

Abstract
Studies in murine models of cancer as well as in cancer patients have demonstrated that the immune response to cancer is often compromised. This paradigm is viewed as one of the major mechanisms of tumor escape. Many therapies focus on employing the professional antigen presenting dendritic cells (DC) as a strategy to overcome immune inhibition in cancer patients. Death receptor 6 (DR6) is an orphan member of the tumor necrosis factor receptor superfamily (TNFRSF21). It is overexpressed on many tumor cells and DR6−/− mice display altered immunity. We investigated whether DR6 plays a role in tumorigenesis by negatively affecting the generation of anti-tumor activity. We show that DR6 is uniquely cleaved from the cell surface of tumor cell lines by the membrane-associated matrix metalloproteinase (MMP)-14, which is often overexpressed on tumor cells and is associated with malignancy. We also demonstrate that >50% of monocytes differentiating into DC die when the extracellular domain of DR6 is present. In addition, DR6 affects the cell surface phenotype of the resulting immature DC and changes their cytokine production upon stimulation with LPS/IFN-γ. The effects of DR6 are mostly amended when these immature DC are matured with IL-1β/TNF-α, as measured by cell surface phenotype and their ability to present antigen. These results implicate MMP-14 and DR6 as a mechanism tumor cells can employ to actively escape detection by the immune system by affecting the generation of antigen presenting cells.
Keywords: Dendritic cells, Death receptor 6, Matrix metalloproteinase 14, Tolerance, Tumor immunity
Introduction
Dendritic cells (DC) play a key role in shaping and regulating immune responses in part by their ability to present antigen to T lymphocytes [1, 2]. They are derived from bone marrow, developing first into immature DC (iDC) and, upon further activation, into mature DC (mDC). Mature DC will process and present antigen in conjunction with co-stimulatory molecules and produce a range of cytokines, leading to the initiation of a specific immune response. In recent years, there has been a growing appreciation for the complex and dynamic interactions between the immune system and emerging tumor cells [3]. In order to persist, tumor cells are forced to develop mechanisms to evade immunological pressure. This can be achieved in a variety of ways, including induction of T cell hypo-responsiveness, extrinsic suppression by regulatory T cell populations, and/or production of immunomodulatory cytokines (e.g., TGF-β, IL-10) [4]. One or a combination of such strategies then allow the tumor to evade the immune system, and together with the acquisition of additional features (e.g., self-sufficiency in growth signals, resistance to apoptosis) can result in malignancy. Novel therapies for cancer vaccination revolve around the idea that mDC primed with tumor antigens can be used to specifically activate a patient’s immune system to eliminate tumor cells [5]. In contrast to immune activation induced by mDC, iDC have been demonstrated to induce tolerance, and as such can promote the deletion of antigen specific T cells [6]. Therefore, it appears that tumor cells could affect the efficacy of the immune response to the tumor through manipulation of the DC population. In fact, it has been shown that tumor cells can induce DC apoptosis [7] and influence DC differentiation and/or maturation in vitro [8, 9]. These studies do not, however, elucidate underlying molecular mechanisms leading to these changes.
Death receptor 6 (DR6) is a member of the TNF receptor superfamily (TNFRSF21) [10, 11]. To date, no ligand for DR6 has been identified and most of what we know about its function is derived from DR6−/− mice. These mice are hyper-reactive to antigen favoring the production of Th2 type cytokines, while maintaining normal Th1 cytokine production [12, 13]. In addition, B cell proliferation to a variety of stimuli is enhanced in DR6−/− mice resulting in increased immunoglobulin titers in vivo [14]. In addition to its apparent role in regulating immune responses, DR6 expression is increased in cancerous tissue biopsies from patients with late stage prostate or breast cancer compared with levels in normal tissue [15]. These authors also demonstrate that DR6 expression is regulated through activation of NF-κB and suggest that elevated levels of anti-apoptotic proteins such as Bcl-XL are necessary to allow for higher levels of DR6 expression. It is unclear at present what advantage tumor cells may have from the relative overexpression of a death receptor on their cell surface.
Matrix metalloproteinases (MMPs) are frequently overexpressed in human tumors, contributing to tumor proliferation, invasion and metastasis [16, 17]. In a recently reported random search for novel targets for the membrane-associated MMP-14, DR6 was identified as a substrate [18]. This finding, together with the observed overexpression of DR6 on tumor cells [15], led us to hypothesize that DR6 is providing a means for tumor cells to escape detection by the immune system. In the current investigation, we demonstrate that DR6 is cleaved from tumor cells in a uniquely MMP-14-dependent manner. Furthermore, the cleaved DR6 extracellular domain alters normal differentiation of monocytes into iDC as measured by expression of cell surface molecules and stimulated cytokine production. During this differentiation, DR6 also induces monocytic cell death, effectively reducing the number of iDC by >50%. Interestingly, we found that additional activation of iDC by cytokines, which results in the generation of mDC, is not affected by the presence of the extracellular domain of DR6. Taken together, our results suggest a role for tumor cell-derived DR6 in immune modulation via impeding the development of an effective anti-tumor DC population.
Materials and methods
Reagents
DR6-Fc fusion protein and anti-DR6-biotin were obtained from R&D Systems. Recombinant human DR6 (extracellular domain) with C-terminal FLAG and HIS tags was expressed transiently in 293 cells and purified on an IMAC column. Mouse monoclonal and rabbit polyclonal anti-DR6 antibodies were generated by immunizing with human DR6 (extracellular domain) using standard immunization protocols. Rabbit serum was affinity-purified prior to use. The broad-spectrum MMP inhibitor GM6001 and Furin inhibitor I (Decanoyl-RVKR-CMK) were purchased from Calbiochem. Recombinant human MMP2 and MMP14 and anti-MMP-14 were purchased from Chemicon. Phycoerythrin (PE)-conjugated F(ab′)2 fragment donkey anti-rabbit IgG (H + L), PE-conjugated streptavidin and peroxidase-conjugated streptavidin was acquired from Jackson Immunoresearch Laboratories. Recombinant human interleukin (IL)-4, human IL-1β, and human tumor necrosis factor (TNF)-α were obtained from R&D systems. Granulocyte macrophage colony stimulating factor (GM-CSF), Sargramostim Leukine®, was purchased from Immunex.
Monocyte differentiation and DC maturation
Monocytes were isolated from heparinized peripheral blood from healthy human volunteers. Following Ficoll separation, monocytes were magnetically tagged using anti-CD14 magnetic beads (Miltenyi Biotech). Positive cells were recovered upon passing the tagged mononuclear cells through an autoMACS device (Miltenyi Biotech). Cells (1 × 106 cells/ml) were resuspended in complete RPMI (cRPMI: RPMI 1640 with 10% fetal calf serum, 2 mM l-glutamine, 1% non-essential amino acids, 1% sodium pyruvate, 50 mM HEPES, 50 μM β-mercaptoethanol, and 1% penicillin/streptomycin; all supplements were acquired from Invitrogen). Immature DC were obtained following incubation for 4 days at 37°C, 5% CO2 with 4 ng/ml of recombinant human IL-4 and 8 ng/ml recombinant human GM-CSF in the presence or absence of various concentrations of DR6-Fc. Following incubation, an aliquot of cells were analyzed for cell surface markers by flow cytometry. To further induce DC maturation, half of the media was replaced and fresh cRPMI containing IL-4 (8 ng/ml), GM-CSF (16 ng/ml), IL-1β (20 ng/ml) and TNF-α (60 ng/ml). Cultures were incubated for an additional 3 days at 37°C, 5% CO2. At this point, the cells were counted, analyzed by flow cytometry for cell surface markers, and used in a mixed lymphocyte reaction.
Cell lines
MDA-MB-231, MCF-7, and PC-3 cells were obtained from American Type Culture Collection (ATCC). The cells were cultured in RPMI 1640 + 10 % FBS and 1% penicillin/streptomycin. MMP-14 cDNA was acquired from Origene (TC116990). The cDNA was modified via restriction digestion to remove approximately 1,000 base pairs in the 3′-UTR. The cDNA then was subcloned into a pcDNA3.1 expression vector (Invitrogen). The MMP-14 construct was transfected into MCF-7 cells using Lipofectamine 2000 (Invitrogen) according to manufacturer’s protocol. Stable transfectants were obtained via selection with G418 (Invitrogen). Cells expressing MMP-14 were enriched by two round of cell sorting using an anti-MMP-14 specific rabbit polyclonal antibody (Chemicon).
Immunoblotting
Cells were lysed in RIPA lysis buffer (1% triton X100, 0.1% SDS, 0.5% sodium deoxycholate, 150 mM NaCl, 10 mM Tris, pH 7.2, 25 mM β-glycerophosphate, 10 mM sodium pyrophosphate, 2 mM sodium orthovanadate, 1 mM PMSF, 10 mM NaF, 0.4 mM EDTA, 1 mM aprotinin, 1 mM α-1-antitrypsin, 1 mM leupeptin). Lysates were kept on ice for 30 min and spun at 14,000 rpm for 10 min in an Eppendorf centrifuge at 4°C. Supernatants were used for immunoprecipitation using DR6 specific monoclonal or polyclonal antibodies directly coupled to CNBr-activated sepharose beads (Amersham) for 1 h at 4°C. The extracellular domain of DR6 was immunoprecipitated from tissue culture supernatant using the same protocol. Beads were washed three times, mixed with SDS reducing sample buffer and boiled for 5 min. Proteins were separated by SDS-PAGE, transferred to nitrocellulose or PVDF, and visualized by blotting using specific antibodies and enhanced chemiluminescence.
DR6 ELISA
Anti-human DR6 affinity purified rabbit polyclonal antibody (pAb) was coated on an ELISA plate (Greiner) at 0.5 μg/ml and incubated overnight. Plates were blocked with 1% BSA/PBS solution. Samples (tissue culture supernatant) and a standard (recombinant human DR6) were added to the plate and incubated for 2 h. Anti-hDR6 pAb-biotin (R&D Systems) was used as the detection antibody, followed by streptavidin-HRP. Plates were developed with OPD for 10 min, stopped with 1 N HCl and absorbance was read at 405 nm. MDA-MB-231 cells were treated with a titration of GM6001 or Furin inhibitor for 48 h and cell culture supernatants were collected. DR6 released into the cell culture supernatant was measured with DR6 ELISA. Cell density was measured by MTS assay (CellTiter AQueous, Promega).
MMP activity assay
Recombinant human DR6-Fc was incubated with MMP-2 or MMP-14 in a 20:1 molar ratio with or without 10 nM GM6001 inhibitor overnight at room temperature. The reactions were separated by SDS-PAGE and stained with Coomassie Blue.
Phenotypical analysis
Primary cells (monocytes, iDC, or mDC) were resuspended in PBS containing 2% FBS and 0.1% sodium azide and incubated with an optimal concentration of directly labeled antibodies directed against the following molecules: CD11c, CD14, CD40, CD80, CD86, CD83, CD206, HLA-DR (all obtained from BD-Pharmingen), CD1a (Immunotech), CD16 or CD209 (Beckman-Coulter). Cells were incubated at 4°C for 30 min, washed in PBS/FBS/sodium azide, and a minimum of 10,000 live cells (those excluding propidium iodide (Molecular Probes)) per sample were analyzed on a flow cytometer. DR6 expression on PC-3, MDA-MB-231 and MCF-7 cells was assessed as above using 1 μg/ml biotinylated or unbiotinylated mouse anti-DR6 antibody with PE labeled appropriate secondary reagent.
Mixed lymphocyte reaction
To assess the competency of DC to stimulate lymphocytes in an allogeneic mixed lymphocyte reaction, mDC treated with or without DR6-Fc were harvested. DC were diluted to 12,500, 6,250, and 3,125 cells/ml in cRPMI and 100 μl of the appropriate dilution was added to wells of a 96-well plate in triplicate. Responder lymphocytes (post-Ficoll, CD14 depleted), were resuspended to 1 × 106 cells/ml in cRPMI and 100 μl added to each well. Spontaneous proliferation from dendritic cells and lymphocytes was assessed by incubating cells in media only. Cells were incubated for 3 days at which time the cells were pulsed with 1 μCi [methyl-3H] thymidine (Amersham). Cells were returned to the incubator and were harvested 18 h later. 3H-thymidine incorporation was measured on a TopCount NXT microplate scintillation counter (Packard).
Cytokine production
Monocytes were differentiated as above for 4 days in the presence or absence of 10 μg/ml DR6-Fc, harvested, re-plated (0.5 × 106 cells/ml) and stimulated with LPS (1 ng/ml) and IFN-γ (10 ng/ml) for 24 h. Cytokine levels were determined in tissue culture supernatant using a human cytokine multiplex assay (Linco), following the manufacturer’s instructions.
Statistical analyses
Paired t-tests (2-tailed) were used to determine statistical differences. Where necessary, data were log-transformed prior to analysis; P values less than 0.05 were considered significant.
Results
DR6 is released from the cell surface of cancer cell lines
A wide range of tumor cell lines express relatively high levels of DR6 mRNA [15]. To explore this further at the protein level, tumor cells were analyzed for cell surface expression of DR6. Both PC-3 and MDA-MB-231 cells were found to express DR6 on the cell surface (Fig. 1a). Although MCF-7 cells have previously been reported to be negative for DR6 mRNA [15], we observed a low level of DR6 expression by flow cytometry (Fig. 1a). These results were corroborated by immunoprecipitation experiments, where a clear immuno-reactive band was found in PC-3 and MDA-MB-231 cells, while a low intensity band was found in MCF-7 cells (Fig. 1b).
Fig. 1.

DR6 expression on cancer cell lines. a Expression was analyzed by flow cytometry. Cells were stained using DR6-specific antibodies with secondary control shown (dashed lines). b DR6 was immunoprecipitated from lysates derived from 12.5 × 106 cells, separated on SDS-PAGE, transferred to PVDF and probed for DR6. Recombinant DR6-Fc fusion protein (10 ng) was used as a positive control. c Extracellular domain of DR6 in tissue culture supernatant. DR6 was immunoprecipitated from 10 ml of tissue culture supernatant of confluent PC-3, MDA-MB-231, MCF-7 cells or medium (negative control)
Since TNFRSF members can be cleaved from the cell surface [10, 18], we also analyzed the supernatants of these cell lines for the presence of DR6 and/or fragments thereof. Using immunoprecipitation, we found a fragment of DR6 in PC-3 and MDA-MB-231 cell supernatant, but not in the supernatant from MCF-7 cells (Fig. 1c). We confirmed that this protein corresponded to the extracellular domain of DR6 by using MALDI-MS and subsequent amino acid sequence analysis following tryptic digest and liquid chromatography (data not shown). After deglycosylation (DR6 has 6 predicted N-linked glycosylation sites [11]), we found a ∼36 kDa protein, suggesting membrane-proximal cleavage of DR6 (data not shown).
DR6 is cleaved via an MMP-14-dependent mechanism
It has been previously reported that DR6 is a substrate for MMP-14 [18]. Since many tumor cells typically express multiple MMPs, we first investigated whether these are involved in the shedding of DR6 from the cell surface. MDA-MB-231 cells were grown in the presence of either GM6001 or a furin inhibitor (both broad-spectrum MMP inhibitors) for 48 h. Cell culture supernatants were collected, spun down to remove any cell debris and assayed for the extracellular domain of DR6 by ELISA. Results show a decrease of the levels of DR6 in the culture supernatant in the presence of increasing concentrations of either GM6001 or the furin inhibitor (Fig. 2a). We did not observe any significant cell death as a result of these treatments (data not shown). These data demonstrate the involvement of MMPs in the cleavage of DR6 from MDA-MB-231 cells.
Fig. 2.

MMP-14 is responsible for cleaving DR6 from the cell surface of tumor cells. a DR6 released into the cell culture supernatant of MDA-MB-231 cells was decreased following treatment with varying doses of GM6001 or Furin inhibitor for 48 h, as measured by ELISA. b MMP-14, but not MMP-2, cleaves DR6. DR6-Fc was treated with recMMP-2 or recMMP-14 in the presence or absence of the generic MMP inhibitor GM6001 for 18 h at room temperature. Proteins were separated on SDS-PAGE and visualized by Coomassie stain. c MMP-14 expression in MCF-7 cells before and after transfection with MMP-14, analyzed by flow cytometry. d The extracellular domain of DR6 is cleaved following transfection of MMP-14. DR6 was immunoprecipitated from 10 ml of tissue culture supernatant of MCF-7 cells transfected with MMP-14 or vector control
MMP-14 mediates the degradation of fibrillar collagen and fibronectin, but also is responsible for the activation of MMP-2 [17, 19]. DR6-Fc was used to determine whether DR6 could be cleaved directly by either recombinant MMP-2 or MMP-14 in vitro. Results demonstrate that MMP-14 readily cleaves DR6-Fc, whereas MMP-2 does not (Fig. 2b). The enzymatic cleavage by MMP-14 is completely inhibited by GM6001.
While many tumor cells express a variety of MMPs, it is documented that MCF-7 cells do not express MMP-14 [20]. Since we observed low levels of DR6 on these cells, we transfected MMP-14 into MCF-7 cells and monitored whether cleaved DR6 would appear in the supernatant. Using flow cytometry, we observed clear expression of MMP-14 following transfection but not in the vector-control parent cells (Fig. 2c). We then performed DR6 immunoprecipitation experiments from the supernatant of either parent or MMP-14-transfected MCF-7 cells. Results demonstrate that cleaved DR6 was present only in the supernatant of MMP-14 transfected cells (Fig. 2d).
Taken together, these results demonstrate that the extracellular domain of DR6 can be cleaved from the surface of cancer cell lines and that MMP-14 is sufficient for this process.
DR6 blocks monocyte to iDC differentiation
It has been reported that tissue culture supernatant from tumor cell lines can significantly inhibit monocyte differentiation into DC [8, 9], thereby potentially compromising the initiation of an immune reaction. Therefore, we questioned whether tumor-derived DR6 alone could prevent the differentiation of monocytes into DC, leading to a compromised development of a Th1 response. To investigate this hypothesis, we isolated CD14+ monocytes from human peripheral blood and cultured them in the presence of IL-4 and GM-CSF to induce differentiation into iDC in the presence or absence of DR6-Fc. In the absence of DR6-Fc, monocytes lost CD14 expression (data not shown) and differentiated into iDC as judged by their expression of cell surface molecules (CD11c+CD1a+CD40+CD80+CD83−CD206+CD209+HLA-DR+; Figs. 3a, 5a). Cells grown in the presence of DR6-Fc were also negative for CD14 and had cell surface expression of CD11c, CD40, CD206, and CD209 comparable to the control condition (Figs. 3a, 5a). In contrast, CD1a, CD16, CD83, CD86 and HLA-DR expression differed between the control and DR6-Fc conditions. Expression of CD1a was significantly reduced when cells differentiated in the presence of DR6-Fc (13.5% vs 73.1% in control (n = 14), P < 0.0001, Fig. 3a). While CD86 was only expressed on a limited number of cells in the control condition, more than 80% of cells in the DR6-Fc condition expressed CD86 (85.6% vs 35.3% in control (n = 14), P < 0.001, Fig. 3a). A significant portion of cells expressed low levels of CD83 in the presence of DR6-Fc and expression was found exclusively on CD1a negative cells (35.8% vs 8.9% in control (n = 14), P < 0.001, Fig. 3a). In the control condition, a small fraction (<5%) of CD1a+ cells co-expressed CD16, while CD16 expression was completely absent in the DR6-Fc condition (P = 0.049; Fig. 3a). While there was no difference in the percentage of HLA-DR positive cells, the expression level was somewhat higher in the presence of DR6-Fc (Fig. 5a), but this did not reach statistical significance (control MFI = 53 ± 5.1; DR6-Fc MFI = 69 ± 5.4, n-12, P = 0.078).
Fig. 3.

DR6 treatment inhibits monocyte differentiation into immature DC. a Phenotypic characterization of monocytes grown for 4 days in IL-4/GM-CSF in the presence or absence of DR6-Fc (10 μg/ml). Cell surface expression of a number of molecules on live cells (those excluding propidium iodide) was evaluated. One representative donor out of 4 is shown. b Monocytes were differentiated as described in (a) in the absence (media) or presence of DR6-Fc, denatured DR6-Fc, or an Fc control protein (10 μg/ml). After 4 days, CD1a expression was analyzed by flow cytometry (CD1a expression on undifferentiated monocytes is shown in the left histogram). One representative experiment out of 4 is shown
Fig. 5.
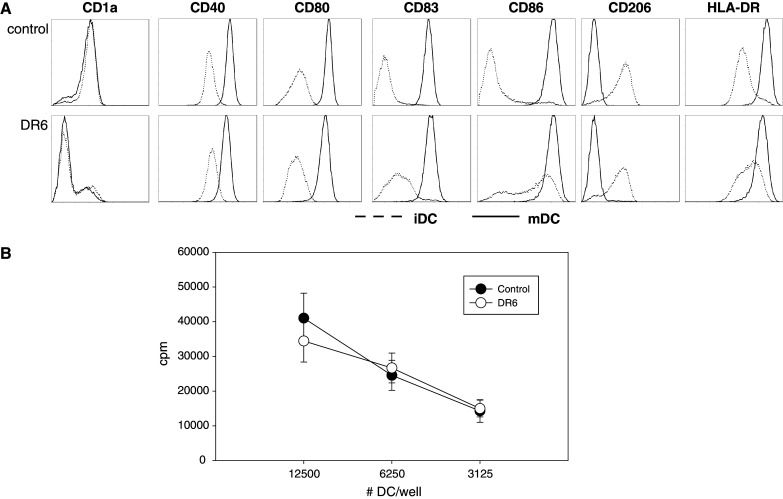
Characterization of DC following maturation. Monocytes were grown for 4 days in IL-4/GM-CSF (iDC), followed by 3 days of IL-1β/TNF-α to induce maturation (mDC), in the presence or absence of DR6-Fc (10 μg/ml). a Cells were analyzed by flow cytometry and expression of cell surface molecules on iDC (dotted lines) and mDC (solid lines) were compared on live cells (those excluding propidium iodide). One representative donor out of 3 is shown. b MLR results using mDC grown in the presence (open circles) or absence (closed circles) of DR6. Responder lymphocytes from one donor were incubated with a standardized number (12,500, 6,250, or 3,125 per well) of mDC from an unrelated donor for 4 days and proliferation was measured using 3H thymidine for the final 18 h. Results (expressed in cpm) of 5 donor pairs (mean ± SEM) are shown
The inhibition of monocyte differentiation into iDC by DR6, as exemplified by monitoring CD1a expression, was not observed with Fc control protein (Fig. 3b). To ensure that the activity was related to the structure of the DR6-Fc protein, we denatured the protein (95°C for 30 min). This completely abrogated the effect of DR6-Fc on monocyte differentiation (Fig. 3b). These effects of DR6-Fc were dose-dependent, as incubation with a 10-fold lower dose resulted in partial inhibition of CD1a expression (data not shown). While conducting these experiments, we observed that incubation of monocytes with DR6-Fc resulted in a >50% loss of cells after the 4 day culture (cell yield 2.98 ( ± 0.27) × 105 for control conditions and 1.29 (± 0.16) × 105 for DR6-Fc (n = 8), P < 0.0001). This reduction in cell number was also dose dependent (∼20% cell loss with 1 μg/ml DR6-Fc) and was observed as early as 24 h into the culture (data not shown). These data demonstrate that DR6 not only influences monocyte differentiation into iDC, but also affects their survival. Furthermore, our data imply that monocytes express a DR6 binding partner on their cell surface.
DR6 alters the cytokine production profile by DC
We studied the cytokine production profile of iDC generated in the presence or absence of DR6. After harvest, an equal number of iDC were stimulated for 24 h with LPS and IFN-γ, supernatants were collected and cytokine levels were determined using a multiplex assay. Results demonstrate that levels of IL-6, IL-8, and IL12p70 were significantly reduced in stimulated DC grown in the presence of DR6 (Fig. 4). In contrast, stimulated levels of CCL2 were significantly enhanced in DR6-grown DC, with no difference in production of TNF-α or CCL3 (Fig. 4). We observed low level production of a number of other cytokines upon stimulation, including IL-10, IL-7, IL-1α, and G-CSF, that was not different between control and DR6-treated cells (data not shown). This demonstrates that incubation with DR6 does not cause a general suppression in the ability of DC to produce cytokines but rather appears to quantitatively influence production of selected cytokines.
Fig. 4.

DR6 alters cytokine production induced by LPS/IFN-γ. iDC generated in the presence or absence of 10 μg/ml DR6-Fc were harvested, counted and stimulated with LPS and IFN-γ for 24 h. Concentrations of cytokines were determined using a multiplex assay. Each symbol represents a different donor (group mean is represented by the horizontal line). P values are indicated where paired t-test showed a significant difference; ns = not significant
Influence of DR6 on DC maturation and function
Since antigen presentation is most effectively done by mature rather than immature DC, we explored the impact of DR6 on mDC function. To this end, mDC were generated by incubating monocytes with IL-4 and GM-CSF for 4 days, followed by a maturation step using IL-1β and TNF-α for an additional 3 days in the continuous presence or absence of DR6-Fc (10 μg/ml). After this 7 day period, cells were harvested, counted and analyzed by flow cytometry. In the presence of DR6, the total number of mDC was reduced on average by 55.2 ± 4.2 % (n = 10, P < 0.0001). This result confirms the observation of the effect of DR6-Fc treatment on cell yield during monocyte differentiation into iDC. Flow cytometric analysis revealed that most of the differences induced by DR6 observed in iDC diminished or disappeared (Fig. 5a). Maturation with TNF-α/IL-1β resulted in increased expression levels of a number of molecules (CD40, CD80, CD83, CD86), whereas CD206 expression was lost and CD1a expression remained unchanged (all relative to levels on iDC). The density of HLA-DR molecules following maturation increased more in the control condition than with DR6, resulting in a significantly higher expression in control cells relative to DR6 treated cells (MFI: 228 ± 44 vs 154 ± 22 for control and DR6 treatment respectively (n = 5); P = 0.04). The expression of CD16 on a small subset of cells in the control iDC (Fig. 3a) disappeared following maturation (data not shown). Maturation eliminated most of the phenotypic differences induced by DR6 observed in iDC, with reduced CD1a expression as a notable exception.
These matured cells were used in a mixed lymphocyte reaction to assess their functionality. The number of DC (control or DR6 treated) was normalized (12,500, 6,250, or 3,125 DC per well) and a fixed number of responder cells from a different donor were added to each well. Cells were incubated for 4 days and proliferation was measured by 3H-thymidine incorporation. The results demonstrate that there was no statistically significant difference in the capacity of mDC generated under control conditions or treated with DR6 to induce lymphocyte proliferation (Fig. 5b). Together the results show that maturation signals eliminate most DR6-induced phenotypic differences observed in iDC and that the ability of the resulting mDC to present antigen was similar on a per cell basis.
Discussion
The role of DR6 in physiology is still largely unknown. DR6 has a cytoplasmic death domain and ectopic overexpression can lead to apoptosis [11]. Therefore it seems counterintuitive that many tumor cells [11, 15] and tumorigenic tissue from prostate and breast cancer patients [15] have elevated DR6 expression. In fact, these cells can only survive if they simultaneously express NF-κB regulated survival proteins such as Bcl-2, Bcl-XL or survivin [15]. Our results suggest that elevated DR6 expression can provide an advantage to tumor cells by influencing the immune system. DR6 is cleaved from the cell surface by the metalloproteinase MMP-14 and the extracellular domain of DR6 induces cell death in differentiating monocytes. In addition, DR6 affects the cell surface phenotype of the surviving iDC and changes their cytokine production profile following stimulation. We propose that this is one of the tumor evasion mechanisms by which emerging tumors inhibit the development and level of anti-tumor activity in the tumor microenvironment and the sentinel lymph nodes.
An additional hypothesis regarding the role for DR6 in shaping anti-tumor immunity is that the extracellular domain of DR6 prevents the generation of an anti-tumor Th1 response. This is based on the phenotype of the DR6−/− mice which clearly shows that DR6 plays an important role in the regulation of immune system activation, directing T cell differentiation towards Th2 responses [12–14]. Interestingly enough however, production of Th1 cytokines in DR6−/− cells is equivalent to that in WT cells [12, 13], demonstrating that DR6−/− mice do not have a deficiency in generating Th1 responses. We show here that the cytokine profile of iDC generated in the presence of DR6 is quantitatively altered compared to control iDC. DR6−grown iDC stimulated with LPS/IFN-γ produce lower levels of IL-6, IL-8, and IL-12p70, a pattern suggestive of creating a reduced inflammatory milieu. Others have generated DC that produced virtually no IL-12 following stimulation with LPS/IFN-γ and went on to demonstrate that these cells promoted Th2 differentiation [21]. However, these cells also produced greatly increased levels of IL-10, something we did not observe even though we used the same stimulatory conditions. Given that IL-12 is considered the dominant factor in directing differentiation towards Th1 cells [22], the ∼50% reduction we observed by DR6 is likely insufficient to completely inhibit Th1 generation. In addition, we found that DR6-grown iDC produced significantly greater levels of CCL2 while production of TNF-α and CCL3 was not different. Although CCL2 has been associated with a Th2 phenotype in mice, this link has not been observed in human DC [23]. While we used LPS/IFN-γ to stimulate cytokine production, other stimuli may affect production in different ways. It is known that microbial compounds can selectively cause either Th1- or Th2-promoting DC [24]. In addition, the potential influence of DR6 on DC cytokine production in the tumor microenvironment is likely patient dependent, making the end result on the local immune formation difficult to predict.
We observed that DR6 treatment resulted in a number of changes in expression of cell surface molecules. Most notably, we observed that DR6 induced a significant reduction in CD1a expression, while other DC markers (e.g., CD11c, CD209, CD206) were expressed normally. Chang et al. described the generation of CD1a negative iDC by varying culture conditions [21]. In their studies, they found an association of the CD1a-phenotype with a Th2 cytokine profile (elevated IL-10, no IL-12), something we did not observe. They also did not describe some of the additional differences we observed, namely the increased expression of CD86, HLA-DR, and CD83. Upregulation of CD86 and HLA-DR could indicate in vitro activation of the iDC, possibly a result of the large numbers of dead cells in our culture. Cell death, in particular necrosis, can induce a pro-inflammatory milieu [25] and result in DC maturation [26]. CD83 has been described as a marker of DC maturation, rather than one of cell activation [27]. All three molecules were expressed at an intermediate level in the DR6-containing cultures, suggestive of low-level activation. Additional stimulation with IL-1β/TNF-α resulted in much higher levels of expression, equal to that observed in control cultures. These findings suggest that CD83 can also be seen as an activation marker, which would fit with its proposed role in T cell activation [27]. The relative lack of CD1a expression on the majority of mDC differentiated in the presence of DR6 was the only difference that remained. This result implies that it is possible to generate DC that lack CD1a expression, possibly resulting in DC with an altered ability to present lipids.
Cancer patients often are incapable of mounting an immune response to their tumor cells. Since anti-tumor specific immune cells are present in many patients, it appears that dominant mechanisms of resistance exist downstream of immune priming that prevent the elimination of the tumor [4]. The reasons for this can be multifaceted. One possible scenario is that the local DC population is functionally impaired, leading to a lack of T cell activation. In support of this, it has been reported that there is a paucity of DC in the tumor microenvironment of cancer patients [28, 29]. Moreover, DC found in sentinel lymph nodes from melanoma patients were phenotypically and morphologically immature [29, 30]. These observations are in agreement with results demonstrating that tumor cells can induce DC apoptosis [7] and influence DC differentiation and/or maturation in vitro ([8, 9] and DCD, PJR, and RJB, unpublished observations). The reduction in DC frequency in sentinel lymph nodes compared to normal lymph nodes could easily be the result of decreased DC survival [30]. It is well documented that DC are highly mobile and, upon encountering antigen (e.g., in the tumor microenvironment), will relocate to secondary lymphoid organs to induce T cell responses [31]. Our data identify tumor-derived DR6 as one such immuno-modulatory molecule preventing effective local DC formation, by inducing cell death and reduced DC differentiation, resulting in fewer activating DC trafficking from the tumor to the sentinel lymph nodes. It has been reported that the frequency of DC in sentinel lymph nodes is not always reduced [32] and this may depend on the factors the tumor is producing. It would be interesting to determine the correlation between DR6 expression on various tumors and DC frequency in sentinel lymph nodes.
Dendritic cells not only play a critical role in immune activation, but can also induce peripheral T cell tolerance [6]. This is clearly helpful in a steady state situation (i.e., in the absence of inflammation) to prevent autoimmunity as a result of the uptake of dying autologous cells by DC [33]. To exemplify this concept, dying cells loaded with OVA resulted in the induction of tolerance to OVA in the absence of inflammatory mediators [34]. It is therefore easy to envision that interference with DC maturation is advantageous for tumor cells and can be employed to prevent activation of the immune system. Tolerance can also be achieved via the induction and/or expansion of regulatory T cells and their prevalence is increased in cancer patients [35, 36]. Interestingly, it has been demonstrated that tumor-derived regulatory T cells can directly suppress immunity by maintaining DC in an immature state [37]. Therefore, targeting regulatory T cells has been proposed as a potential therapeutic approach in cancer although this has to be balanced to avoid autoimmunity [38]. It has also been demonstrated that regulatory T cells with a high affinity for autoantigens divide extensively in the physiological steady state [39] and it is plausible that iDC are involved in driving this proliferation. Even though tumor cells express some neo-antigens, most molecular entities derived from tumors are self antigens and as such induce tolerance. Tumor cells could use DC to promote tolerance induction to evade the immune system and our data suggests a role for DR6 in this process. Therefore, interfering with mechanisms tumor cells exploit to manipulate the DC population might be equally effective as a therapy as targeting regulatory T cells.
Expression of MMP-14 correlates with malignancy in a variety of tumor types [17]. MMP-14 is directly responsible for breaking down type I collagen and indirectly (via the activation of proMMP-2) for the degradation of type IV collagen. Expression of MMP-14 into the non-metastatic MCF-7 cells allows these cells to become invasive and induces the rapid development of highly vascularized tumors in nude mice [20]. In addition to the established role of MMP-14 in allowing tumor cells to metastasize, we describe an additional role for MMP-14 in aiding establishment of the tumor by preventing the development of local immunity via the manipulation of DC formation. Given that many different tumor types over-express both MMP-14 and DR6, it is intriguing to speculate that such co-expression is a risk factor for tumor metastasis. Additional analyses, which should include an analysis of the local DC population, are necessary to confirm this hypothesis.
Our results show that, under the right circumstances, functionally effective mDC can develop even in the presence of DR6. Even though iDC formation in the presence of DR6 was very inefficient, these cells were readily matured by TNF-α and IL-1β and indistinguishable from control cells in their ability to present antigen. Whiteside et al. reported that tumor cells can have a significant suppressive effect on DC, but that DC from cancer patients can be matured and activated normally ex vivo [40]. These findings indicate that suppression of DC by tumor cells is reversible. In the tumor microenvironment additional suppressive factors are present, most prominently IL-10, prostaglandin E2, and TGF-β [4, 41]. IL-10 can also inhibit the IL-4- and GM-CSF-induced differentiation of monocytes into iDC [42, 43]. However, unlike the induction of cell death by DR6, IL-10 re-directs the differentiation of monocytes from iDC into macrophages without significant cell death. Thus the therapeutic hurdle is to administer treatment at the site of the tumor and boost anti-tumor immunity in the presence of the various local immunosuppressive mechanisms. One approach that is being investigated to overcome immune inertia in patients is to combine chemotherapy with adjuvants that stimulate the innate immune system [5]. Again, the DC compartment is central to this type of treatment and success would be compromised if tumor cells are able to inhibit DC differentiation and survival. Therefore, effectively blocking the function of tumor-derived factors such as DR6 could protect DC differentiation and survival and would thus contribute to the success of these DC-based therapies.
References
- 1.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 2.Pulendran B, Palucka K, Banchereau J. Sensing pathogens and tuning immune responses. Science. 2001;293:253–256. doi: 10.1126/science.1062060. [DOI] [PubMed] [Google Scholar]
- 3.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6:836–848. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- 4.Gajewski TF, Meng Y, Harlin H. Immune suppression in the tumor microenvironment. J Immunother. 2006;29:233–240. doi: 10.1097/01.cji.0000199193.29048.56. [DOI] [PubMed] [Google Scholar]
- 5.Schuler G, Schuler-Thurner B, Steinman RM. The use of dendritic cells in cancer immunotherapy. Curr Opin Immunol. 2003;15:138–147. doi: 10.1016/S0952-7915(03)00015-3. [DOI] [PubMed] [Google Scholar]
- 6.Steinman RM, Nussenzweig MC. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci USA. 2002;99:351–358. doi: 10.1073/pnas.231606698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Esche C, Lokshin A, Shurin GV, et al. Tumor’s other immune targets: dendritic cells. J Leukoc Biol. 1999;66:336–344. doi: 10.1002/jlb.66.2.336. [DOI] [PubMed] [Google Scholar]
- 8.Aalamian M, Pirtskhalaishvili G, Nunez A, et al. Human prostate cancer regulates generation and maturation of monocyte-derived dendritic cells. Prostate. 2001;46:68–75. doi: 10.1002/1097-0045(200101)46:1<68::AID-PROS1010>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 9.Katsenelson NS, Shurin GV, Bykovskaia SN, et al. Human small cell lung carcinoma and carcinoid tumor regulate dendritic cell maturation and function. Mod Pathol. 2001;14:40–45. doi: 10.1038/modpathol.3880254. [DOI] [PubMed] [Google Scholar]
- 10.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/S0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 11.Pan G, Bauer JH, Haridas V, et al. Identification and functional characterization of DR6, a novel death domain-containing TNF receptor. FEBS Lett. 1998;431:351–356. doi: 10.1016/S0014-5793(98)00791-1. [DOI] [PubMed] [Google Scholar]
- 12.Liu J, Na S, Glasebrook A, et al. Enhanced CD4+ T cell proliferation and Th2 cytokine production in DR6-deficient mice. Immunity. 2001;15:23–34. doi: 10.1016/S1074-7613(01)00162-5. [DOI] [PubMed] [Google Scholar]
- 13.Zhao H, Yan M, Wang H, et al. Impaired c-Jun amino terminal kinase activity and T cell differentiation in death receptor 6-deficient mice. J Exp Med. 2001;194:1441–1448. doi: 10.1084/jem.194.10.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidt CS, Liu J, Zhang T, et al. Enhanced B cell expansion, survival, and humoral responses by targeting death receptor 6. J Exp Med. 2003;197:51–62. doi: 10.1084/jem.20020617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kasof GM, Lu JJ, Liu D, et al. Tumor necrosis factor-alpha induces the expression of DR6, a member of the TNF receptor family, through activation of NF-kappaB. Oncogene. 2001;20:7965–7975. doi: 10.1038/sj.onc.1204985. [DOI] [PubMed] [Google Scholar]
- 16.Overall CM, Lopez-Otin C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer. 2002;2:657–672. doi: 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- 17.Seiki M. Membrane-type 1 matrix metalloproteinase: a key enzyme for tumor invasion. Cancer Lett. 2003;194:1–11. doi: 10.1016/S0304-3835(02)00699-7. [DOI] [PubMed] [Google Scholar]
- 18.Tam EM, Morrison CJ, Wu YI, et al. Membrane protease proteomics: Isotope-coded affinity tag MS identification of undescribed MT1-matrix metalloproteinase substrates. Proc Natl Acad Sci U S A. 2004;101:6917–6922. doi: 10.1073/pnas.0305862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sato H, Takino T, Okada Y, et al. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature. 1994;370:61–65. doi: 10.1038/370061a0. [DOI] [PubMed] [Google Scholar]
- 20.Sounni NE, Devy L, Hajitou A, et al. MT1-MMP expression promotes tumor growth and angiogenesis through an up-regulation of vascular endothelial growth factor expression. Faseb J. 2002;16:555–564. doi: 10.1096/fj.01-0790com. [DOI] [PubMed] [Google Scholar]
- 21.Chang CC, Wright A, Punnonen J. Monocyte-derived CD1a+ and CD1a− dendritic cell subsets differ in their cytokine production profiles, susceptibilities to transfection, and capacities to direct Th cell differentiation. J Immunol. 2000;165:3584–3591. doi: 10.4049/jimmunol.165.7.3584. [DOI] [PubMed] [Google Scholar]
- 22.O’Garra A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity. 1998;8:275–283. doi: 10.1016/S1074-7613(00)80533-6. [DOI] [PubMed] [Google Scholar]
- 23.de Jong EC, Smits HH, Kapsenberg ML. Dendritic cell-mediated T cell polarization. Springer Semin Immunopathol. 2005;26:289–307. doi: 10.1007/s00281-004-0167-1. [DOI] [PubMed] [Google Scholar]
- 24.Kapsenberg ML. Dendritic-cell control of pathogen-driven T-cell polarization. Nat Rev Immunol. 2003;3:984–993. doi: 10.1038/nri1246. [DOI] [PubMed] [Google Scholar]
- 25.Rovere-Querini P, Capobianco A, Scaffidi P, et al. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep. 2004;5:825–830. doi: 10.1038/sj.embor.7400205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dumitriu IE, Baruah P, Valentinis B, et al. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J Immunol. 2005;174:7506–7515. doi: 10.4049/jimmunol.174.12.7506. [DOI] [PubMed] [Google Scholar]
- 27.Prechtel AT, Steinkasserer A. CD83: an update on functions and prospects of the maturation marker of dendritic cells. Arch Dermatol Res. 2007;299:59–69. doi: 10.1007/s00403-007-0743-z. [DOI] [PubMed] [Google Scholar]
- 28.Laguens G, Coronato S, Laguens R, et al. Human regional lymph nodes draining cancer exhibit a profound dendritic cell depletion as comparing to those from patients without malignancies. Immunol Lett. 2002;84:159–162. doi: 10.1016/S0165-2478(02)00172-4. [DOI] [PubMed] [Google Scholar]
- 29.Polak ME, Johnson P, Di Palma S, et al. Presence and maturity of dendritic cells in melanoma lymph node metastases. J Pathol. 2005;207:83–90. doi: 10.1002/path.1809. [DOI] [PubMed] [Google Scholar]
- 30.Cochran AJ, Huang RR, Lee J, et al. Tumour-induced immune modulation of sentinel lymph nodes. Nat Rev Immunol. 2006;6:659–670. doi: 10.1038/nri1919. [DOI] [PubMed] [Google Scholar]
- 31.Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol. 2005;5:617–628. doi: 10.1038/nri1670. [DOI] [PubMed] [Google Scholar]
- 32.Ishigami S, Natsugoe S, Uenosono Y, et al. Infiltration of antitumor immunocytes into the sentinel node in gastric cancer. J Gastrointest Surg. 2003;7:735–739. doi: 10.1016/S1091-255X(03)00076-3. [DOI] [PubMed] [Google Scholar]
- 33.Iyoda T, Shimoyama S, Liu K, et al. The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J Exp Med. 2002;195:1289–1302. doi: 10.1084/jem.20020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu K, Iyoda T, Saternus M, et al. Immune tolerance after delivery of dying cells to dendritic cells in situ. J Exp Med. 2002;196:1091–1097. doi: 10.1084/jem.20021215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liyanage UK, Moore TT, Joo HG, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169:2756–2761. doi: 10.4049/jimmunol.169.5.2756. [DOI] [PubMed] [Google Scholar]
- 36.Ormandy LA, Hillemann T, Wedemeyer H, et al. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005;65:2457–2464. doi: 10.1158/0008-5472.CAN-04-3232. [DOI] [PubMed] [Google Scholar]
- 37.Larmonier N, Marron M, Zeng Y, et al. Tumor-derived CD4(+)CD25(+) regulatory T cell suppression of dendritic cell function involves TGF-beta and IL-10. Cancer Immunol Immunother. 2007;56:48–59. doi: 10.1007/s00262-006-0160-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–352. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 39.Fisson S, Darrasse-Jeze G, Litvinova E, et al. Continuous activation of autoreactive CD4+ CD25+ regulatory T cells in the steady state. J Exp Med. 2003;198:737–746. doi: 10.1084/jem.20030686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whiteside TL, Stanson J, Shurin MR, et al. Antigen-processing machinery in human dendritic cells: up-regulation by maturation and down-regulation by tumor cells. J Immunol. 2004;173:1526–1534. doi: 10.4049/jimmunol.173.3.1526. [DOI] [PubMed] [Google Scholar]
- 41.Kim R, Emi M, Tanabe K. Cancer cell immune escape and tumor progression by exploitation of anti-inflammatory and pro-inflammatory responses. Cancer Biol Ther. 2005;4:924–933. doi: 10.1158/1535-7163.MCT-04-0293. [DOI] [PubMed] [Google Scholar]
- 42.Allavena P, Piemonti L, Longoni D, et al. IL-10 prevents the differentiation of monocytes to dendritic cells but promotes their maturation to macrophages. Eur J Immunol. 1998;28:359–369. doi: 10.1002/(SICI)1521-4141(199801)28:01<359::AID-IMMU359>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 43.Buelens C, Verhasselt V, De Groote D, et al. Interleukin-10 prevents the generation of dendritic cells from human peripheral blood mononuclear cells cultured with interleukin-4 and granulocyte/macrophage-colony-stimulating factor. Eur J Immunol. 1997;27:756–762. doi: 10.1002/eji.1830270326. [DOI] [PubMed] [Google Scholar]