

Abstract
Adoptive cell transfer (ACT), either using rapidly expanded tumor infiltrating lymphocytes or T-cell receptor transduced peripheral blood lymphocytes, can be considered one of the most promising approaches in cancer immunotherapy. ACT results in the repopulation of the host with high frequencies of tumor-specific T cells; however, optimal function of these cells within the tumor micro-environment is required to reach long-term tumor clearance. We and others have shown that ongoing anti-tumor immune responses can be impaired by the expression of ligands, such as PD-L1 (B7-H1) on tumor cells. Such inhibitory molecules can affect T cells at the effector phase via their receptor PD-1. PD-L1/PD-1 interaction has indeed been shown crucial in inducing T-cell anergy and maintaining peripheral tolerance. In order to maximize anti-tumor responses, antibodies that target the PD-1/PD-L1 axis are currently in phase I/II trials. Alternatively, a more refined approach could be the selective targeting of PD-1 in tumor-specific T cells to obtain long-term resistance against PD-1-mediated inhibition. We addressed whether this goal could be achieved by means of retroviral siRNA delivery. Effective siRNA sequences resulting in the reduction of surface PD-1 expression led to improved murine as well as human T-cell immune functions in response to PD-L1 expressing melanoma cells. These data suggest that blockade of PD-1-mediated T-cell inhibition through siRNA forms a promising approach to achieve long-lasting enhancement of tumor-specific T-cell function in adoptive T-cell therapy protocols.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-010-0842-0) contains supplementary material, which is available to authorized users.
Keywords: siRNA, T cell, PD-1, PD-L1, B7-H1
Introduction
In addition to established anti-proliferative approaches, targeted therapies and immunotherapies are unraveling new ways to fight cancer [1, 2]. In particular, adoptive cell transfer (ACT) of T cells has gained attention due to the possibility to select, manipulate, and re-infuse tumor associated antigen (TAA) specific T cells, resulting in long-term persistence of the transferred T cells and high response rates against melanoma as demonstrated in recent phase I studies [1, 3–5]. However, despite significant induction of anti-tumor immune responses, tumor progression remains frequent [1, 3]. Mechanisms postulated to cause unresponsiveness or tumor immune escape include impairment of efficient antigen-presentation, insufficient co-stimulation, and/or presence of co-inhibitory molecules that inhibit T-cell responses.
The programmed death-1 (PD-1, CD279) receptor, that interacts with programmed death ligand-1 (PD-L1, B7-H1, CD274), is one of such co-inhibitory molecules. PD-1 is a member of the CD28 family and is up-regulated on T and B cells upon activation. Binding of PD-1 to one of its ligands (PD-L1 or PD-L2) in combination with T-cell receptor (TCR) signaling leads to inhibition of T-cell function including cytokine production, cytolytic activity, and proliferation [6–8]. Finally, as PD-L1 is up-regulated on a broad variety of human cancer cells [9–11], it is likely that the PD-1/PD-L1 signaling pathway may be involved in the immune escape of tumor cells [11, 12]. Multiple studies have demonstrated that PD-L1 expression on tumors is correlated with an unfavorable prognosis in kidney, ovarian, bladder, breast, gastric, and pancreatic cancers [13]. Moreover, in animal models PD-L1 over-expression on tumor cells results in impaired tumor control [11]. Thus, blocking the PD-1/PD-L1 signaling pathway may form a promising approach to improve the efficacy of anti-tumor immune therapy [12].
With the aim to enhance the function of tumor-reactive T cells, two blocking anti-PD-1 monoclonal antibodies have been developed and are currently tested in Phase I and II clinical trials ([13, 14] and Bramer et al., poster 3018, ASCO 2009). However, systemic disruption of the PD-1 gene in different mice strains led to strain-specific autoimmune diseases, like autoimmune cardiomyopathy, lupus-like arthritis, glomerulonephritis, and autoimmune diabetes [15, 16]. Although no side effects have been reported following administration of blocking antibodies during short-term tumor experiments, the knock-out mice suggest that long-term interference with PD-1/PD-L1 (e.g. during clinical maintenance therapies) could lead to unwanted autoimmune responses as observed in clinical trials testing anti-CTLA-4 monoclonal antibodies [17].
To develop a more targeted approach, a selective disruption of PD-1 signaling restricted to the TAA-specific T cells could prevent PD-1 inhibition mediated by the tumor. Here, we examined the possible use of short-hairpin double stranded silencing RNA (siRNA) [18] to restrain PD-1 surface expression on tumor-specific T cells.
We found that long-term siRNA-mediated reduction of PD-1 surface expression could be achieved on murine and human T cells by retroviral transduction, resulting in improved immune functions as measured by cytokine production, cytolytic degranulation, and proliferation.
Materials and methods
Animals
C57BL/6 2C TCRtg;RAG2−/− [19] and 2C;RAG2−/−;PD-1−/− mice [11] were kindly provided by Thomas F. Gajewski, University of Chicago, and Tasuko Honjo, University of Kyoto. The animals were maintained and used in agreement with the Institutional Animal Care and Use Committee, according to guidelines of the Society for Laboratory Animal Science, Germany, and national guidelines.
Tumor cell lines and culture
Tumor cell lines were cultured in complete medium as described before [9]. The murine lymphoma cell line EL4, the murine B lymphoma cell line A20, the murine plasmacytoma cell line J558, the P815.B7.1 mastocytoma cell line [20], the human B cell line CIRA2, and the murine melanoma cell line B16.SIY [9] have been described before as indicated.
The B16.SIY E12 cell line was obtained by transfection of B16.F10 murine melanoma with a variant of the pLEGFP-SIY vector [21] in which the GFP sequence was excised.
The producer cell line Phoenix-ECO was purchased from LGC/ATCC (Teddington, UK). The human melanoma cell lines Mel MTO and Mel GBU, as well as the producer cell line FlyRD, were kept at the NKI. Human melanoma cell lines were cultured in RPMI, 10% FCS (Greiner bio one), and penicillin/streptomycin (Roche).
To augment PD-L1 expression, murine and human tumor cell lines were cultured for 48 h in the presence of 20 ng/ml rMU IFNγ or 200 ng/ml rHU IFNγ, respectively (PromoKine, Germany). Cells were washed 3 times after incubation and no detectable IFNγ was present in supernatant of thus treated tumor cells.
Antibodies
Antibodies against the following murine molecules coupled to the indicated fluorochromes were purchased from BD Pharmingen (San Diego, CA): anti-CD4-PE, anti-CD8α-APC, rat-IgG2a,κ-PE, rat-IgG2a,κ-APC, and eBioscience (San Diego, CA): anti-PD-1-PE, anti-PDL1-PE, golden Syrian hamster-IgG-PE.
The mAb 1B2, recognizing the murine 2C TCR [22], was prepared from a hybridoma provided by Thomas F. Gajewski, University of Chicago.
Capture/detector antibody pairs for ELISA detecting the murine or human cytokines IL-2 or IFNγ were purchased from BD Pharmingen and used according to the manufacturer’s protocol.
Antibodies recognizing the following human molecules coupled to the indicated fluorochromes were purchased from BD biosciences: anti-CD4-PE/FITC, anti-CD8-PE/FITC/PercP.Cy5.5, anti-CD8-APC, anti-CD107a-PE, anti-HLA-A2-FITC, anti-INF-γ-PE, and eBioscience: anti-PD-1-AF647, anti-PDL1-PE, mIgG-AF647/PE.
Surface expression of TCR-transduced PBMCs was measured by staining with MHC-tetramers, using MHC-monomers generated by ultraviolet-induced peptide exchange [23].
Flow cytometric analysis was performed using FACSCaliburTM (Becton-Dickinson) flow cytometers and FlowJo software (TreeStar, San Carlos, CA).
T-cell purification
Spleens were harvested from 2C/RAG2−/− and 2C/RAG2−/−/PD-1−/− mice and prepared into single cell suspensions. CD8+ T cells were purified by negative selection using SpinSep Kit according to the manufacturer’s instructions (StemCell, Vancouver, Canada). An aliquot of purified cells was routinely stained with 1B2-FITC, CD8α-APC, and CD4-PE for analysis by flow cytometry. T-cell purity was generally >90%.
Human PBMCs
Peripheral blood mononuclear cells (PBMCs) from anonymous healthy donors were obtained from the local blood bank (Sanquin, Amsterdam) and isolated from buffy-coats and subsequent Ficoll-Isopaque density centrifugation.
Plasmids, siRNA sequences, and other reagents
pSUPER.retro.puro and pSUPER.retro.neo.GFP (Oligoengine, Seattle) were, respectively, used to express the siRNA sequences, and the plasmid pMX encoding the 1D3 hmCys TCR, previously described [24] and modified at the NKI-AVL was used to generate MART-1-specific T cells.
The siRNA sequences targeting PD-1 (supplemental Table 1) were cloned into the pSUPER plasmids according to the instructions of the manufacturer (oligoengine, Seattle). siRNA-mu1, 2, and 3 and siRNA-hu1, 2, and 3 were designed by Ambion using their in house algorithm, siRNA-mu4 to -10 were designed using the sfold-software (http://sfold.wadsworth.org/). SiRNA-shuffle was generated by shuffling the sequence of siRNA-mu2.
pSUPER.retro.puro transduced T cells were cultured in the presence of 2.5 μg/ml puromycin (Sigma-Aldrich, Taufkirchen, Germany).
Amaxa electroporation
The murine lymphoma cell line EL4 and primary 2C TCRtg T cells were transfected using the AMAXATM electroporator and the Mouse T-cell NucleofectorTM kit according to the manufacturer’s protocol with the program setting X-01 (Amaxa, Köln, Germany). CIRA2 tumor cells were transfected with the Human T cell NucleofectorTM kit with the program setting U-15.
Retroviral transduction of murine T cells
The producer cell line Phoenix-ECO was double-transfected with pSUPER.retro.puro-siRNA and the packaging vector pCL-ECO (Imgenex, San Diego, CA) using FuGENE6 lipofection (Roche, USA) following manufacturer’s protocol. As much as 24 h after transfection, the medium was refreshed. The viral supernatant was harvested on day 2 and added to 2C TCRtg splenocytes, which had been activated with P815.B7.1 mastocytoma cells the day before. Cells were transfected via spinfection (90 min, 430 g) on two consecutive days [25] using polybrene (Sigma-Aldrich, Germany) (8 μg/ml final concentration). After the second spinfection 2C TCRtg T cells were repetitively stimulated with P815.B7.1 mastocytoma cells in the presence of puromycin in order to select for pSUPER transduced cells.
Retroviral transduction of human T cells
FLYRD18 packaging cells were plated in 10 cm plates at 1.2 × 106 cells/well. After one day, cells were transfected with 10 μg retroviral vector DNA using FuGENE-6 (Roche Diagnostics, Indianapolis, IN). After 48 h, retroviral supernatant was pooled, centrifuged, and frozen at −80°C. PBMCs were activated with 20 U/mL IL-2 and 2 μg/ml phytohemagglutinin, at 106 cells/ml. After 48 h of stimulation, PBMCs were resuspended in retroviral supernatant, transferred to RetroNectin-coated plates at 0.5 × 106 cells/ml, and centrifuged for 90 min at 430 g. For co-transductions, the indicated retroviral supernatants were mixed at a ratio of 1:1. Following transduction, PBMCs were cultured in Yssel’s medium supplemented with 20 U/mL IL-2 (Proleukin; Chiron, CA) and stimulated with irradiated JY cells and allogeneic PBMCs, plus 100 ng/ml phytohemagglutinin and 20 U/mL IL-2. Prior to functional assays, transduced PBMCs were stimulated for 48 h with 2 μg/ml phytohemagglutinin to induce PD-1 up-regulation.
Cytokine and proliferation assay
IL-2 or IFNγ production was detected in supernatants by ELISA after coculturing siRNA transfected or retrovirally transduced 2C TCRtg T cells with mitomycin C-treated B16.SIY melanoma cells for 18 h. To detect proliferative capacity of the siRNA transduced 2C TCRtg T cells, the cells were co-cultured with B16.SIY-IFNγ for 18 h, subsequently H3-thymidine (1 μCi/well) was added and cells were harvested 6 h later and radiolabel integration measured with a TopCount-NXT instrument (Perkin Elmer).
In vitro PD-L1 blockade
During co-culture of T cells and tumor cells, 10 μg/ml functional grade purified anti-mouse B7-H1 (PD-L1) antibody or functional grade purified rat IgG2a isotype control (eBiosience, San Diego, CA) was added to murine T cells and 10 μg/ml anti-PD-L1 antibody (clone 5-496, previously described, [6, 26]) or IgG control = sandoglobulin (Novartis, Arnhem) was added to human T cells.
Intracellular IFNγ staining and LAMP-test
Activated human T cells were co-cultured with tumor cells (E:T = 1) in 200 μl Yssel’s medium in the presence of 2 μl Golgi Plug (BD). After 4 h of incubation the cells were stained extracellularly with anti-CD107a (LAMP)-PE or intracellularly with anti-IFNγ-PE using the Cytofix/Cytoperm Kit (BD) according to the manufacturer’s instructions.
Statistical analysis
Two-tailed Student t tests were performed where indicated, differences between datasets with a p-value of ≤0.05 were considered significant.
Results
Identification of functional siRNA sequences to suppress murine PD-1 surface expression
PD-1 has been shown to be absent on naïve T cells and to be up-regulated transiently upon activation [9]. As a result, primary T cells were inadequate for initial screening of potential siRNA sequences concerning their efficacy to down-regulate PD-1 expression. Therefore, we analyzed various murine lymphoid cell lines by flow cytometry for constitutive PD-1 surface expression and found the murine lymphoma cell line EL4 to express PD-1 at high expression levels (Fig. 1a).
Fig. 1.

Transient reduction of PD-1 surface expression on murine T cells. a The murine tumor cell lines EL4, J558, and A20 were analyzed for PD-1 expression by flow cytometry (filled PD-1 staining; fine line isotype staining). b 2 × 106 EL4 cells were Amaxa transfected with 3 μg PD-1-specific siRNA-mu1, -mu2, or -mu3 and analyzed after 48 h by flow cytometry (filled PD-1-stained untransfected EL4; bold line PD-1-stained siRNA-transfected EL4; fine line IgG-stained untransfected EL4). c Freshly isolated 2C TCRtg CD8+ T cells were stimulated with P815.B7.1 (E:T 1:5). On day 4, 2 × 106 primed T cells were transfected with 3 μg siRNA-mu1, -mu2, or -mu3. After resting 4 h at 37°C, the transfected T cells were stimulated with IFNγ pretreated B16.SIY (E:T = 1:1). As much as 48 h after transfection, the stimulated and transfected 2C TCRtg T cells were analyzed for PD-1 expression via flow cytometry (filled untransfected 2C TCRtg T cells; bold line transfected 2C TCRtg T cells; fine line 2C TCRtg PD1−/− T cells). 2C TCRtg T cells were transfected with the indicated murine siRNAs and stimulated with IFNγ pretreated B16.SIY for 18 h (E:T = 1:1). Co-culture supernatants were analyzed for IL-2 (d) and IFNγ (e) production. Data are representative of two independent experiments. *P < 0.05; n.s. non-significant
We tested several siRNAs (siRNA-mu1, -mu2, -mu3; Ambion) directed against murine PD-1 for their capacity to down-regulate PD-1 surface expression on this cell line. Transfection efficiency into EL4 cells reached up to 95% of surviving cells (data not shown) and transfected cells were analyzed by flow cytometry 24, 48, 72, and 96 h after transfection. SiRNA-mu2 and to a lesser extent siRNA-mu1 were capable of reducing PD-1 surface expression (Fig. 1b). Compared to cells transfected with the FAM-labeled negative control siRNA (Ambion) (Mean fluorescence intensity, MFI, for PD-1 = 684), cells transfected with siRNA-mu1 and siRNA-mu2 displayed a 1.9 and 3-fold reduction in PD-1 expression (MFI 370 and 225). Transfection of cells with siRNA-mu3 did not reduce the PD-1 expression (MFI 744). The reduction of PD-1 expression obtained upon transfection with siRNA-mu1 or siRNA-mu2 was observed up to 72 h (data not shown).
Improved T-cell function induced by siRNA-mediated PD-1 surface suppression in activated TCR transgenic T cells
Having established efficient reduction of PD-1 surface expression in EL4 cells, we then tested the siRNAs in primary T cells. CD8+ T cells were purified from splenocytes of 2C TCR transgenic (tg) mice. As PD-1 is strongly up-regulated on activated T cells, the naïve CD8+ T cells were stimulated with P815.B7.1 mastocytoma cells expressing the p2CA antigen recognized by the 2C TCR as well as B7.1 to provide co-stimulatory signals (described previously [25]). After 4–5 days, primed T cells were harvested and transfected by electroporation with siRNAs. In parallel, P815.B7.1-stimulated 2C TCRtg T cells and PD1−/− 2C TCRtg T cells were mock transfected to obtain T cells with unaltered PD-1 suppression and a control for complete absence of PD-1 surface expression.
Four hours after transfection, the T cells were co-cultured with B16.SIY-INFγ, a murine melanoma cell line that expresses the cognate SIY (SIYRRYGL) antigen [22, 27] and PD-L1 at high surface levels induced by prior IFNγ exposure [9]. Flow cytometric analysis 48 h after transfection (44 h after re-stimulation) showed that transfection of primary T cells with PD-1 siRNAs resulted in a suppression of PD-1 surface expression similar to that observed on EL4 lymphoma cells. SiRNA-mu2 was again most efficient with a 3.1-fold suppression of PD-1 (MFI = 206) as compared to the mock transfected 2C TCRtg T cells (MFI = 647), while siRNA-mu1 and siRNA-mu3 led to a 1.5 (MFI = 443) and 1.1 (MFI = 590) fold suppression of PD-1 expression, respectively. A fraction of the siRNA-mu2 transfected 2C TCRtg T cells presented complete PD-1 surface suppression identical to PD-1 deficient 2C T cells (Fig. 1c).
Subsequently, we addressed whether the transient knock-down of PD-1 surface expression was sufficient to enhance T-cell responsiveness. To this purpose, previously activated 2C TCRtg T cells were transfected with the indicated siRNAs and co-cultured for 18 h with PD-L1 expressing B16.SIY-IFNγ melanoma cells. Supernatants were analyzed for IL-2 and IFNγ content by ELISA. We measured an increase of 34% IL-2 and 75% IFNγ production when T cells were transfected with siRNA-mu2 as compared to mock transfected 2C TCRtg T cells (Fig. 1d, e). These results demonstrated that a transient siRNA-induced PD-1 surface suppression was able to improve cytokine production of TCRtg T cells exposed to melanoma targets expressing PD-L1.
Retroviral transduction with siRNA targeting PD-1 results in stable reduction of PD-1 surface expression and improved T-cell function
As mentioned above, siRNA transfection was transient and PD-1 surface suppression was no longer detectable after 96 h (data not shown). We thus tested stable transduction using the retroviral pSUPER RNAi vector system to generate T cells with sustained PD-1 knock-down. To broaden our panel of siRNAs, seven additional siRNA sequences were created using the sfold-software.
Three out of the seven newly designed siRNA showed relevant reduction of PD-1 surface expression (pSUPER-siRNA-4, -7, and -10; Supplemental Fig. 1); however, siRNA-mu2 remained the most efficient sequence. pSUPER-siRNA-mu4 was slightly less efficient than pSUPER-siRNA-mu2 and was chosen for further experiments as an intermediate between highly functional siRNA-mu2 and the negative control.
After retroviral transduction and puromycin selection of 2C TCRtg T cells, flow cytometric analysis showed a potent reduction of PD-1 surface expression with pSUPER-siRNA-mu2 (MFI 73), whereas T cells transduced with pSUPER-siRNA-mu4 (MFI 168) were comparable to pSUPER-empty transfected T cells (MFI 170). Shuffle siRNA did not alter PD-1 surface expression resulting in an identical MFI as the empty vector transduced T cells (Fig. 2a). These effects were stable as we could find sufficient PD-1 suppression even after up to 7 rounds of stimulation (Supplemental Fig. 2). Moreover, the stable alteration of PD-1 surface expression had no impact on the T-cell phenotype. Other than the decrease in PD-1 expression, untransduced or pSUPER-siRNA-shuffle transduced 2C TCRtg T cells had a similar phenotype to that of pSUPER-siRNA-mu2 transduced T cells (Supplemental Fig. 2).
Fig. 2.

Stable reduction of PD-1 surface expression on murine T cells. a 2C TCRtg T cells were retrovirally transduced with pSUPER-siRNA-vectors containing the indicated siRNAs and restimulated with P815.B7 (E:T = 1:5) in the presence of puromycin (2.5 μg/ml) for at least three stimulation rounds. Flow cytometry analysis was performed on day 2 after re-stimulation (filled untransduced 2C TCRtg T cells; bold line siRNA transduced 2C TCRtg T cells). b Total RNA was isolated from 106 T cells of each group: naïve 2C TCR transgenic cells, untransduced (=activated), pSuper-empty, pSuper-siR-mu4, pSuper-siR-mu2, and PD1−/− 2C TCR transgenic T cells (stimulated as in a). RNA was retro-transcribed into cDNA and subsequently analyzed with PD-1-specific primers by PCR. The intensity of the bands was measured via densitometry and normalized against β-actin. c As in a, 3 times stimulated, transduced 2C TCRtg T cells were co-cultured with IFNγ pretreated B16.SIY.E12 (E:T = 1:1) in the presence of 3H-thymidine. Radioactive uptake was measured after 6 h. d As in c, transduced 2C TCRtg T cells were co-cultured with IFNγ pretreated B16.SIY.E12 (E:T = 1:1) and IFNγ was measured in 18 h-culture supernatant by ELISA. e 2C TCRtg T cells, transduced with indicated siRNAs and selected with puromycin for several rounds of restimulation were co-cultured with IFNγ pretreated B16.SIY.E12 (E:T = 1:1) in the presence of no antibody, isotype control (10 μg/ml), or anti-PD-L1 mAb (10 μg/ml). IFNγ was measured by ELISA after 18 h. Data are representative of two independent experiments. **P < 0.01; n.s.: non-significant
We confirmed the impact of retroviral delivery of anti-PD-1 siRNA directly at the mRNA expression level by RT-PCR. Analysis of the results through densitometry, normalized against the housekeeping gene β-actin, confirmed the ability of pSUPER-siRNA-mu4 and -mu2 to induce PD-1 mRNA degradation (Fig. 2b).
We then addressed the benefits of stable PD-1 knock-down on the proliferative capacity and cytokine production of retrovirally modified 2C T cells. pSUPER-siRNA-shuffle, -siRNA-mu2 or -siRNA-mu4 transduced T cells were incubated with B16.SIY-IFNγ in the presence of H3-Thymidine and radioactive uptake was measured after 6 h. pSUPER-siRNA-mu2 transduced cells showed a 2.6-fold higher uptake compared to pSUPER-siRNA-shuffle and a 1.5-fold higher uptake compared to pSUPER-siRNA-mu4 (Fig. 2c). The improved proliferative capacity of pSUPER-siRNA-mu2 transduced 2C T cells was accompanied by an increased ability to produce IFNγ in response to antigen recognition of a similar magnitude (2.4-fold) (Fig. 2d).
Flow cytometry results of pSUPER-siRNA-mu2 transduced T cells showed reduced PD-1 expression on the cells but no complete loss of expression (Fig. 2a). This posed the question whether knock-down of PD-1 on 2C TCRtg T cells was sufficient to block all signaling via the PD-1/PD-L1 pathway or whether there was residual signaling. To address this, pSUPER-siRNA-mu2 or pSUPER-siRNA-mu4 transduced 2C T cells were co-cultured with B16.SIY-IFNγ in the presence of functional anti-PD-L1 blocking antibody or control IgG.
As expected siRNA-mu2 transduced T cells reached higher levels of IFNγ production than cells transduced with siRNA-mu4 in absence of any antibody or presence of the control IgG. The addition of anti-PD-L1 blocking antibody, however, led to a further increase in IFNγ production from siRNA-mu2 transduced T cells, indicating that the residual expression of PD-1 upon transduction with anti-PD-1 siRNA was associated with residual inhibitory activity (Fig. 2e).
Identification of siRNA sequences targeting human PD-1
Based on the proof of principle that retroviral siRNA transduction can lead to stable alteration of PD-1 surface expression in murine T cells, we sought to find PD-1-targeting siRNA sequences effective in human T cells.
Analogous to our approach in the murine system, we tested pre-designed siRNAs (Ambion; siRNA-hu1, -hu2, -hu3) transiently upon transfection of the human B-cell line CIRA2, which expresses PD-1 constitutively (Fig. 3a). Compared to shuffle-siRNA transfected B cells (MFI 121), siRNA-hu1 or -hu3 transfected cells showed similar reduction in PD-1 surface expression (MFI 52 or 45), whereas siRNA-hu2 had no effect on the PD-1 expression (MFI 129). SiRNA-hu1 was chosen for further experiments.
Fig. 3.

Reduction of PD-1 surface expression on human T cells. a 2 × 106 CIRA2 cells were transfected with the indicated siRNAs and analyzed after 45 h by flow cytometry (filled PD-1stained untransfected CIRA2; bold line PD-1-stained siRNA-transfected CIRA2; fine line IgG-stained untransfected CIRA2, isotype). Human PBL were retrovirally transduced with pMP71-1D3hmcys-TCR alone (upper panel) or in combination with pSUPER-siRNA-hu1-GFP (lower panel). As much as 4 days after transduction the transduction efficiency was analyzed by flow cytometry on CD4+ T cells (b) and CD8+ T cells (c). d Human PBLs were co-transduced with pMP71-1D3-hmcys-TCR and pSUPER-siRNA-hu1-GFP or pSUPER-shuffle-GFP and restimulated with 2 μg/ml PHA to induce PD-1 surface expression. On day 2, PD-1 expression was analyzed by flow cytometry (cells were pre-gated for 1D3 TCR+ GFP+CD4+ or 1D3 TCR+GFP+CD8+; filled siRNA-shuffle-GFP transduced control; bold line siRNA-hu1-GFP transduced). Data are representative of four independent experiments
To establish stable PD-1 knock-down in human T cells, to avoid puromycin selection, and to identify double-transduced T cells, we integrated siRNA-hu1 and siRNA-shuffle into the pSUPER.retro.neo.GFP vector, thus allowing analysis of siRNA transduced T cells by flow cytometry.
To generate tumor-specific T cells with a defined antigen reactivity we transduced human PBLs with the MART-1-specific TCR 1D3 [24]. Single transduction of PBL with the vector pMP71-1D3hmcys encoding the TCRα and β chain of the 1D3 TCR resulted in gene-modification efficacy of 56% for CD4+ and 50% for CD8+ (identified by MART-1-MHC tetramer staining). Double transduction with the 1D3 TCR and pSUPER-siRNA-GFP yielded on average 22 ± 5% GFP+1D3+CD4+ and 15 ± 3% double transduced CD8+ T cells (Fig. 3b, c).
Subsequently, we analyzed the PD-1 expression on 1D3 TCR+ GFP+ CD4+ or CD8+ T cells co-transduced with either pSUPER-siRNA-hu1-GFP or the control pSUPER-siRNA-shuffle-GFP. Although PD-1 expression on the CD4+ (MFI = 22 for siRNA-shuffle) and CD8+ (MFI = 18 for siRNA-shuffle) T cells was modest, 1D3+ GFP+ cells containing siRNA-hu1 showed approximately 50% down-regulation of their surface PD-1 (MFI = 7 for CD4+, MFI = 11 for CD8+ T cells) (Fig. 3d). The stable PD-1 knockdown in human T cells could be maintained for at least 6 rounds of stimulation (data not shown).
Retroviral transduction with siRNA targeting PD-1 improves human melanoma-specific T-cell functions
The seemingly low PD-1 surface suppression obtained led us to assess to what extent PD-1-targeting siRNAs can improve human T-cell functionality. In order to specifically focus on the impact of PD-1 interference with the function of the tumor-specific T cells (and to ignore single transduced T cells present in the culture) we used a flow cytometry-based approach consisting in measuring intracellular IFNγ and extracellular LAMP-1 expression (=CD107a, a marker of cytotoxic degranulation of T cells). To this purpose, double-transduced PBL were co-incubated with melanoma cells that were positive for the cognate antigen of the 1D3 TCR, MART-1 and were pretreated with IFNγ in order to induce up-regulation of PD-L1 surface expression (MTO IFNγ) (Supplemental Fig. 3). Anti-PD-L1 mAb (MTO IFNγ + αPD-L1) or IgG control (MTO IFNγ + IgG) were used to assess the relative effects of siRNA as compared to mAb blockade of PD-L1 (Fig. 4). In control conditions, the T cells were left alone. The 1D3 TCR transduced PBLs were either co-transduced with a vector coding an irrelevant siRNA control (siR-shuffle) or the relevant anti-PD-1 siRNA (siR-hu1) or without an additional vector (mock). In first instance, when analyzing IFNγ expression by 1D3+ CD8+ T cells (Fig. 4a), we found that siR-hu1 had a slight but significant effect on the increased frequency of IFNγ producing T cells. Although the use of anti-PD-L1 antibody had a very low effect on improving IFNγ secretion, no improvement in T-cell responsiveness was obtained when combining siR-hu1 and anti-PD-L1 blocking antibodies (Fig. 4a). Similar conclusions could be drawn regarding the frequency of T cells displaying cytotoxic degranulation (Fig. 4b). Interestingly, the impact of cell surface PD-1 reduction by siR-hu1 compared to that of mock transduction or siR-shuffle transduction was more prominent in 1D3+ CD4+ T cells, where it resulted in a 2-fold increase in the frequency of MART-1 specific IFNγ expression (Fig. 4c). Addition of anti-PD-L1 blocking antibodies could not improve the T-cell response. Similar effects of siR-hu1 and anti-PD-L1 blocking antibodies were observed on the MART-1-specific cytotoxic responses of CD4+ T cells (Fig. 4d). Of note, the intensity of fluorescence reflecting the level of IFNγ secretion of either CD8+ or CD4+ T cells was comparable whether the cells were transduced with anti-PD-1 siRNA or not (data not shown). Comparable results were obtained when performing an ELISpot assay (Supplemental Fig. 4).
Fig. 4.
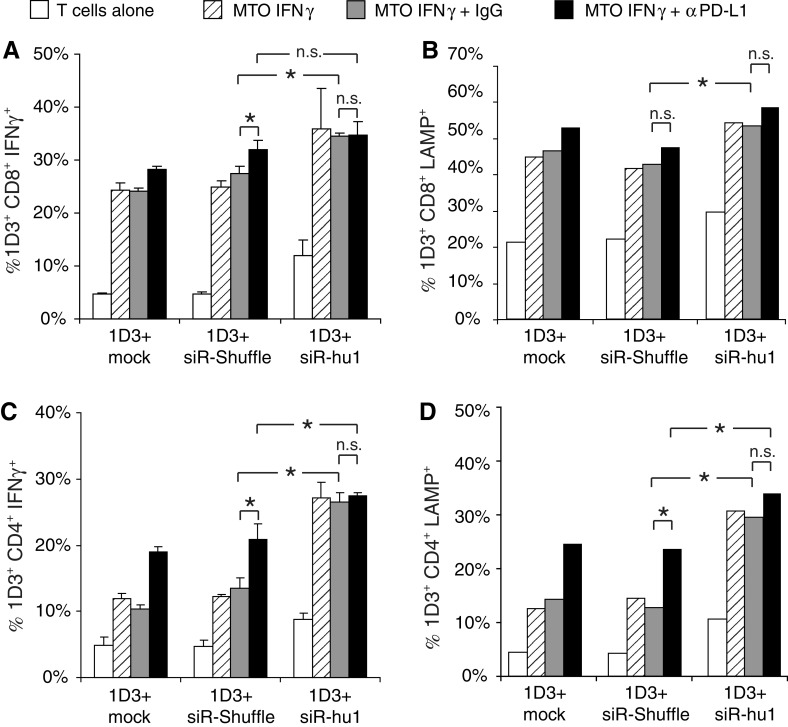
Functional analysis of human T cells with reduced PD-1 surface expression. a The melanoma cell line MTO was pretreated with 200 ng/ml IFNγ for 48 h to induce PD-L1 surface expression (MTO IFNγ) before starting the co-culture. Single and double 1D3 TCR/siRNA-hu1 (or -shuffle) co-transduced human PBLs were activated for 48 h with 2 μg/ml PHA to induce PD-1 surface expression. 2 × 104 pre-activated T cells were co-cultured (E:T = 1:5) for 4 h in the following settings: no tumor cells, MTO IFNγ, MTO IFNγ + 10 μg/ml sandoglobulin (IgG), or MTO IFNγ + 10 μg/ml anti-PD-L1 mAb (αPD-L1). Cells were stained with a MART-1-tetramer, CD8, and intracellular IFNγ for flow cytometry analysis. Shown are frequencies of CD8+ lymphocytes that were MART-1-tet+, GFP+ (siRNA transduction reporter), and IFNγ+. b For the analysis of the cytolytic activity via the LAMP (CD107a) expression on the T cells during stimulation, anti-LAMP-PE antibody was added prior to incubation. After 4 h of co-culture, the cells were stained with MART-1-tetramer and CD8 and analyzed by flow cytometry, following the same gating scheme as in a. c IFNγ production and d cytolytic degranulation of MART-1-specific CD4+ T cells were assessed similar to CD8+ T cells. Data are representative of two independent experiments. *P < 0.05; n.s. non-significant
Thus, in contrast to the siRNA sequences identified to target murine PD-1, the human siRNA sequence improved the frequency of cytokine producing human CD4+ T cells as effectively as the antibody blockade.
In summary, these results demonstrate that reduction of PD-1 surface expression on gene-modified human melanoma-specific T cells leads to improved cytokine production and cytotoxicity of the T cells.
Discussion
PD-L1 is broadly expressed on human tumors and its high expression is correlated with poor clinical prognosis [13]. Moreover, PD-1/PD-L1 signaling blockade has been shown to improve anti-tumor immune responses in animal models [9–11] and for human T cells in in vitro studies [6, 10]. Such signaling blockade may be a promising approach to improve T-cell function and is currently tested with two different mAb in clinical phase I and II studies ([14] and Bramer et al., poster 3018, ASCO 2009).
However, PD-1/PD-L1 signaling plays a pivotal role in the preservation of peripheral tolerance [15]. In order to circumvent the need for long-term systemic suppression of the PD-1/PD-L1 axis on the open repertoire adaptive immunity, a selective T-cell interference of the PD-1 signaling might enable specific long-lasting immune responses without unwanted side effects. Therefore, the possible selective knock-down of PD-1 surface expression on tumor-specific T cells using siRNA technology was investigated in this study.
We cloned validated siRNAs targeting murine PD-1 (siRNA-mu2 and -mu4) into the pSUPER-vector system [28] to allow stable siRNA expression within transduced T cells. Retroviral siRNA transduction resulted in mRNA degradation and sustained suppression of PD-1 surface expression, which led to increased proliferation and IFNγ production.
Similar to the approach in the murine setting, we developed siRNAs targeting PD-1 in human T cells. Various siRNA sequences were screened by transient transfection into a B-cell line that displays constitutive PD-1 expression (CIRA2) and we identified two siRNA sequences that interfere efficiently with PD-1 surface expression. We co-transduced the most effective siRNA-hu1 with the melanoma antigen (MART-1)-specific 1D3 TCR [24] into human PBLs. The combination of two retroviral vectors containing the 1D3 TCR genes and the siRNA targeting human PD-1 led to relatively low transduction efficiencies that were higher in CD4+ T cells than in CD8+ T cells (respectively 26% and 16% double transduced T cells). However, for a possible clinical application it would be favorable to achieve better co-transduction efficiencies and exclude single transduced T cells. This could be obtained by expressing the TCRα and β chain together with the siRNA within one single retroviral vector. Such multicistronic vectors have been described recently [29, 30].
To focus on TCR-transduced T cells expressing the siRNA, analyses were performed by flow cytometry on double-transduced T cells. Therefore, the effects of PD-1 knock-down on the function of human T cells was measured by intracellular IFNγ expression and extracellular presentation of the lysosomal molecule LAMP (CD107a), and not by Chromium release assay. The impact of the siRNAs was greater in CD4+ than in CD8+ T cells, resulting in more significant frequency of IFNγ producing cells as well as CD107a expression within the CD4+ compartment as compared to the shuffle transduced T cells. These observations are in line with previous data showing that human CD4+ T cells are (more) susceptible for PD-L1-mediated T-cell inhibition [6, 7]. Moreover, we have observed that PD-1-mediated inhibition is more pronounced at relatively low antigen density (manuscript in preparation). As the 1D3 TCR is MHC-I restricted, the CD4+ T cells that recognize the MART-1 antigen do not benefit from the costimulatory molecule CD8. Such lack of costimulation might be another reason for the increased susceptibility to PD-1 inhibition observed in transduced CD4+ T cells.
In contrast to what was observed for murine T cells, addition of anti-PD-L1 mAb to human siRNA transduced T cells did not result in a further increase in cytokine production or cytolytic activity. This observation suggests that the identified human siRNA sequences are more effective than that of the mouse sequences or that the available anti-human PD-L1 antibodies are unable to completely block PD-1/PD-L1 interaction.
Another advantage of the presently described method may result from the fact that T cells mediating anti-tumor immunity release IFNγ within the tumor microenvironment. This IFNγ can in turn induce up-regulation of PD-L1 at the tumor cell surface, leading to an increased delivery of inhibitory signals to the tumor-specific T cells. It is within this context that siRNA-mediated PD-1 targeting in tumor-specific T cells may prove most beneficial to optimize the long-term anti-tumor function of adoptively transferred gene-engineered T cells. Further in vivo experiments will have to be performed to support such hypotheses.
In summary, we functionally defined siRNAs capable of knocking-down co-inhibitory molecules at the surface of mouse and human T cells. This method may be an alternative approach to modulate the function of tumor-specific T cells generated by TCR transduction, or TIL expansion for adoptive T-cell therapy.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by the Wilhelm Sander-Stiftung, grant 2005.020.01 and the Dutch Cancer Society (KWF) grant, NKI 2008-3988 to C.B. We thank Bianca Heemskerk for carefully reviewing this manuscript.
Abbreviations
- MFI
Mean fluorescence intensity
- PD-1
Programmed death-1 receptor (CD279)
- PD-L1
Programmed death ligand-1 (B7-H1, CD274)
- PBL
Peripheral blood leukocytes
- TIL
Tumor infiltrating lymphocytes
- TCR
T cell receptor
- mAb
Monoclonal antibody
References
- 1.Rosenberg SA, Dudley ME. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr Opin Immunol. 2009;21:233–240. doi: 10.1016/j.coi.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.June CH. Adoptive T cell therapy for cancer in the clinic. J Clin Invest. 2007;117:1466–1476. doi: 10.1172/JCI32446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mackensen A, Meidenbauer N, Vogl S, Laumer M, Berger J, Andreesen R. Phase I study of adoptive T-cell therapy using antigen-specific CD8+ T cells for the treatment of patients with metastatic melanoma. J Clin Oncol. 2006;24:5060–5069. doi: 10.1200/JCO.2006.07.1100. [DOI] [PubMed] [Google Scholar]
- 4.Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, Greenberg PD. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA. 2002;99:16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Schallmach E, Kubi A, Shalmon B, Hardan I, Catane R, Segal E, Markel G, Apter S, Nun AB, Kuchuk I, Shimoni A, Nagler A, Schachter J. Minimally cultured or selected autologous tumor-infiltrating lymphocytes after a lympho-depleting chemotherapy regimen in metastatic melanoma patients. J Immunother. 2009;32:415–423. doi: 10.1097/CJI.0b013e31819c8bda. [DOI] [PubMed] [Google Scholar]
- 6.Blank C, Kuball J, Voelkl S, Wiendl H, Becker B, Walter B, Majdic O, Gajewski TF, Theobald M, Andreesen R, Mackensen A. Blockade of PD-L1 (B7–H1) augments human tumor-specific T cell responses in vitro. Int J Cancer. 2006;119:317–327. doi: 10.1002/ijc.21775. [DOI] [PubMed] [Google Scholar]
- 7.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, Ling V, Bowman MR, Carreno BM, Collins M, Wood CR, Honjo T. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, Greenfield EA, Bourque K, Boussiotis VA, Carter LL, Carreno BM, Malenkovich N, Nishimura H, Okazaki T, Honjo T, Sharpe AH, Freeman GJ. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 9.Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, Gajewski TF. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64:1140–1145. doi: 10.1158/0008-5472.CAN-03-3259. [DOI] [PubMed] [Google Scholar]
- 10.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 11.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA. 2002;99:12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blank C, Gajewski TF, Mackensen A. Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: implications for tumor immunotherapy. Cancer Immunol Immunother. 2005;54:307–314. doi: 10.1007/s00262-004-0593-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berger R, Rotem-Yehudar R, Slama G, Landes S, Kneller A, Leiba M, Koren-Michowitz M, Shimoni A, Nagler A. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res. 2008;14:3044–3051. doi: 10.1158/1078-0432.CCR-07-4079. [DOI] [PubMed] [Google Scholar]
- 15.Nishimura H, Honjo T. PD-1: an inhibitory immunoreceptor involved in peripheral tolerance. Trends Immunol. 2001;22:265–268. doi: 10.1016/S1471-4906(01)01888-9. [DOI] [PubMed] [Google Scholar]
- 16.Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1−/− mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci USA. 2005;102:11823–11828. doi: 10.1073/pnas.0505497102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weber J. Ipilimumab: controversies in its development, utility and autoimmune adverse events. Cancer Immunol Immunother. 2009;58:823–830. doi: 10.1007/s00262-008-0653-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hannon GJ. RNA interference. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 19.Sha WC, Nelson CA, Newberry RD, Kranz DM, Russell JH, Loh DY. Selective expression of an antigen receptor on CD8-bearing T lymphocytes in transgenic mice. Nature. 1988;335:271–274. doi: 10.1038/335271a0. [DOI] [PubMed] [Google Scholar]
- 20.Gajewski TF. B7-1 but not B7-2 efficiently costimulates CD8+ T lymphocytes in the P815 tumor system in vitro. J Immunol. 1996;156:465–472. [PubMed] [Google Scholar]
- 21.Spiotto MT, Yu P, Rowley DA, Nishimura MI, Meredith SC, Gajewski TF, Fu YX, Schreiber H. Increasing tumor antigen expression overcomes “ignorance” to solid tumors via crosspresentation by bone marrow-derived stromal cells. Immunity. 2002;17:737–747. doi: 10.1016/S1074-7613(02)00480-6. [DOI] [PubMed] [Google Scholar]
- 22.Kranz DM, Tonegawa S, Eisen HN. Attachment of an anti-receptor antibody to non-target cells renders them susceptible to lysis by a clone of cytotoxic T lymphocytes. Proc Natl Acad Sci USA. 1984;81:7922–7926. doi: 10.1073/pnas.81.24.7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toebes M, Coccoris M, Bins A, Rodenko B, Gomez R, Nieuwkoop NJ, van de Kasteele W, Rimmelzwaan GF, Haanen JB, Ovaa H, Schumacher TN. Design and use of conditional MHC class I ligands. Nat Med. 2006;12:246–251. doi: 10.1038/nm1360. [DOI] [PubMed] [Google Scholar]
- 24.Jorritsma A, Gomez-Eerland R, Dokter M, van de Kasteele W, Zoet YM, Doxiadis II, Rufer N, Romero P, Morgan RA, Schumacher TN, Haanen JB. Selecting highly affine and well-expressed TCRs for gene therapy of melanoma. Blood. 2007;110:3564–3572. doi: 10.1182/blood-2007-02-075010. [DOI] [PubMed] [Google Scholar]
- 25.Gajewski TF, Markiewicz MA, Uyttenhove C (2001) The p815 mastocytoma tumor model. Curr Protoc Immunol. Chapter 20:Unit 20.4. doi:10.1002/0471142735.im2004s43 [DOI] [PubMed]
- 26.Selenko-Gebauer N, Majdic O, Szekeres A, Hofler G, Guthann E, Korthauer U, Zlabinger G, Steinberger P, Pickl WF, Stockinger H, Knapp W, Stockl J. B7-h1 (programmed death-1 ligand) on dendritic cells is involved in the induction and maintenance of T cell anergy. J Immunol. 2003;170:3637–3644. doi: 10.4049/jimmunol.170.7.3637. [DOI] [PubMed] [Google Scholar]
- 27.Udaka K, Wiesmuller KH, Kienle S, Jung G, Walden P. Self-MHC-restricted peptides recognized by an alloreactive T lymphocyte clone. J Immunol. 1996;157:670–678. [PubMed] [Google Scholar]
- 28.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 29.Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, Braich R, Manoharan M, Soutschek J, Skare P, Klein LO, Davis MM, Chen CZ. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129:147–161. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 30.Uckert W, Schumacher TN. TCR transgenes and transgene cassettes for TCR gene therapy: status in 2008. Cancer Immunol Immunother. 2009;58:809–822. doi: 10.1007/s00262-008-0649-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.