarek RA, Traverso LW. Interferon-based adjuvant chemoradiation therapy after pancreaticoduodenectomy for pancreatic adenocarcinoma. Am J Surg. 2003;185:476. doi: 10.1016/S0002-9610(03)00051-5. [DOI] [PubMed] [Google Scholar]
- 23.Raraty MG, Magee CJ, Ghaneh P, Neoptolemos JP. New techniques and agents in the adjuvant therapy of pancreatic cancer. Acta Oncol. 2002;41:582. doi: 10.1080/028418602321028184. [DOI] [PubMed] [Google Scholar]
- 24.Solorzano CC, Hwang R, Baker CH, Bucana CD, Pisters PW, Evans DB, Killion JJ, Fidler IJ. Administration of optimal biological dose and schedule of interferon alpha combined with gemcitabine induces apoptosis in tumor-associated endothelial cells and reduces growth of human pancreatic carcinoma implanted orthotopically in nude mice. Clin Cancer Res. 2003;9:1858. [PubMed] [Google Scholar]
- 25.Tsai JY, Iannitti DA, Safran H. Combined modality therapy for pancreatic cancer. Semin Oncol. 2003;30:71. doi: 10.1016/S0093-7754(03)00273-2. [DOI] [PubMed] [Google Scholar]
- 26.Van den Eynde BJ, Morel S. Differential processing of class-I-restricted epitopes by the standard proteasome and the immunoproteasome. Curr Opin Immunol. 2001;13:147. doi: 10.1016/S0952-7915(00)00197-7. [DOI] [PubMed] [Google Scholar]
- 27.von Bernstorff W, Voss M, Freichel S, Schmid A, Vogel I, Johnk C, Henne-Bruns D, Kremer B, Kalthoff H. Systemic and local immunosuppression in pancreatic cancer patients. Clin Cancer Res. 2001;7:925s. [PubMed] [Google Scholar]
- 28.Wang L, Wu WZ, Sun HC, Wu XF, Qin LX, Liu YK, Liu KD, Tang ZY. Mechanism of interferon alpha on inhibition of metastasis and angiogenesis of hepatocellular carcinoma after curative resection in nude mice. J Gastrointest Surg. 2003;7:587. doi: 10.1016/S1091-255X(03)00072-6. [DOI] [PubMed] [Google Scholar]