

Abstract
Background
Alloreactive T-cell responses are known to result in the production of large amounts of proinflammatory cytokines capable of activating and maturing dendritic cells (DC). However, it is unclear whether these allogeneic responses could also act as an adjuvant for concurrent antigen-specific responses.
Objective
To examine effects of simultaneous alloreactive and antigen-specific T-cell responses induced by semi-allogeneic DC.
Methods
Semi-allogeneic DC were generated from the F1 progeny of inbred strains of mice (C57BL/6 and C3H, or C57BL/6 and DBA). We directly primed antigen-specific CD8+ and CD4+ T-cells from OT-I and OT-II mice, respectively, in the absence of allogeneic responses, in vitro, and in the presence or absence of alloreactivity in vivo.
Results
In vitro, semi-allogeneic DC cross-presented ovalbumin (OVA) to naïve CD8+ OT-I transgenic T-cells, primed naïve CD4+ OT-II transgenic T-cells and could stimulate strong alloreactive T-cell proliferation in a primary mixed lymphocyte reaction (MLR). In vivo, semi-allogeneic DC migrated efficiently to regional lymph nodes but did not survive there as long as autologous DC. In addition, they were not able to induce cytotoxic T-lymphocyte (CTL) activity to a target peptide, and only weakly stimulated adoptively transferred OT-II cells. The CD4+ response was unchanged in allo-tolerized mice, indicating that alloreactive T-cell responses could not provide help for concurrently activated antigen-specific responses. In an EL4 tumour-treatment model, vaccination with semi-allogeneic DC/EL4 fusion hybrids, but not allogeneic DC/EL4 hybrids, significantly increased mouse survival.
Conclusion
Expression of self-Major histocompatibility complex (MHC) by semi-allogeneic DC can cause the induction of antigen-specific immunity, however, concurrently activated allogeneic bystander responses do not provide helper or adjuvant effects.
Keywords: Semi-allogeneic, Dendritic cell vaccination, Adjuvant, DC fusion, Antigen specific T-cell, Alloreactivity
Introduction
Advances in the generation of large numbers of dendritic cells (DC) from blood monocytes, stem cells or bone marrow, allowed DC to be generated and directly loaded with antigen ex vivo, and induced powerful antigen-specific immune responses upon reintroduction into the patient [1–3, 42]. Increasingly, however, the importance of DC maturation in vaccination strategies has been realized [12, 14, 25, 31], with the use of immature DC in vaccination strategies highlighted as a cause of antigen-specific inhibition of immune responses in both humans and mice [13, 23]. In addition, it has been shown in mice that immature DC, when matured in vivo in adjuvant-pre-treated sites, were more effective at inducing anti-tumour immunity than DC matured ex vivo [33].
Recent studies have found that the proinflammatory cytokines IL-1β, TNF-α, IL-6 and IFN-γ were released by T-cells during the course of an allo-immune response, and that the combination of these cytokines led to the activation and maturation of bystander DC in humans [27, 39]. In response to alloreactive T-cell-derived cytokines, it was shown that the costimulatory molecules CD80, CD83, CD86 and CD40, chemokine receptor CCR7, adhesion molecule ICAM-1, DC-lysosomal associated membrane protein (DC-LAMP) and HLA-DR were all up-regulated [27, 39]. These studies suggest that the proinflammatory cytokines induced by direct allorecognition could aid concurrently activated, antigen-specific T-cell responses to exogenous antigens or indirectly presented alloantigens. It has been suggested that CD40L expression by activated alloreactive CD4+ T-cells [7, 9] could condition DC via CD40 and enable DC to facilitate antigen-specific CD8+ T-cell priming. This would bypass the need for simultaneous presentation of tumour peptides by both Major histocompatibility complex (MHC) classes I and II molecules and increase the proportion of cancer patients in whom a cytotoxic CD8+ T-cell response would be possible [15, 17]. This form of T-cell help could prove particularly potent as it has been estimated that between 1 and 10% of a host’s T-cells are potentially alloreactive [37]. All these studies suggest that the presence of a concurrent allo-response could induce a “cytokine storm” conducive for the maturation of immature DC in vivo. Such a micro-environment would be ideal for the maturation of DC as part of an immunotherapy strategy, if the response by T-cells could be shown to remain tumour antigen-specific.
In this study, we determined whether it would be possible to generate antigen-specific immune responses using semi-allogeneic (partially MHC-matched) DC, thus providing a source of allo-derived cytokines and co-stimulation to help drive a concurrent antigen-specific immune response. This would replicate the partial mismatch situation seen in allogeneic stem cell transplantation and provide insight into whether allo-enhanced DC-induced immune responses can aid the in vivo maturation needed for effective DC-driven anti-tumour immunotherapy.
Materials and methods
Mice and cell lines
C57BL/6 (H-2b), C3H/HeN (H-2k) [C3H/HeN × C57BL/6] F1 (B6C3F1, H-2k/b), DBA/2 (H-2d) and [DBA/2 × C57BL/6] F1 (B6D2F1, H-2d/b) 6-8-week-old female mice were purchased from Harlan, Oxford, UK. OT-I [22] and OT-II [6] T-cell receptor (TCR) transgenic mice (both H-2b) were bred in our facility. CD8+ OT-I T-cells recognize Chicken Ovalbumin (OVA)257-264 peptide (SIINFEKL) in association with MHC class I (H-2Kb), whereas CD4+ OT-II T-cells recognize OVA323–339 peptide (ISQAVHAAHAEINEAGR) in association with MHC class II (I-Ab). All animal studies were carried out in accordance with UK Home Office regulations and were approved by our local ethical committee.
EL4, an H-2b thymoma, and the E.G7 derivative line, which stably expresses chicken OVA on its cell surface [32], were maintained in X-VIVO-15 (Biowhittaker, Walkersville, MD, USA), 100 U/ml penicillin and 100 μg/ml streptomycin (both from Sigma, Poole, UK). The B3Z T-cell hybridoma [24], which is specific for the H-2Kb/SIINFEKL complex, was maintained in RPMI (Sigma) supplemented with 10% FCS (Sigma), 100 U/ml penicillin and 100 μg/ml streptomycin. EL4, E.G7 and B3Z were a kind gift from Dr J. Morrow, Department of Immunology, St. Bartholomew’s and the Royal London School of Medicine, London, UK.
Antibodies and other reagents
FITC-conjugated monoclonal antibodies AF6-88.5 (anti-H-2Kb), 36-7-5 (anti-H-2Kk), SF1-1.1 (anti-H-2Kd) 28-18-8S (anti-I-Ab) and PE-conjugated B20.1 (anti-Vα2) were purchased from BD Biosciences, Oxford, UK. Biotinylated MR9-4 (Vβ5.1, 5.2; BD) was detected using Streptavidin Qdot-800 conjugate (Cambridge Bioscience, Cambridge, UK). APC-conjugated RM4-5 (anti-CD4) and unlabelled CT-17.1/CT-17.2 (mouse anti-mouse FcRγII/III-CD16/CD32) were from Caltag-Medsystems, Silverstone, UK. SIINFEKL peptide (OVA257–264) was purchased from ProImmune Ltd., Oxford, UK. OVA323–339 peptide (ISQAVHAAHAEINEAGR) was synthesized by Mimotopes Ltd., Wirral, UK. Purified Grade V OVA was from Sigma. Irrelevant control peptide GAD65171–190 (IKTGHPRYFNQLSTGLDMVG) was donated by Dr. Tim Tree, King’s College London, and control peptide Vesicular Stomatitis Virus (VSV)52–59 (RGYVYQGL) was purchased from the Department of Molecular Biology and Biotechnology, University of Sheffield, UK. Anti-CD4 (L3T4), -CD8 (Ly-2) and -CD11c (HL3) Microbeads for MACS were purchased from Miltenyi Biotec, Bisley, UK.
Bone marrow-derived dendritic cells and phenotypic analysis
Bone marrow cells were cultured under serum-free conditions in the presence of 5 ng/ml of GM-CSF (R&D systems, Abingdon, UK) and 10 ng/ml of IL-4 (PeproTech EC Ltd., London, UK) as described previously [40]. Non-adherent or loosely adherent DCs were harvested for use on day 7. Tolerogenic (immature) C3H DC were generated following a modified version of the “OX Method” [29]. C3H bone marrow cells were cultured at 2 × 106 cells/ml in X-VIVO-15 containing 1 ng/ml of GM-CSF at 37°C/5% CO2. On day 2, the plate was swirled gently and half the medium was removed and replaced with the same volume of medium containing 1 ng/ml GM-CSF. On day 4, the contents of each well were divided into two fresh wells, which were topped up to 4 ml/well with medium containing 1 ng/ml GM-CSF. Immature DC were harvested on day 6, washed twice in PBS, and injected i.v. at 106 cells/mouse to induce tolerance. Mice were immunized with semi-allogeneic DC i.d. 7 days later. DC were analysed by FACS (BD Bioscience) following Fc-receptor-blocking using anti-CD16/CD32 to prevent non-specific antibody staining. DC were then incubated with H-2K antibodies for 15 min, washed twice, and analysed.
Allogeneic mixed lymphocyte reaction
Dendritic cells were plated out in triplicate in 96-well U bottomed plates (Greiner bio-one, Stonehouse, UK), serially diluted 1:2 from 104 cells/well down to 6.25 × 102 cells/well. Splenocytes were harvested from the spleens of naïve mice and depleted of red-blood cells using Red Blood Cell Lysing Buffer (0.155 M ammonium chloride in 0.01 M Tris–HCl buffer, Sigma), and B-cells using B220 Dynabeads (Dynal, Wirral, UK) according to the manufacturer’s instructions. The splenocytes were then added to the plate at 105 cells/well (final volume: 200 μl/well). After 5 days at 37°C, splenocyte proliferation was assessed by [methyl-3H] thymidine (1 μCi/well, Amersham Pharmacia Biotech, Little Chalfont, UK) incorporation over 6 h using a Liquid Scintillation Analyzer (TRI-CARB 2200CA, Packard). Secondary mixed lymphocyte reactions (MLR’s) were performed over 3 days using Inguinal LN cells from immunized mice as responders.
B3Z colorimetric assay
Dendritic cells were pulsed for 4 h with various concentrations of SIINFEKL peptide or VSV52–59 irrelevant peptide, washed twice and resuspended in phenol-red free RPMI (Sigma) containing 100 U/ml penicillin and 100 μg/ml streptomycin, 1% FCS and 2 mM l-Glutamine. DC (104 cells/well) were then co-cultured in a 96-well U bottomed plate with 5 × 104/B3Z cells/well, also suspended in phenol-red free RPMI, overnight at 37°C (final well volume: 200 μl). 150 μl of supernatant was taken from each well and replaced with 150 μl of PBS containing 5 mM ONPG (Sigma) and 0.5% Nonidet-P40 (BDH, Bristol, UK). The plate was incubated at 37°C for 2 h and optical density measured using a Precision Microplate Reader (Molecular Devices, Sunnyvale, CA, USA) at 450 nm with wavelength correction set at 650 nm.
OVA presentation assays
Dendritic cells (104 cells/well) and various concentrations of OVA323–339 peptide or whole OVA protein were co-cultured with 3 × 104 naïve OT-II CD4+ T-cells [from the spleen, inguinal and mesenteric lymph nodes of OT-II mice, purified using CD4+ Microbeads (Miltenyi Biotec) by autoMACS™ selection, >95% purity]. GAD65171–190 peptide (10 μg/ml) and bovine serum albumin (BSA) protein (1,000 μg/ml) were used as negative controls. After 90 (peptide) or 96 h (protein) lymphocyte proliferation was assessed by 3H-thymidine incorporation over 6 (protein) or 18 h (peptide) of culture.
CD11c− CD8+ OT-I cells were purified using Microbeads (Miltenyi Biotec) and autoMACS selection (>98% purity) and cultured (105/well) with DC (104/well) and OVA for 3 days at 37°C. BSA (1 mg/ml) was used as negative control. Lymphocyte proliferation was assessed by 3H-thymidine incorporation over the final 6 h of culture.
DC migration studies
Dendritic cell migration was quantified as described previously [26] with slight modifications. Briefly, DC were washed twice in PBS, labelled with 2.5 μM carboxy fluorescein succinimidyl ester (CFSE) (Invitrogen, Paisley, UK; 10 min 37°C) and injected into naïve mice (106 DC/flank i.d.). 20, 44 and 68 h later the inguinal lymph nodes were removed, pooled, counted and analysed by flow cytometry.
DC competition experiments
OT-II CD4 cells or OT-I CD8 cells were labelled with CFSE as above. 6 × 104 labelled cells were cultured with 6 × 103 C3H × C57 F1 DC in 1 ml, with or without a 100-fold excess (6 × 106) of CD11c-depleted C3H × C57 F1 (autologous) or C57 (alloreactive) CD4 or CD8 T-cells. OVA peptides (2 μg/ml) were added as above. After 3 days, cells were washed, stained for CD4/CD8 and the entire sample was analysed by flow cytometry with gating on CFSE+ events.
Activation of CD4+ OT-II T-cells in vivo
OVA-specific CD4+ T-cells from OT-II mice were washed twice in PBS, labelled with 2.5 μM CFSE, washed and transferred into tail veins of mice (∼5 × 106 each in 100 μl PBS). About 24 h later mice received DC previously pulsed with 150 μg/ml of OVA for 1 h (washed extensively) at 106 DC/flank i.d. For allo-tolerization experiments mice received 106 immature C3H DC i.v. 6 days before OT-II transfer. About 3 days following i.d. immunization the inguinal lymph nodes were stained with CD4, Vα2 and Vβ5 antibodies and analysed by flow cytometry.
DC–EL4 fusion hybrid formation
Dendritic cell and EL4 (irradiated at 100 Gy) tumour cells were mixed together at a ratio of 1:1, washed in Mg2+/Ca2+-free PBS (ICN, Irvine, CA, USA) at 37°C, and pelleted tightly together by centrifugation at 400g for 10 min. The supernatant was carefully removed and fusion induced by gently stirring the pellet with the tip of a pipette adding 1 ml of HYBRI-MAX® PEG/DMSO solution (also at 37°C, Sigma) over the course of 1 min. The cells were gently stirred for a further minute, and then 10 ml of X-VIVO-15 medium (at 37°C) was added drop-wise over the following 3 min. Cell hybrids were then washed; hybrids were not isolated prior to immunization. To determine DC–EL4 fusion efficiency by flow cytometry, DC were labelled with the red fluorescent membrane dye PKH26 (Sigma) and EL4 cells with the green fluorescent intracellular dye 5-chloromethylfluorescein diacetate (CMFDA, Molecular Probes, Eugene, OR, USA). When DC fused to DC and EL4 cells fused to EL4 cells were mixed together they formed a small number of cell aggregates that resulted in false “double positives”. When this had been taken into account, the efficiency of successful DC to EL4 cell fusion was estimated to be 4.2%.
EL4 tumour experiments
For tumour prevention experiments, DC from C57, C3H, DBA, C3H × C57 F1 or DBA × C57 F1 mice were fused with irradiated EL4 cells, and injected s.c. into the flanks of naive C57BL/6 mice (n = 8 per group, 106 starting DC/mouse) on days −16 and −7. On day 0, mice were challenged with 5 × 104 viable EL4 cells injected s.c. The control group was not immunized prior to tumour challenge. Tumour growth was monitored at regular intervals and mice were culled when tumour diameters reached 16–17 mm. For tumour treatment experiments, naïve mice were challenged s.c with 5 × 104 viable EL4 cells on day 0, and then immunized on both flanks on days 3 and 7 with DC/EL4 fusion hybrids.
Cytotoxicity assay
Dendritic cells were pulsed with 10 μg/ml of SIINFEKL peptide and 5 μg/ml human β2-microglobulin (Sigma) for 3 h at 37°C, then washed and injected i.d. (106 cells/flank) into the right and left flanks of naïve C57BL/6 mice. About 7 days after immunization splenocytes from these mice were co-cultured at a ratio of 10:1 with EG7 cells (irradiated at 50 Gy) in X-VIVO-15 medium supplemented with 50 μM β-mercaptoethanol for 5 days. After 2 days 10 U/ml of rmIL-2 (R&D) was added. On day 5 the ability of the splenocytes (effectors) to kill 51Chromium (51Cr)-labelled EG7 or EL4 cells (5 × 103cells/well, targets) was then assessed over 4 h. Background 51Cr release was measured from 51Cr labelled cells alone. Total 51Cr-released was measured following treatment of 51Cr-labelled cells with Triton X100. Target cell lysis was calculated using mean counts per minute (cpm) of triplicate wells in the following equation:
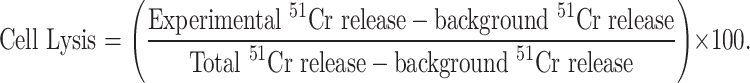 |
If
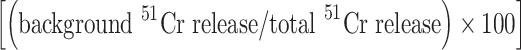 was >25%, the experimental data were considered invalid.
was >25%, the experimental data were considered invalid.
Statistical analysis
Standard error of mean (SEM) and significance values were calculated using GraphPad Prism Version 3.02 for Windows, GraphPad Software, San Diego, CA, USA. Statistical comparisons of mean values were performed using unpaired Student’s t-test. Statistical comparisons of survival curves were performed using the logrank test with the null hypothesis that treatments did not change survival. P < 0.05 (*) were considered significant. P < 0.005 (**) and P < 0.001 (***) are indicated.
Results
Semi-allogeneic (F1) DC induce potent alloreactive immune responses from both parental strains
Dendritic cells can induce particularly strong responses in MHC-mismatched T-cell populations when combined in primary MLRs in vitro. To determine whether semi-allogeneic DC could induce equally strong alloreactions we co-cultured C3H/HeN × C57BL/6 F1 DC (C3H × C57 F1, H-2k/b) with either C57BL/6 (C57, H-2b) or C3H/HeN (C3H, H-2k) naïve B-cell-depleted splenocytes (Fig. 1). Semi-allogeneic DC were found to induce a similar alloreactive response to that induced by fully allogeneic DC (C3H DC and C57 DC, respectively), when co-cultured with either C57 or C3H parental splenocytes. Therefore, the presence of “self”-MHC on the surface of semi-allogeneic DC does not appear to affect their capacity to induce allo-responses. As a negative control, semi-allogeneic DC (as well as C57 and C3H DC) were co-cultured with semi-allogeneic splenocytes and were shown not to induce a response as expected. These data demonstrate the potential of semi-allogeneic DC to induce alloreactive immune responses.
Fig. 1.

C3H × C57 F1 DC are just as potent as allogeneic DC in their ability to stimulate alloreactive proliferation in a primary MLR. C57 (open squares), C3H (open triangles), or C3H × C57 F1 DC (solid circles) were co-cultured with naïve C57, C3H or C3H × C57 F1 B-cell-depleted splenocytes as indicated for 5 days at 37°C. Proliferation was measured by the uptake of 3H-thymidine over 6 h. Graphs represent combined data from two separate sets of experiments carried out in triplicate. Error bars represent SEM
Semi-allogeneic (F1) DC prime naïve antigen-specific T-cells in vitro
To determine whether semi-allogeneic C3H × C57 F1 DC would be able to induce antigen-specific immune responses on an H-2b background, we analysed the expression of H-2Kb and I-Ab by flow cytometry (Fig. 2a, b). Semi-allogeneic DC were found to express both parental H-2K haplotypes (H-2Kb and H-2Kk), although the level of H-2Kb expression was just over half of that expressed by C57 DC as determined by the mean fluorescence intensity. The MHC Class II positive cells in the C3H × C57 F1 DC population expressed around 25% less I-Ab than C57 DC. The level of H-2Kk expressed by C3H × C57 F1 DC however, was very similar to that expressed by C3H DC. A similar H-2K expression pattern was observed between C57 DC, DBA DC and semi-allogeneic DBA × C57 F1 DC following staining for H-2Kb or H-2Kd (data not shown).
Fig. 2.

C3H × C57 F1 DC induce antigen-specific MHC Classes I and II—restricted T-cell activation in vitro. a Semi-allogeneic DC express H-2Kb (filled histograms) and H-2Kk (open histograms). Numbers represent the Mean Fluorescence Intensity of H-2Kb (top row) and H-2Kk (bottom row) staining. b Semi-allogeneic DC express I-Ab. Numbers represent the Mean Fluorescence Intensity in the I-Ab positive-staining gate as indicated. ND not detectable. c DC were pulsed for 4 h with SIINFEKL peptide, washed and co-cultured with the SIINFEKL/H-2Kb-restricted B3Z hybridoma for 24 h. The cells were then lysed and monitored for LacZ expression by the introduction of ONPG substrate. d DC were co-cultured with CD11c− CD8+ OT-I cells and the indicated concentration of OVA for 72 h with proliferation assessed over the final 6 h. e DC were co-cultured with naïve CD4+ OT-II lymphocytes for 90 h (peptide) or f 96 h (protein). Proliferation was measured over the final 6 h. Error bars represent SEM
To analyse functional presentation of antigen, C3H × C57 F1 DC were pulsed with various concentrations of the H-2Kb-presented SIINFEKL peptide and assayed for their ability to stimulate the B3Z (SIINFEKL/H-2Kb-specific) T-cell hybridoma. Activation through the TCR induces the expression of the reporter gene product β-galactosidase, which is assayed after lysis. As shown in Fig. 2c, semi-allogeneic C3H × C57 F1 DC but not allogeneic C3H DC were able to present SIINFEKL peptide to the H-2b B3Z hybridoma in vitro. However, semi-allogeneic C3H × C57 F1 DC induced significantly weaker TCR-mediated β-galactosidase production at each concentration of SIINFEKL peptide tested as compared with autologous C57 DC. As the B3Z hybridoma only requires the presentation of SIINFEKL in the context of H-2Kb to become activated, and does not require co-stimulatory molecules, this result was most likely due to reduced expression of H-2Kb by C3H × C57 F1 DC as compared with C57 DC.
OT-I and OT-II transgenic T-cells were used to investigate whether semi-allogeneic DC were capable of priming naïve antigen-specific T-cell responses in vitro. DC were co-cultured with CD11c-depleted CD8+ OT-I T-cells and the indicated concentration of whole OVA protein (Fig. 2d). Semi-allogeneic C3H × C57 F1 DC and autologous C57 DC, but not allogeneic C3H DC, stimulated naïve CD8+ OT-I T-cells to proliferate. Interestingly, there was no significant difference in the ability of semi-allogeneic C3H × C57 F1 DC to cross-present OVA peptides to CD8+ OT-I T-cells as compared with autologous C57 DC. OT-I T-cells did not respond to the control protein, BSA. When pulsed with OVA323–339 peptide (Fig. 2e) or whole OVA protein (Fig. 2f), semi-allogeneic DC were also capable of priming naïve MHC class II-restricted CD4+ OT-II T-cells in vitro. However, semi-allogeneic DC were less efficient at stimulating CD4+ OT-II T-cells than autologous DC, particularly at low peptide/protein concentrations. Together these data show that semi-allogeneic C3H × C57 F1 DC are capable of processing and presenting antigen in the context of H-2Kb, as well as I-Ab, in order to drive both MHC classes I- and II-restricted antigen-specific T-cell priming in vitro.
Alloreactive T-cells do not compete with, or enhance proliferation of antigen-specific T-cells responding to the same DC in vitro
To determine the effect of alloreactive T-cells on OVA presentation by semi-allogeneic DC, C3H × C57 F1 DC were cultured with CFSE labelled OT-I CD8 or OT-II CD4 T-cells and OVA peptides (Fig. 3). The proliferation of both CD4 and CD8 cells was unaffected by the addition of a 100-fold excess of either autologous C3H × C57 F1 T-cells (no alloresponse), or alloreactive C57 T-cells. The data suggest that competition for access to DC by T-cells responding to different antigenic determinants does not reduce antigen-dependent proliferation.
Fig. 3.

Alloantigen-reactive T-cells do not interfere with T-cell responses to an exogenous antigen in vitro. OT-I CD8 T-cells (a) or OT-II CD4 T-cells (b) were labelled with CFSE and cultured with C3H × C57 F1 DC + OVA peptides, in the presence of a 100-fold excess of unlabelled autologous (F1) or alloreactive (C57) T-cells as indicated. After 3 days cells were stained for CD4 or CD8 and analysed for CFSE dilution. Data indicate % divided cells in gated CFSE+ CD4/CD8+ populations. Similar data were obtained in two further experiments
Semi-allogeneic DC migrate efficiently to regional lymph nodes but do not persist as long as autologous DC
The induction of primary immune responses in vivo requires migration of DC to regional lymph nodes in order to interact with naïve T-cells. The ability of semi-allogeneic DC to migrate to regional lymph nodes is therefore a critical consideration if they are to be successfully utilized in generic DC vaccines. Autologous (C57), semi-allogeneic (C3H × C57 F1) and allogeneic (C3H) DC were labelled with CFSE prior to i.d. injection into the flanks of naïve C57BL/6 mice. At various time-points following injection the inguinal lymph nodes were removed, pooled, and the number of CFSE+ DC in whole pooled lymph node samples was quantified by flow cytometry. As shown in Fig. 4a, autologous, semi-allogeneic and allogeneic DC displayed a similar capacity to migrate to the inguinal lymph nodes 20 h after injection. At 44 h post-injection the number of CFSE+ autologous or semi-allogeneic DC continued to rise to a similar degree, however, the number of CFSE+ allogeneic DC appeared to decrease. At 68 h the numbers of CFSE+ semi-allogeneic or allogeneic DC had significantly declined compared with autologous DC-injected mice. The decrease in resident CFSE+ DC populations in mice injected with semi-allogeneic DC at 68 h correlated with a significant expansion in total lymph node cell counts (Fig. 4b) suggesting that an alloreactive T-cell response was mediating the destruction of semi-allogeneic DC. The data indicate that semi-allogeneic DC, at least during primary immunization, migrate efficiently to the regional lymph nodes but are susceptible to killing mediated by the ensuing alloreactive immune response.
Fig. 4.

C3H × C57 F1 DC migrate to regional LN as efficiently as C57 DC but do not persist as long. a Mice were injected i.d. on both flanks with OVA-pulsed, CFSE-labelled DC. At the time-points indicated inguinal LN were harvested, pooled (for each mouse), digested with collagenase and analysed for the presence of CFSE+ cells by flow cytometry. b Pooled LN counts at each time-point highlight the presence of an active allo-response. Data are representative of two separate experiments with similar results. Error bars represent SD
Semi-allogeneic DC were not capable of inducing cytotoxic T-lymphocyte (CTL)-activity to a target peptide
In order to evaluate the potential of semi-allogeneic DC to induce MHC class I-restricted antigen-specific cytotoxicity, autologous (C57), semi-allogeneic (C3H × C57 F1) and allogeneic (C3H) DC were pulsed with SIINFEKL peptide and adoptively transferred i.d. into the flanks of naïve C57BL/6 mice. After 7 days spleens were removed and cultured with irradiated EG7 cells for 5 days prior to use in a standard 4-h chromium release assay (Fig. 5). Autologous C57 DC were found to induce cytotoxic T-lymphocyte (CTL) activity directed against EG7 target cells, but not against control EL4 target cells, indicating that CTL activity was OVA-specific. No evidence of CTL activity directed at either EG7 or EL4 target cells was seen in mice injected with peptide-pulsed semi-allogeneic (C3H × C57 F1) DC or allogeneic (C3H) DC. Similar results were seen when using pooled draining lymph node (inguinal) and spleen cells as a source of CTL (data not shown). Furthermore, multiple weekly injections (up to four injections) of peptide-pulsed semi-allogeneic DC did not result in induction of OVA-specific CTL activity, suggesting that resident DC were not stimulated to induce CTL activity to a foreign antigen during the course of strong alloreactive immune responses (data not shown). Pre-immunization of recipients with OVA/alum also failed to result in detectable CTL, and similar results were obtained using semi-allogeneic DC derived from DBA × C57 F1 mice (H-2Kd/b, data not shown). These results indicate that semi-allogeneic DC do not directly, nor indirectly, induce antigen-specific CTL activity in naïve C57BL/6 mice. These results suggest that semi-allogeneic DC do not become conditioned to stimulate antigen-specific CD8+ T-cells to become CTL through interactions with activated alloreactive CD4+ T-cells.
Fig. 5.

C3H × C57 F1 DC were not capable of inducing CTL activity to a target peptide (SIINFEKL). Naïve C57 mice were immunized i.d. with C57, C3H or C3H × C57 F1 DC pulsed with 10 μg/ml SIINFEKL peptide. About 7 days later splenocytes from two mice/group were harvested, pooled, restimulated for 5 days with irradiated EG7 cells and then tested for their ability to kill 51Cr-labelled EG7 or EL4 target cells. Data are representative of five independent experiments with similar results
Semi-allogeneic DC induce weak activation of adoptively transferred OT-II CD4 T-cells in vivo
Our data suggested that semi-allogeneic DC were not capable of inducing effective cell-mediated immunity in vivo, despite their effective APC functions in vitro. We investigated whether this might be due to destruction of semi-allogeneic DC or more competition from concurrent alloreactive T-cell responses in vivo. CFSE+-labelled OT-II CD4+ T-cells were adoptively transferred i.v. into groups of C57BL/6 mice. After 24 h the mice were injected i.d. with OVA-pulsed autologous (C57), semi-allogeneic (C3H × C57 F1) or allogeneic (C3H) DC (day 0). One group of mice that received OVA-pulsed semi-allogeneic (C3H × C57 F1) DC had been pre-tolerized to C3H allo-antigens through the i.v. injection of immature C3H DC 7 days prior to the start of the experiment (day 7 [29]). Draining inguinal lymph nodes were harvested 72 h later and pooled, total cell numbers were then noted and each sample analysed for the division of CFSE+ cells by flow cytometry. As shown in Fig. 6a, the total lymph node cell count increased significantly in mice that received semi-allogeneic (C3H × C57 F1) or allogeneic (C3H) DC as compared to mice that received autologous (C57) DC, indicative of alloreactivity. Mice that were pre-tolerized to C3H allo-antigens however, did not show a significant increase in total lymph node cell count in response to immunization with semi-allogeneic (C3H × C57 F1) DC. Furthermore, when lymph node cells from these mice were cultured in a secondary MLR with semi-allogeneic (C3H × C57 F1) DC, they failed to proliferate (Fig. 6b), confirming that these mice were tolerized to C3H alloantigens. In comparison to autologous (C57) DC, semi-allogeneic DC induced weaker OT-II proliferation in vivo, however, this was not unexpected as they have a lower expression of I-Ab. We did not observe a significant difference in OT-II proliferation between naïve and allo-tolerized C57 mice injected with OVA-pulsed semi-allogeneic (C3H × C57 F1) DC (Fig. 6c), indicating that alloreactive T-cell responses do not provide help for antigen-specific stimulation in vivo. Conversely, alloreactivity does not appear to interfere with antigen-specific responses through competition for proinflammatory cytokines or interactions with DC.
Fig. 6.

Ability of C3H × C57 F1 DC to prime immune responses in mice pre-tolerized to alloantigen. a Mice tolerized to C3H allo-antigens do not have evidence of LN expansion in response to injection with C3H × C57 F1 DC. Mice were tolerized to C3H-allo-antigens through the injection of 1 × 106 immature (day 6) C3H DC i.v. After 6 days the mice received 4–5 × 106 naïve CFSE-labelled CD4+ OT-II cells i.v. 24 h later mice were challenged with the indicated DC pulsed with 150 μg/ml OVA i.d. on both flanks. Inguinal LN were harvested 72 h later. Data represent average LN cell counts ± SEM derived from five separate experiments. b LN cells from mice tolerized to C3H allo-antigens do not respond to C3H × C57 F1 DC in a secondary MLR. c OT-II cells adoptively transferred into C3H allo-antigen-tolerized mice proliferate poorly when stimulated by C3H × C57 F1 DC (as shown by CFSE dilution). Numbers refer to the percentage of CFSE+ cells that divided ± SD for three mice/group. Plots represent one experiment of five with similar results
Semi-allogeneic DC fusion hybrids increase survival rates in EL4 tumour models
In previous studies we showed that the direct fusion of DC with tumour cells provides an effective method for loading DC with tumour antigens [19, 20]. We wished to determine whether the presence of autologous MHC class I molecules (H-2Kb) on tumour cells would enable semi-allogeneic DC fusion hybrids to stimulate tumour-specific CD8+ CTL in vivo. In addition we hypothesized that expression of autologous MHC class II molecules (I-Ab) by semi-allogeneic DC might provide CD4 T-cell help for tumour-specific CD8+ T-cells, as DC/tumour cell fusion hybrids have been shown to be able to directly activate tumour-reactive CD4 T-cells [20]. To investigate this we hybridized autologous (C57), semi-allogeneic (C3H × C57 F1 or DBA × C57 F1) or allogeneic (C3H or DBA) DC to irradiated EL4 cells (H-2Kb+ I-Ab−) and used these fusion hybrids in the treatment of tumours induced by a lethal dose of live EL4 cells. C57BL/6 mice were given two identical immunizations of DC/EL4 fusion hybrids a week apart, and were challenged with viable EL4 cells (day 0) 2 weeks after the second immunization. As shown in Fig. 7a, untreated control mice were all culled by day 28. Prophylactic vaccination, regardless of whether the DC used were autologous, semi-allogeneic or allogeneic, resulted in a significant increase in survival time when compared with untreated controls, and in the long-term tumour free survival of some mice within each vaccinated group (other than in mice receiving DBA DC). All tumour-free mice were re-challenged on day 91 with five times the original tumour dose (2.5 × 105 cells/mouse), yet remained tumour-free up to day 138 (data not shown), demonstrating long-lived anti-tumour immunity had been induced in these mice. However, that allogeneic DC/EL4 hybrids offer some protection against a subsequent challenge with viable EL4 cells might be taken to imply that host DC cross-present EL4 antigens to the immune system in this scenario. We therefore evaluated the influence of vaccination in the treatment of mice pre-challenged with tumour cells. Mice were given viable EL4 cells (day 0) and then two immunizations of DC/EL4 fusion hybrids on days 3 and 7 (Fig. 7b). Untreated control mice were all culled by day 27. Mice immunized with allogeneic DBA- or C3H DC/EL4 fusion hybrids did not display a significant difference in survival time (P = 0.9827 and 0.2655, respectively), despite the tumour-free survival of one mouse in each of these groups. However, mice immunized with autologous C57- or semi-allogeneic C3H × C57 F1- or DBA × C57 F1 DC/EL4 fusion hybrids survived significantly (P = 0.0054*, 0.0081* and 0.0034**, respectively), longer than controls. These data suggest that semi-allogeneic DC hybrids present tumour antigens to elicit tumour-specific immunity. Thus it appears that semi-allogeneic DC that express autologous MHC classes I- and II molecules may offer an advantage over allogeneic DC in therapeutic DC/tumour cell fusion hybrid therapy of established tumours.
Fig. 7.

Semi-allogeneic DC/EL4 fusion hybrids increase survival expectancy in EL4 tumour prevention (a) and b tumour treatment experiments. a C57BL/6 mice were immunized twice with C57, DBA, C3H, C3H × C57 F1, or DBA × C57 F1 DC fused to irradiated EL4 cells (n = 8). Untreated mice were used as a control. Two weeks later all the mice were challenged with a subcutaneous injection of 5 × 104 viable EL4 cells. The numbers in brackets in a represent the number of tumour-free mice in each group at the end of the experiment. b C57BL/6 mice were challenged with a subcutaneous injection of 5 × 104 viable EL4 cells and treated on days 3 and 7 with C57, DBA, C3H, C3H × C57 F1 or DBA × C57 F1 DC fused to irradiated EL4 cells (n = 8). Mice were culled when tumour diameters reached 16–17 mm
Discussion
In recent years, the beneficial effects of the alloresponse in cancer immunotherapy have been repeatedly demonstrated in the clinic. Hemopoietic stem cell transplantation and donor lymphocyte infusions have been shown to be particularly effective in the treatment of hematological malignancies such as chronic and acute myeloid leukaemia [4, 18], and also show promise in the therapy of solid tumours [8, 10]. The vigorous induction of anti-tumour T-cell responses, which results following presentation of tumour antigens in the context of foreign MHC molecules, as compared to self-MHC molecules, may in part be explained by the absence of tolerance which normally occurs during development in the thymus [41]. Alloreactive responses in vivo are usually extremely potent and the resultant massive release of cytokines has been well-documented and termed a “cytokine storm” [16].
Our study addressed the role of alloreactivity in the promotion of a concurrently activated antigen-specific T-cell response. By using partially MHC-matched semi-allogeneic DC derived from F1 strains, we were able to examine the induction of antigen-specific CD8 and CD4 T-cell responses in vitro in the presence or absence of allostimulation. We showed that in vitro, semi-allogeneic C3H × C57 F1 (H-2k/b) DC induce alloreactivity in an MLR, present peptide to B3Z hybridoma cells in the context of H-2Kb, prime naïve OT-II CD4 T-cells (H-2b) to proliferate when pulsed with antigen, and are capable of the cross-presentation of OVA peptides to MHC class I-restricted OT-I T-cells. However, semi-allogeneic DC were not as efficient as autologous DC in the presentation of SIINFEKL peptide to the B3Z hybridoma or in the priming of OT-II CD4 T-cells at low doses of protein or peptide, which may result from the reduced expression of H-2Kb and I-Ab by these DC in comparison to autologous DC.
In vivo, we showed that although semi-allogeneic DC migrate efficiently to the regional lymph nodes following adoptive transfer, they were unable to prime CTL to a target peptide in naïve mice. A recent study by Fukuma et al. [17] showed that semi-allogeneic embryonic stem cells (ES cells) engineered to express OVA and then subsequently differentiated into DC were able to activate OVA-specific CTL. The priming capacity of ES cell-derived DC was suggested to be related to the expression of SPI-6, a specific inhibitor of granzyme B not expressed by bone-marrow-derived DC, which would make ES cell-derived DC resistant to cytotoxicity by CTL [17]. However, the use of whole OVA may have provided a source of class II-restricted epitopes, resulting in antigen-specific CD4+ help for CD8+ CTL induction, as ES cell-derived DC pulsed with SIINFEKL were weak stimulators of CTL [17]. Our data suggest that CD4+ T-cells activated by allogeneic MHC class II molecules co-expressed by bone-marrow-derived, class I-peptide-pulsed semi-allogeneic DC do not provide help for the priming of antigen-specific CTL.
To ascertain the influence of allogeneic immune responses on the priming of antigen-specific CD4+ T-cells, we studied the ability of OVA-pulsed semi-allogeneic DC to prime adoptively transferred CD4+ OT-II cells in allo-competent and allo-tolerized hosts. Mice were tolerized to alloantigens with immature allogeneic DC, which are known to induce alloantigen-specific tolerance resulting in long-term survival of cardiac allografts in mice [5, 29]. Alloantigen-tolerized mice did not show lymph node expansion in response to immunization with semi-allogeneic DC, and lymph-node-derived cells did not proliferate in response to semi-allogeneic DC in a secondary MLR, confirming that these mice were tolerized to (C3H) alloantigens. Compared to autologous DC, semi-allogeneic DC induced weak activation of OT-II cells in allo-competent mice. Importantly, the ability of semi-allogeneic DC to induce OT-II activation was not significantly altered in allo-tolerized mice, suggesting that alloreactive T-cell responses do not provide help for antigen-specific CD4+ T-cell responses in vivo, and additionally, that allogeneic immune responses do not inhibit OT-II activation through competition for proinflammatory cytokines, CD4+ growth factors, or interactions with DC. The latter conclusion was reinforced by our in vitro competition experiments. Although the work of Willis et al. [43] demonstrates that T-cells of differing specificities do compete for access to DC in vivo, this study was performed in a syngeneic system with two exogenous peptides. In our study, we observed a loss of semi-allogeneic DC in draining lymph nodes, suggesting that they are killed or receive cytokine signals that prevent their retention in lymphoid tissue.
It is likely that semi-allogeneic DC survive for longer in the lymph nodes of allo-tolerized mice than in the lymph modes of naïve mice [28]. However, allo-tolerization does not appear to have a significant impact on OT-II-proliferation, so it seems unlikely that alloreactive cytotoxicity directed against semi-allogeneic DC in the lymph node can be held solely responsible for the poor stimulation of antigen-specific T-cells. Although in this study we did not look at the possibility that NK cells kill semi-allogeneic DC, it has recently been shown that semi-allogeneic DBA × C57 F1 DC adoptively transferred into Rag−/− (H-2b) mice, which are deficient in T- and B-cells but not NK cells, survive for >2 weeks, yet fully MHC mismatched allogeneic DC are rapidly destroyed [44]. Therefore, the presence of self-MHC molecules on semi-allogeneic DC may well protect these cells from NK cell killing. It seems more probable that the weaker direct stimulation of antigen-specific CD4+ and CD8+ T-cell responses by semi-allogeneic DC compared to autologous DC is due to low “self”-MHC expression (H-2Kb/I-Ab) by these DC. Clearly not just the presence, but also the level of self-MHC expression is an important consideration when choosing suitable semi-allogeneic DC for vaccination studies.
The fusion of DC to tumour cells results in the production of hybrid cells capable of presenting endogenous tumour antigens (from the tumour partner) in association with the requisite adhesion and co-stimulatory molecules (from the DC partner) to induce tumour-specific T-cell activation. Fusion hybrids created using autologous or allogeneic DC have been shown capable of inducing anti-tumour immune responses [21, 30, 36], suggesting that MHC-restriction may not be as important as the provision of co-stimulatory signals. Our data suggest however, that it is advantageous for the DC partner to express self-MHC molecules, as recipients of semi-allogeneic DC/EL4 fusion hybrids showed significantly increased survival time in an EL4 tumour treatment model, but mice receiving allogeneic DC/EL4 hybrids did not. Since EL4 cells are MHC class II negative, this observation suggests that the expression of self-MHC class II molecules by semi-allogeneic DC, which is absent on allogeneic DC, may be critical for the induction of anti-tumour immune responses. In addition, the role of MHC class I expression by semi-allogeneic DC should not be overlooked. It is possible that semi-allogeneic DC, when hybridized to tumour cells, may also provide the processing machinery for MHC class I presentation of tumour antigens. Our findings that DC/tumour hybrid-immunized mice show increased survival following tumour cell challenge are potentially at odds with our results suggesting a lack of CTL and weak CD4+ T-cell induction in vivo. One possible explanation could be that differences in processing and presentation of endogenously expressed antigens by the hybrids in vivo allowed a more efficient stimulation of the host immune system, compared with exogenous addition of antigen or peptide to DC ex vivo.
Semi-allogeneic DC do not appear to differ from autologous DC in their ability to act as fusion partners for the induction of anti-tumour immunity in a treatment model and may also allow greater flexibility in these protocols. Suitably MHC-matched DC lines or DC generated from healthy volunteers in advance, for example, could negate the requirement to generate DC from ill patients, and may also allow rapid treatment at a considerably reduced cost.
While the induction of tolerance to alloantigens might allow repeated use of the same semi-allogeneic DC population, further work is required to define whether Th1-inducing allogeneic responses [11, 34, 35, 39] would be beneficial in polarizing concurrently activated cancer-specific immune responses towards a Th1 phenotype [38]. The present findings demonstrate that the expression of self-MHC by semi-allogeneic DC can lead to the induction of antigen-specific T-cell responses, yet concurrently activated allogeneic responses do not provide help. The usefulness of partially MHC-matched semi-allogeneic DC in vaccination strategies may therefore arise from the expression of self-MHC molecules, but not the concurrent activation of alloreactivity.
Acknowledgements
This work was supported by Leukaemia Research Fund and a JRC studentship from King’s College London School of Medicine.
Abbreviations
- 51Cr
51Chromium
- CFSE
Carboxy fluorescein succinimidyl ester
- CTL
Cytotoxic T-lymphocyte
- DC
Dendritic cell
- ES
Embryonic stem cells
- MHC
Major histocompatibility complex
- MLR
Mixed lymphocyte reaction
- OVA
Ovalbumin
- SEM
Standard error of the mean
- TCR
T-cell receptor
Footnotes
Joanna Galea-Lauri and Alistair Noble share senior authorship of this work.
References
- 1.Avigan D, Vasir B, Gong J, Borges V, Wu Z, Uhl L, Atkins M, Mier J, McDermott D, Smith T, Giallambardo N, Stone C, Schadt K, Dolgoff J, Tetreault JC, Villarroel M, Kufe D. Fusion cell vaccination of patients with metastatic breast and renal cancer induces immunological and clinical responses. Clin Cancer Res. 2004;10:4699–4708. doi: 10.1158/1078-0432.CCR-04-0347. [DOI] [PubMed] [Google Scholar]
- 2.Banchereau J, Palucka AK, Dhodapkar M, Burkeholder S, Taquet N, Rolland A, Taquet S, Coquery S, Wittkowski KM, Bhardwaj N, Pineiro L, Steinman R, Fay J. Immune and clinical responses in patients with metastatic melanoma to CD34+ progenitor-derived dendritic cell vaccine. Cancer Res. 2001;61:6451–6458. [PubMed] [Google Scholar]
- 3.Banchereau J, Schuler-Thurner B, Palucka AK, Schuler G. Dendritic cells as vectors for therapy. Cell. 2001;106:271–274. doi: 10.1016/S0092-8674(01)00448-2. [DOI] [PubMed] [Google Scholar]
- 4.Barber LD, Madrigal JA. Exploiting beneficial alloreactive T cells. Vox Sang. 2006;91:20–27. doi: 10.1111/j.1423-0410.2006.00775.x. [DOI] [PubMed] [Google Scholar]
- 5.Beriou G, Peche H, Guillonneau C, Merieau E, Cuturi MC. Donor-specific allograft tolerance by administration of recipient-derived immature dendritic cells and suboptimal immunosuppression. Transplantation. 2005;79:969–972. doi: 10.1097/01.TP.0000158277.50073.35. [DOI] [PubMed] [Google Scholar]
- 6.Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 7.Bingaman AW, Ha J, Durham MM, Waitze SY, Tucker-Burden C, Cowan SR, Pearson TC, Larsen CP. Analysis of the CD40 and CD28 pathways on alloimmune responses by CD4+ T cells in vivo. Transplantation. 2001;72:1286–1292. doi: 10.1097/00007890-200110150-00018. [DOI] [PubMed] [Google Scholar]
- 8.Bregni M, Ueno NT, Childs R. The second international meeting on allogeneic transplantation in solid tumors. Bone Marrow Transplant. 2006;38:527–537. doi: 10.1038/sj.bmt.1705479. [DOI] [PubMed] [Google Scholar]
- 9.Buhlmann JE, Gonzalez M, Ginther B, Panoskaltsis-Mortari A, Blazar BR, Greiner DL, Rossini AA, Flavell R, Noelle RJ. Cutting edge: sustained expansion of CD8+ T cells requires CD154 expression by Th cells in acute graft versus host disease. J Immunol. 1999;162:4373–4376. [PubMed] [Google Scholar]
- 10.Childs R, Chernoff A, Contentin N, Bahceci E, Schrump D, Leitman S, Read EJ, Tisdale J, Dunbar C, Linehan WM, Young NS, Barrett AJ. Regression of metastatic renal-cell carcinoma after nonmyeloablative allogeneic peripheral-blood stem-cell transplantation. N Engl J Med. 2000;343:750–758. doi: 10.1056/NEJM200009143431101. [DOI] [PubMed] [Google Scholar]
- 11.Conrad R, Remberger M, Cederlund K, Hentschke P, Sundberg B, Ringden O, Barkholt L. Inflammatory cytokines predominate in cases of tumor regression after hematopoietic stem cell transplantation for solid cancer. Biol Blood Marrow Transplant. 2006;12:346–354. doi: 10.1016/j.bbmt.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 12.Dhodapkar MV, Steinman RM. Antigen-bearing immature dendritic cells induce peptide-specific CD8+ regulatory T cells in vivo in humans. Blood. 2002;100:174–177. doi: 10.1182/blood.V100.1.174. [DOI] [PubMed] [Google Scholar]
- 13.Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. 2001;193:233–238. doi: 10.1084/jem.193.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dubsky P, Ueno H, Piqueras B, Connolly J, Banchereau J, Palucka AK. Human dendritic cell subsets for vaccination. J Clin Immunol. 2005;25:551–572. doi: 10.1007/s10875-005-8216-7. [DOI] [PubMed] [Google Scholar]
- 15.Fabre JW. The allogeneic response and tumor immunity. Nat Med. 2001;7:649–652. doi: 10.1038/89008. [DOI] [PubMed] [Google Scholar]
- 16.Ferrara JL. Cytokine dysregulation as a mechanism of graft versus host disease. Curr Opin Immunol. 1993;5:794–799. doi: 10.1016/0952-7915(93)90139-J. [DOI] [PubMed] [Google Scholar]
- 17.Fukuma D, Matsuyoshi H, Hirata S, Kurisaki A, Motomura Y, Yoshitake Y, Shinohara M, Nishimura Y, Senju S. Cancer prevention with semi-allogeneic ES cell-derived dendritic cells. Biochem Biophys Res Commun. 2005;335:5–13. doi: 10.1016/j.bbrc.2005.06.096. [DOI] [PubMed] [Google Scholar]
- 18.Galea-Lauri J. Immunological weapons against acute myeloid leukaemia. Immunology. 2002;107:20–27. doi: 10.1046/j.1365-2567.2002.01497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galea-Lauri J, Darling D, Mufti G, Harrison P, Farzaneh F. Eliciting cytotoxic T lymphocytes against acute myeloid leukemia-derived antigens: evaluation of dendritic cell-leukemia cell hybrids and other antigen-loading strategies for dendritic cell-based vaccination. Cancer Immunol Immunother. 2002;51:299–310. doi: 10.1007/s00262-002-0284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galea-Lauri J, Wells JW, Darling D, Harrison P, Farzaneh F. Strategies for antigen choice and priming of dendritic cells influence the polarization and efficacy of antitumor T-cell responses in dendritic cell-based cancer vaccination. Cancer Immunol Immunother. 2004;53:963–977. doi: 10.1007/s00262-004-0542-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gong J, Nikrui N, Chen D, Koido S, Wu Z, Tanaka Y, Cannistra S, Avigan D, Kufe D. Fusions of human ovarian carcinoma cells with autologous or allogeneic dendritic cells induce antitumor immunity. J Immunol. 2000;165:1705–1711. doi: 10.4049/jimmunol.165.3.1705. [DOI] [PubMed] [Google Scholar]
- 22.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 23.Jiang HR, Muckersie E, Robertson M, Forrester JV. Antigen-specific inhibition of experimental autoimmune uveoretinitis by bone marrow-derived immature dendritic cells. Invest Ophthalmol Vis Sci. 2003;44:1598–1607. doi: 10.1167/iovs.02-0427. [DOI] [PubMed] [Google Scholar]
- 24.Karttunen J, Sanderson S, Shastri N. Detection of rare antigen-presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proc Natl Acad Sci USA. 1992;89:6020–6024. doi: 10.1073/pnas.89.13.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, Gabrilovich D. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63:4441–4449. [PubMed] [Google Scholar]
- 26.Lappin MB, Weiss JM, Delattre V, Mai B, Dittmar H, Maier C, Manke K, Grabbe S, Martin S, Simon JC. Analysis of mouse dendritic cell migration in vivo upon subcutaneous and intravenous injection. Immunology. 1999;98:181–188. doi: 10.1046/j.1365-2567.1999.00850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laurin D, Kanitakis J, Bienvenu J, Bardin C, Bernaud J, Lebecque S, Gebuhrer L, Rigal D, Eljaafari A. Allogeneic reaction induces dendritic cell maturation through proinflammatory cytokine secretion. Transplantation. 2004;77:267–275. doi: 10.1097/01.TP.0000101006.39475.41. [DOI] [PubMed] [Google Scholar]
- 28.Lee WC, Zhong C, Qian S, Wan Y, Gauldie J, Mi Z, Robbins PD, Thomson AW, Lu L. Phenotype, function, and in vivo migration and survival of allogeneic dendritic cell progenitors genetically engineered to express TGF-beta. Transplantation. 1998;66:1810–1817. doi: 10.1097/00007890-199812270-00040. [DOI] [PubMed] [Google Scholar]
- 29.Lutz MB, Suri RM, Niimi M, Ogilvie AL, Kukutsch NA, Rossner S, Schuler G, Austyn JM. Immature dendritic cells generated with low doses of GM-CSF in the absence of IL-4 are maturation resistant and prolong allograft survival in vivo. Eur J Immunol. 2000;30:1813–1822. doi: 10.1002/1521-4141(200007)30:7<1813::AID-IMMU1813>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 30.Marten A, Renoth S, Heinicke T, Albers P, Pauli A, Mey U, Caspari R, Flieger D, Hanfland P, Von Ruecker A, Eis-Hubinger AM, Muller S, Schwaner I, Lohmann U, Heylmann G, Sauerbruch T, Schmidt-Wolf IG. Allogeneic dendritic cells fused with tumor cells: preclinical results and outcome of a clinical phase I/II trial in patients with metastatic renal cell carcinoma. Hum Gene Ther. 2003;14:483–494. doi: 10.1089/104303403321467243. [DOI] [PubMed] [Google Scholar]
- 31.McIlroy D, Gregoire M. Optimizing dendritic cell-based anticancer immunotherapy: maturation state does have clinical impact. Cancer Immunol Immunother. 2003;52:583–591. doi: 10.1007/s00262-003-0414-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moore MW, Carbone FR, Bevan MJ. Introduction of soluble protein into the class I pathway of antigen processing and presentation. Cell. 1988;54:777–785. doi: 10.1016/S0092-8674(88)91043-4. [DOI] [PubMed] [Google Scholar]
- 33.Nair S, McLaughlin C, Weizer A, Su Z, Boczkowski D, Dannull J, Vieweg J, Gilboa E. Injection of immature dendritic cells into adjuvant-treated skin obviates the need for ex vivo maturation. J Immunol. 2003;171:6275–6282. doi: 10.4049/jimmunol.171.11.6275. [DOI] [PubMed] [Google Scholar]
- 34.Noble A, Leggat JA, Inderberg EM. CD8+ immunoregulatory cells in the graft-versus-host reaction: CD8 T cells activate dendritic cells to secrete interleukin-12/interleukin-18 and induce T helper 1 autoantibody. Immunology. 2003;109:476–486. doi: 10.1046/j.1365-2567.2003.01687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng Q, Li K, Patel H, Sacks SH, Zhou W. Dendritic cell synthesis of C3 is required for full T cell activation and development of a Th1 phenotype. J Immunol. 2006;176:3330–3341. doi: 10.4049/jimmunol.176.6.3330. [DOI] [PubMed] [Google Scholar]
- 36.Siders WM, Vergilis KL, Johnson C, Shields J, Kaplan JM. Induction of specific antitumor immunity in the mouse with the electrofusion product of tumor cells and dendritic cells. Mol Ther. 2003;7:498–505. doi: 10.1016/S1525-0016(03)00044-3. [DOI] [PubMed] [Google Scholar]
- 37.Suchin EJ, Langmuir PB, Palmer E, Sayegh MH, Wells AD, Turka LA. Quantifying the frequency of alloreactive T cells in vivo: new answers to an old question. J Immunol. 2001;166:973–981. doi: 10.4049/jimmunol.166.2.973. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki T, Fukuhara T, Tanaka M, Nakamura A, Akiyama K, Sakakibara T, Koinuma D, Kikuchi T, Tazawa R, Maemondo M, Hagiwara K, Saijo Y, Nukiwa T. Vaccination of dendritic cells loaded with interleukin-12-secreting cancer cells augments in vivo antitumor immunity: characteristics of syngeneic and allogeneic antigen-presenting cell cancer hybrid cells. Clin Cancer Res. 2005;11:58–66. [PubMed] [Google Scholar]
- 39.Wallgren AC, Andersson B, Backer A, Karlsson-Parra A. Direct allorecognition promotes activation of bystander dendritic cells and licenses them for Th1 priming: a functional link between direct and indirect allosensitization. Scand J Immunol. 2005;62:234–242. doi: 10.1111/j.1365-3083.2005.01663.x. [DOI] [PubMed] [Google Scholar]
- 40.Wells JW, Darling D, Farzaneh F, Galea-Lauri J. Influence of interleukin-4 on the phenotype and function of bone marrow-derived murine dendritic cells generated under serum-free conditions. Scand J Immunol. 2005;61:251–259. doi: 10.1111/j.1365-3083.2005.01556.x. [DOI] [PubMed] [Google Scholar]
- 41.Whitelegg AM, Oosten LE, Jordan S, Kester M, van Halteren AG, Madrigal JA, Goulmy E, Barber LD. Investigation of peptide involvement in T cell allorecognition using recombinant HLA class I multimers. J Immunol. 2005;175:1706–1714. doi: 10.4049/jimmunol.175.3.1706. [DOI] [PubMed] [Google Scholar]
- 42.Wierecky J, Mueller M, Brossart P. Dendritic cell-based cancer immunotherapy targeting MUC-1. Cancer Immunol Immunother. 2006;55:63–67. doi: 10.1007/s00262-005-0673-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Willis RA, Kappler JW, Marrack PC. CD8 T cell competition for dendritic cells in vivo is an early event in activation. Proc Natl Acad Sci USA. 2006;103:12063–12068. doi: 10.1073/pnas.0605130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu G, Xu X, Vu MD, Kilpatrick ED, Li XC. NK cells promote transplant tolerance by killing donor antigen-presenting cells. J Exp Med. 2006;203:1851–1858. doi: 10.1084/jem.20060603. [DOI] [PMC free article] [PubMed] [Google Scholar]