

Abstract
Aims: The association between tissue factor (TF) expression and increased rate of tumour metastasis is well established. In this study, we have examined the hypothesis that the expression of TF by disseminated tumour cells confers protection against immune recognition and cytotoxicity. Materials and methods: A hybrid EGFP-TF protein was expressed in HT29 colon carcinoma and K562 lymphoblast cell lines. To assess the cytotoxic activity against tumour cells over-expressing TF, a novel method was used, based on the direct measurement of fluorescently labelled HT29 or K562 target cells. Results: Upon challenge with peripheral blood mononuclear cells (PBMC), tumour cells expressing TF partially evaded cellular cytotoxicity (Δ=15–40% reduction in cytotoxicity). Moreover, the influence of TF was not primarily dependent on its procoagulant function, although the inclusion of 20% (v/v) plasma did lower the rate of cytotoxicity against untransfected cells. However, expression of a truncated form of TF, devoid of the cytoplasmic domain, did not mediate any degree of inhibition of cytotoxicity, suggesting that the protective function of TF is principally due to this domain. Conclusions: We conclude that TF can promote immune evasion in tumour cells expressing this protein leading to increased survival and therefore metastatic rate in such cells.
Keywords: Tissue factor, Metastasis, Cytotoxicity
Introduction
Tissue factor (TF) is a 47 kD transmembrane glycoprotein that forms a complex with circulating factor VIIa to initiate the extrinsic pathway of coagulation [1]. Moreover, the presence of high levels of TF on the surface of many tumour cells may be one of the underlying mechanisms giving rise to the hypercoagulable state associated with malignancy [2, 3]. In support of this, direct correlations between elevated TF expression and advanced stages of malignancy have been reported in several types of cancer, including non-small cell lung cancer [4], breast [5, 6], pancreatic [7], glioma [8], prostate [9] and colorectal cancer [5, 10, 11]. These studies have suggested that TF may also play a major role in growth, invasion and dissemination of the tumour cells.
Emerging experimental data in the past two decades suggest a close interaction between elements of the haemostatic and immune systems [12]. The presence of fibrinoid material in cancer tissue has been reported by many researchers [13–15]. However, the biological significance of the interactions between the haemostatic and immune systems remains obscure. Furthermore, there is conflicting data suggesting a possible mechanism by which the tumour cells form a ‘coat’ of the host’s own proteins on the surface shielding the tumour cells from cytotoxic immune recognition [16–20]. This coat is composed of fibrin together with a polymeric form of human serum albumin and by contrast to fibrin, is resistant to fibrinolytic degradation. Hence, as one of the main initiators of blood coagulation, TF could also play a vital role in allowing the tumour cells to escape immune recognition. In this study, by transfection and expression of TF in tumour cells, we examined the hypothesis that tumour cells producing high levels of TF possess an advantage in evading immune recognition. In order to elucidate further the mechanism by which TF aids tumour cell evasion of immune recognition, we investigated the involvement of fibrin in this process by incubation in the presence and absence of plasma. Finally, we expressed a truncated form of TF, without the cytoplasmic domain, to investigate the specific role of this domain.
Materials and methods
Cell culture and fluorescent labelling of cells
Colorectal adenocarcinoma cell line (HT29) and lymphoblast cell line (K562) were obtained from the ATCC (Teddington, UK) and maintained in RPMI medium [90% (v/v)], foetal calf serum [10% (v/v)], containing 1% (v/v) antibiotic/antimycotic solution (Sigma Chemical Company Ltd., Poole, UK). The cells (5×104 per well) were labelled by resuspension in 200 μl of serum-free RPMI media and incubation with 5 μM 5-(and -6)-chloromethyl seminapthorthohodaflour (SNARF)-1 acetate (Invitrogen, Paisley, UK) for 2 h.
Transient transfection of HT29 and K562 cells
The cDNA sequence for full-length human TF was cloned into a mammalian expression vector (pEGFP-C3) (BD Biosciences, Oxford, UK), downstream of the sequence for green fluorescent protein. Cells (5×105 per well) pre-adapted to OptiMEM-1 media were transfected with pEGFP-TF construct (1 μg), using Lipofectin (Invitrogen, Paisley, UK), in accordance with the manufacturer’s instructions for 6 h. Following 6 h incubation, the medium was replaced and cells were allowed to express the hybrid protein for at least 72 h prior to assaying. Following transfection, only cell populations with >60% transfectants were selected for further investigations. All assays were carried out against two sets of controls, one transfected with the empty pEGFP-C3 plasmid and another untransfected cells. The values obtained in all the assays, for these two sets of controls were identical and therefore the control using pEGFP-C3 vector have been included throughout.
One-stage prothrombin time assay
A standard curve was prepared using serial dilutions of recombinant human TF (Dade Behring, Milton Keynes, UK). Cells were resuspended in PBS (100 μl) prior to assaying. 25 mM CaCl2 (100 μl) and the cell suspension (4×105 cells in 100 μl) were incubated for 30 s before the addition of normal human plasma (Helena Laboratories, Sunderland, UK) (100 μl). The time taken for the clot formation was recorded using a Cascade-M coagulometer (Helena Laboratories, Sunderland, UK). In order to prove that the increased procoagulant activity was as a result of overexpression of TF, a murine MAb capable of blocking human TF (Axis-Shield, Cambridge, UK) was used to neutralise TF activity.
Western blot analysis of total cellular TF antigen
Total protein was extracted by lysing the transfected HT29 and untransfected HT29 in 200 μl denaturing buffer (Sigma Chemical Co. Ltd. Poole, UK) and separated on a 12% (w/v) SDS-PAGE gel and transferred to a nitrocellulose membrane. Membranes were blocked in 5% (w/v) skimmed milk in PBS-Tween 20 and probed with a murine MAb anti human TF (Axis-Shield, Cambridge, UK) diluted 1:1,000, for 1 h at room temperature. The membrane was then developed with anti-mouse IgG alkaline phosphatase conjugated secondary antibody (Promega Corp. Southampton, UK) diluted at a 1:2,000 in PBS-Tween 20 for 1 h at room temperature. Finally, the membrane was washed three times with PBS-Tween 20, for 10 min each time and developed using TMB stabilised-substrate (Promega Corp. Southampton, UK).
Enzyme-linked immunosorbent assay measurement of cell surface TF antigen
The expression of TF antigen on the tumour cells was measured by employing a TF antigen enzyme-linked immunosorbent assay (ELISA) kit (Affinity Biologicals, Ancaster, Canada). The capture antibody was diluted 1/100 in coating buffer (15 mM Na2CO3 buffer, pH 9.6), added (100 μl) to each microtitre plate well and incubated overnight at 4°C to allow adherence. The wells were then blocked with 150 mM phosphate buffered saline (PBS) buffer pH 7.4, containing 1% (w/v) bovine serum albumin (BSA) for 90 min followed by four washes with PBS. Intact cells (2×105) in serum-free medium (100 μl) were added to the wells and incubated for 60 min at room temperature to allow capture, and then washed carefully four times with PBS. The detecting antibody (anti-TF-HRP) was diluted 1/100 in conjugate buffer (100 mM HEPES buffer pH 7.4, 10 mM NaCl, 1% (w/v) BSA and 1% (w/v) Tween 20) and added (100 μl) to each well. The plate was incubated for a further 60 min at room temperature, washed four times with PBS and developed by adding 100 μl of TMB substrate (3,3′,5,5′ Tetramethylbenzidine; Vector Laboratories Ltd., Peterborough, UK) for 15 min at room temperature. The reaction was quenched by the addition of concentrated (18 M) H2SO4 (100 μl) to the wells and quantified at 450 nm on an Anthos 2010 microplate reader (Anthos Labtec Instruments, Wals, Austria).
Flow cytometric and Western blot measurement of cell surface TF antigen
To assess the surface TF expression, cells (2×105) were resuspended with PBS (100 μl) containing FITC labelled murine MAb against human TF (directed against epitope area aa: 203–214; Axis-Shield, Cambridge, UK; final concentration 1 μg/ml) and incubated for 30 min at 4°C in the dark. The cells were then centrifuged, washed twice with PBS and resuspended in PBS (100 μl) and incubated with F(Ab′)2 rabbit anti-mouse IgG:RPE (Serotec Ltd., Oxford, UK) (10 μl) for 60 min at 4°C. Finally, the cells were then centrifuged at 400g for 5 min, the pellet washed twice with PBS, resuspended in PBS (300 μl) and measured on a FACSCalibur flow cytometer running CellQuest software version 3.3 (Becton Dickinson, Oxford, UK).
Site-directed mutagenesis
To express a truncated form of TF, devoid of the cytoplasmic domain, mutation of Ser240 (on the human TF protein) was carried out using the QuickChange® site-directed mutagenesis kit (Stratagene, Amsterdam, Netherlands). Amplification of the pEGFP-TF construct was carried out by polymerase chain reaction at an annealing temperature of 55°C and an extension time of 16 min, using the following primers (The specific mutation TCT→TAA is underlined).
Forward primer: 5′ CATCCTGATATAACTACACAAGTGTAG
Reverse primer: 5′ CTACACTTGTGTAGTTATATCAGGATG
Following the reaction, the wild type DNA was digested by incubation with DpnI. The construct coding for truncated TF was selected and confirmed prior to use. The truncated EGFP-TFtrunc protein was expressed in cells as described above.
Preparation of peripheral blood mononuclear cells
Blood (29 ml) was withdrawn from forearm vein into syringes containing 1 ml of heparin (1,000 unit/ml) and placed into two 50 ml polypropylene tubes containing an equal volume of PBS. This mixture was then layered onto Histopaque-1077 (Sigma Chemical Company Ltd., Poole, UK). The tubes were then centrifuged at 400g for 30 min and the middle layer, containing the peripheral blood mononuclear cells (PBMC) was transferred into a fresh tube. An equal quantity of PBS was then added and centrifuged at 400g for 10 min to sediment the PBMC. The supernatant was discarded, the pellet resuspended in 10 ml of PBS and cell density was adjusted to 106 cells/ml in DMSO freeze medium (TCS CellWorks, Botolph Claydon, UK) and cryopreserved until use.
Visualisation of TF expression in K562 cells by confocal laser scanning microscopy
K562 cells transfected with pEGFP-TF were resuspended in pre-warmed PBS (1 ml) prior to analysis using a Radiance 2100 scanning confocal microscope (Bio-Rad Laboratories Inc, Hemel Hempstead, UK). In order to permit nuclear visualisation, the cells were incubated with 4′,6-Diamidino-2-phenylindole (DAPI) (10 μg/ml) (Sigma Chemical Company Ltd., Poole, UK) at 37°C for 1 h. Subsequently, the cells were incubated for 15 min at 37°C with 1,1′-dioctadecy1-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) (10 μg/ml) (Sigma Chemical Company Ltd., Poole, UK) to identify the cell membrane. The cells were washed three times with PBS and resuspended in pre-warmed culture media (1 ml) prior to analysis on the confocal microscope.
Measurement of the cytotoxic activity of PBMC by flow cytometry
PBMC (5×105) and target K562 human lymphoblast leukaemia cells (5×104) labelled with 5 μM SNARF-1 were mixed together in serum-free medium (200 μl) and incubated for 4 h at 37°C. The cells were centrifuged at 400g for 5 min, the cells washed twice and resuspended in PBS (300 μl) and analysed by flow cytometry. A gate was placed to include 100% of the labelled and exclude all of the unlabelled control cells. The number of cells in this quadrant before and after challenging with PBMC was used to calculate the cytotoxic activity, and was defined as the reduction in the cell number in this quadrant as a percentage of the original number.
Measurement of the cytotoxic activity of PBMC by fluorescence imaging
HT29 cells were seeded out (5×104 per well) in 12 well plates and transfected with the pEGFP-TF as described above. Only successfully transfected cells, as determined by flow cytometry or ELISA and the prothrombin time assays, were used for subsequent investigations. Transfected HT29 and untransfected HT29 cells were labelled with red fluorescence (SNARF-1) and placed in the wells in serum-free medium (200 μl) and incubated for 4 h. Ten random sectors within each well covering approximately 50% of the well, were imaged on a TS100 microscope (Nikon, Kingston-upon-Thames, UK) equipped with fluorescence attachments and camera. PBMC were added to the wells containing the target cells (HT29) at 10:1 (PBMC: HT29) and incubated for 4 h at 37°C. Subsequently, ten random sectors were recorded as before. Analysis was carried out using the ImagePro plus program (Media Cybernetics, Berkshire, UK) and the data calculated as the percentage cell loss after incubation with PBMC against the initial number of viable HT29 as follows:
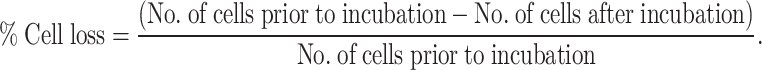 |
Results
In order to investigate the influence of TF on immune cytotoxicity towards tumour cells, an active form of human EGFP-TF protein was expressed in two separate tumour cell lines (HT29 and K562) and the cytotoxicity mediated by PBMC was measured. Flow cytometric analysis confirmed the surface expression of TF indicated by a significant increase in fluorescence in the transfected HT29 cells labelled with the FITC-conjugated anti-TF antibody (not shown) and stained with a phycoerythrin conjugated secondary antibody and compared to untransfected and control cells (Fig. 1). Moreover, Western blot analysis of the TF antigen showed a clear increase in the cells transfected with pEGFP-TF (not shown). The presence of surface TF was further confirmed by ELISA with a mean increase (SD) from 36±3 ng/106 to 68±4.5 ng/106 cells (n=3). Thirdly, the expression of EGFP-TF protein in these cells resulted in a marked decrease in the clotting time from >180 to 73±5 s (n=3), demonstrating the presence of active TF at the surface of the transfected cells. The procoagulant activity was diminished proportionally upon incubation with increasing concentrations of a TF-blocking antibody (results not shown). The expression of EGFP-TF in K562 cells was confirmed by flow cytometry, although these cells did not exhibit any TF pro-coagulant activity. However, lymphoblastic cells such as K562 cells, possess mechanisms to prevent surface exposure of TF, particularly by manipulating the cell surface phosphotidylserine [21] and explains the lack of procoagulant activity in these cells. Finally, confocal microscope analysis of both cell lines indicated the presence of EGFP-TF intracellularly, localised at the inner leaf of the cell membrane (Fig. 2) probably associated with membrane bodies such as caveolae [22, 23].
Fig. 1.

Flow cytometric assessment of TF antigen on the surface of HT29 cells. HT29 cells were transfected with pEGFP-TF and allowed to express the hybrid protein over 72 h. The cells were harvested and labelled with a mouse anti-human TF antibody. The cells were then washed and stained with a phycoerythrin-conjugated secondary antibody. The analysis of transfected and un-transfected cells was carried out by flow cytometry. A marker was set containing 5% of the control sample; 20% of untransfected cells and 49% for pEGFP-TF-transfected cells were in this region. Mean cell fluorescence intensity for the two samples were 3.7 and 7.5, respectively. The data is representative of three independent experiments
Fig. 2.
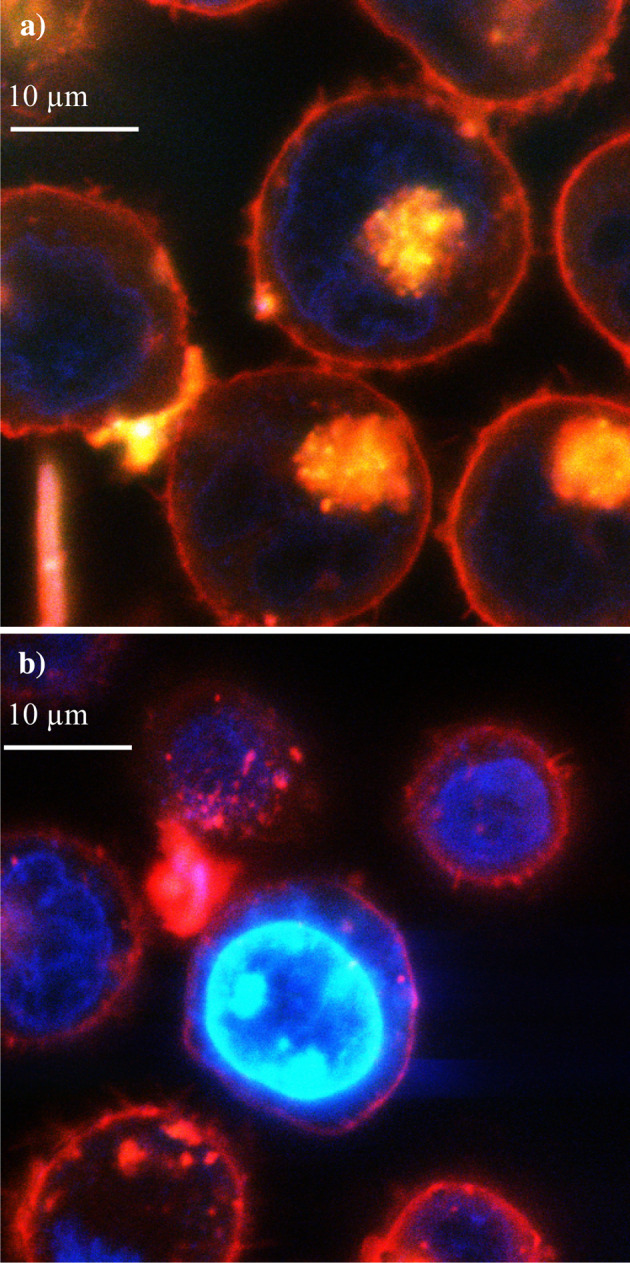
Confocal laser scanning microscopic localisation of EGFP-TF in K562 cells. K562 cells were transfected with pEGFP-TF and allowed to express the hybrid protein over 72 h. The cells were then labelled with 4′,6-Diamidino-2-phenylindole (DAPI) (10 μg/ml) and 1,1′-dioctadecy1-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) (10 μg/ml) to visualise the nucleus (blue) and the cell membrane (red), respectively. Following washing, the cells were re-suspended in PBS (1 ml) prior to visualisation with the confocal microscope. The co-localisation of EGFP-TF (green) with the cell membrane resulted in yellow-coloured foci on the cell membrane (a) which are absent in the untransfected cells (b). All experiments were done in duplicate
In order to investigate the influence of TF in aiding tumour cells to evade immune recognition, the cytotoxic capability of PBMC against HT29 cells expressing EGFP-TF was assessed. The target cells were labelled with SNARF-1 and ten fields of view analysed in each sample, using fluorescence image microscopy, before and after challenge with PBMC. The percentage of surviving (labelled) cells was calculated in each sample following incubation with PBMC, against the initial number of cells. The samples contained parallel sets of TF-overexpressing or control HT29 cells and were challenged separately with PBMC obtained from five donors in duplicate. Overall, there was a 15–40% reduction in cytotoxicity against TF-overexpressing cells in comparison to untransfected cells (Fig. 3). Moreover, PBMC were incubated with pEGFP-TF transfected K562 cells and cellular toxicity was measured by flow cytometry. Overall, although no increase in TF function had been observed with these cells, a 15–20% decrease in cytotoxicity was seen against TF-overexpressing K562 cells in comparison to untransfected cells (data not shown).
Fig. 3.

Measurement of the cytotoxic activity of PBMC against TF-overexpressing and control HT29 cells. pEGFP-TF transfected and un-transfected HT29 cells (5×104 per well) were labelled with 5 μM SNARF-1 in culture medium (200 μl final volume). Ten random views within each well were recorded using a fluorescence microscope. The cells were then challenged separately with five preparations of isolated PBMC at a ratio of 10:1 (PBMC: HT29). After 4 h incubation at 37°C, ten random sectors were analysed as before. Analysis was carried out using the Image Pro plus program and the data were calculated as the percentage reduction in cell number after incubation with PBMC against the initial number of viable HT29 cells. The data represent the mean of duplicates±standard deviation
To investigate whether the mechanism of TF-mediated suppression in immune reactivity involved the formation of fibrin coats on the tumour cells, parallel sets of control or EGFP-TF-expressing HT29 cells were pre-incubated with plasma for 15 min to allow fibrin formation on the cells prior to challenge with PBMC. Pre-incubation of EGFP-TF expressing cells, with 10 and 20% (v/v) plasma prior to challenge with PBMC, resulted in 41 and 34% cell loss, respectively, while the control cells remained at 60 and 40% cell loss (Fig. 4). In the absence of plasma, the cytotoxicity towards cells expressing TF and control cells exhibited 39 and 67% cell loss, respectively. It is also worth noting that the use of citrate as an anticoagulant did not affect the assay at the plasma dilutions used here (not shown).
Fig. 4.

Measurement of the cytotoxic activity of PBMC against TF-overexpressing and control HT29 cells, pre-incubated with plasma. pEGFP-TF transfected and un-transfected HT29 cells (5×104 per well) were labelled with SNARF-1 in culture medium (200 μl final volume). Ten random views within each well were recorded using a fluorescence microscope. Human normal plasma was added to the wells at final concentrations of 0, 10 and 20% (v/v) and incubated for 15 min prior to a 4 h incubation at 37°C, with two preparations of PBMC at a 10:1 ratio (PBMC:HT29). Subsequently, ten random sectors were analysed as before. Analysis was carried out using the Image Pro plus program and the data were calculated as the percentage reduction in cell number after incubation with PBMC cells against the initial number of viable HT29 cells. The data represent the mean of duplicates±standard deviation
Finally, to investigate the involvement of the intracellular domain of TF in the suppression of cytotoxicity towards tumour cells, a truncated form of TF (EGFP-TFtrunc) devoid of the cytoplasmic domain was prepared from the pEGFP-TF plasmid and transfected into HT29 cells. The expression of EGFP-TFtrunc resulted in similar procoagulant activity to those of the parent EGFP-TFwt transfected cells (78±5, 73±5 s, respectively; n=3). These cells and untransfected HT29 cells were labelled and challenged with PBMC from two donors and assessed using fluorescence image microscopy as above. The cytotoxicity observed against cells expressing full-length TF (EGFP-TF) was diminished (59%±10 cell loss), in comparison with the control cells (40%±5 cell loss) (Fig. 5). However, the expression of the truncated form of TF (EGFP-TFtrunc) ablated this protection against PBMC-mediated cytotoxicity (35%±13 cell loss).
Fig. 5.

Measurement of the cytotoxic activity of PBMC against full-length, truncated TF-overexpressing and control HT29 cells. A truncated form of the EGFP-TF was expressed by mutating the Ser240 into a stop codon. HT29 cells transfected with pEGFP-TFwt and pEGFP-TFtrunc transfected and un-transfected HT29 cells (5×104 per well) were labelled with SNARF-1 in culture medium (200 μl final volumes). Ten random views within each well were recorded using a fluorescence microscope. The cells were then challenged with preparations of isolated PBM cells at a ratio of 10:1 (PBMC: HT29). After 4 h incubation at 37°C, ten random sectors were analysed as before. Analysis was carried out using the Image Pro plus program and the data were calculated as the percentage reduction in cell number after incubation with PBMC cells against the initial number of viable HT29 cells. The data represent the mean of duplicates±standard deviation
Discussion
While it has been suggested that more than 95% of cells disseminated from a primary tumour do not survive in the circulation [24], the ability of cancer to metastasise remains a major step in the progression of the disease. One key event in successful tumour cell metastasis is the ability to avoid immune recognition. Previous studies have suggested that fibrin formation may interfere with immune recognition and hence dampen the cytotoxic activity of natural and lymphokine-activated killer cells towards tumour cells [17, 18]. However, other investigations have shown that fibrin formation does not affect immune evasion by tumour cells [19, 20]. The association between blood clots and tumours has been reported since 1878 [25]. Recently, a number of investigations have revealed important associations between TF and tumour pathology, including the upregulation of TF in aggressive tumours [8, 26–28]. Correlations between TF expression and invasiveness [6], metastatic properties [29, 30], susceptibility to doxorubicin [4] and malignant phenotype [28] have all been reported previously. Intriguingly, a number of studies have also reported a requirement for the presence of the intracellular domain of TF for the full pro-metastatic potential of this protein [27, 29, 31].
In this study, we have developed a novel in vitro procedure, employing fluorescence microscopy to measure the cytotoxic activity of PBMC against two separate cell lines. This technique was employed to investigate (1) whether the expression of TF can directly influence tumour cell evasion of immune surveillance, and (2) to examine the underlying mechanisms involved in TF-induced immune avoidance. Studies of cytotoxicity in vitro are usually carried out employing a target cell line that is sensitive to natural cytotoxicity. The determination of the number of targeted cells most commonly relies on a secondary measurement such as 51Cr [32], or other measurable indicator such as an enzyme, fluorescent dye or expressed luciferase [33], released from the lysed cells. These methods rely on the accurate determination of the measurable agent and therefore are prone to errors arising from the background release, poor levels of labelling and inconsistency between experiments. Ultimately, the most accurate measurement of cellular cytotoxicity is by determining the number of viable cells, prior to and following challenge with immune cells.
Here, HT29 and K562 cells were transfected with a vector to express EGFP-TF or EGFP and then labelled with SNARF-1, to easily distinguish the cells from PBMC with which they were incubated. The TF activity measured in the transfected cells was comparable to the ones we have measured in more aggressive TF-expressing tumour cells such as the cell line LoVo. The data indicate a relatively high level of cytotoxicity exhibited by the PBMC against the adherent HT29 cells when measured by fluorescence microscopy (up to 68% cell loss). The assessment of non-adherent K562 cells showed a lower, but still significant, percentage (up to 47% cell loss). Therefore, we have used this means of assessment in order to investigate the mechanism by which TF expression on tumour cells can suppress cellular cytotoxicity.
The transfection of HT29 cells with the pEGFP-TF construct encoding for a hybrid fluorescent protein resulted in the expression of active TF, while K562 lymphoblastic cells expressed an inactive form of the protein. This lack of activity was attributed to the nature of these cells which normally do not present this protein at the surface. Our data clearly show that the expression of TF protects the cells from lysis (Fig. 3). Moreover, the lack of procoagulant activity in K562 cells suggests that TF may be acting via a mechanism not involving the activation of the coagulation pathway. To examine further the influence of the procoagulant extracellular domain of TF, the cells were pre-incubated with either 10 or 20% (v/v) human plasma, to allow fibrin formation, prior to challenge with PBMC. Our data suggest that TF can protect cells from the cytotoxic action of PBMC, independent of fibrin formation (Fig. 4). Furthermore, the protection from PBMC cytotoxicity was not observed in K562 cells (data not shown). In conclusion, our data support the suggestion that fibrin is not the main mechanism by which tumour cells increase their chance of surviving cytotoxic activity.
To examine the possibility that TF may mediate cytotoxic protection through mechanisms involving the cytoplasmic domain of this protein, a truncated TF protein lacking the intracellular region was created. As mentioned previously, the cytoplasmic domain of TF has been associated with increased tumour metastasis and invasiveness [29, 31]. It has recently become clear that this domain possesses signalling capabilities [27, 29, 34–36]. The hybrid EGFP-TFtrunc and EGFP-TFwt proteins were expressed at similar levels in HT29 cells and no difference in the procoagulant activity was observed. This is consistent with current understanding that the cytoplasmic domain is not essential for the procoagulant activity [37, 38]. However, our data also indicate that the deletion of the cytoplasmic domain resulted in tumour cell lysis comparable to that of the control cells, i.e. the protective influence of TF was removed. Hence, we suggest that the ability of TF to protect tumour cells from immune cytotoxicity mainly arises from signalling mechanisms involving the cytoplasmic domain of TF. One feasible explanation would be that TF signalling results in the downregulation of surface antigens that would otherwise allow the killer cells to recognise and target the tumour cells.
In conclusion, we have shown that TF has a direct protective influence on the ability of tumour cells to escape immune recognition by PBMC. Moreover, it is clear that this influence is independent of fibrin coat formation but instead involves the cytoplasmic domain of TF. TF is known to influence cancer cell metastasis by a number of suggested mechanisms including increased angiogenesis [26, 36, 39–42] cellular growth [43–45] and through antiapoptotic influences [34, 46–48]. Therefore, we suggest that in addition to these functions, TF also allows tumour cells to be protected from cellular cytotoxicity, increasing the possibility of successful tumour dissemination and metastasis.
References
- 1.Kirchhofer D, Nemerson Y. Initiation of blood coagulation: the tissue factor/factor VIIa complex. Curr Opin Biotechnol. 1996;7:386–391. doi: 10.1016/S0958-1669(96)80112-1. [DOI] [PubMed] [Google Scholar]
- 2.Callander NS, Varki N, Rao LV. Immunohistochemical identification of tissue factor in solid tumors. Cancer. 1992;70:1194–1201. doi: 10.1002/1097-0142(19920901)70:5<1194::AID-CNCR2820700528>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 3.Kakkar AK, DeRuvo N, Chinswangwatanakul V, Tebbutt S, Williamson RC. Extrinsic-pathway activation in cancer with high factor VIIa and tissue factor. Lancet. 1995;346:1004–1005. doi: 10.1016/S0140-6736(95)91690-3. [DOI] [PubMed] [Google Scholar]
- 4.Koomagi R, Volm M. Tissue-factor expression in human non-small-cell lung carcinoma measured by immunohistochemistry: correlation between tissue factor and angiogenesis. Int J Cancer. 1998;79:19–22. doi: 10.1002/(SICI)1097-0215(19980220)79:1<19::AID-IJC4>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 5.Lawleed BA, Chisholm M, Francis JL. Urinary tissue factor levels in patients with breast and colorectal cancer. J Pathol. 1999;187:291–294. doi: 10.1002/(SICI)1096-9896(199902)187:3<291::AID-PATH213>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 6.Vrana JA., Stang MT, Grande JP, Getz MJ. Expression of tissue factor in tumour stroma correlates with progression to invasive human breast Cancer: paracrine regulation by carcinoma cell-derived members of the transforming growth factor beta family. Cancer Res. 1996;56:5063–5070. [PubMed] [Google Scholar]
- 7.Ueda C, Hirohata Y, Kihara Y, Nakamura H, Abe S, Akahane K, Okamoto K, Itoh H, Otsuki M. Pancreatic cancer complicated by disseminated intravascular coagulation associated with production of tissue factor. J Gastroenterol. 2001;36:845–850. doi: 10.1007/s005350170008. [DOI] [PubMed] [Google Scholar]
- 8.Hamada K, Kuratsu J, Saitoh Y, Takeshima H, Nishi T, Ushio Y. Expression of tissue factor correlates with grade of malignancy in human glioma. Cancer. 1996;77:1877–1883. doi: 10.1002/(SICI)1097-0142(19960501)77:9<1877::AID-CNCR18>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 9.Abdulkadir SA, Carvalhal GF, Kaleem Z, Kisiel W, Humphrey PA, Catalona WJ, Milbrandt J. Tissue factor expression and angiogenesis in human prostate carcinoma. Hum Pathol. 2000;31:443–447. doi: 10.1053/hp.2000.6547. [DOI] [PubMed] [Google Scholar]
- 10.Nakasaki T, Wada H, Shigemori C, Miki C, Gabazza EC, Nobori T, Nakamura S, Shiku H. Expression of tissue factor and vascular endothelial growth factor is associated with angiogenesis in colorectal cancer. Am J Hematol. 2002;69:247–254. doi: 10.1002/ajh.10061. [DOI] [PubMed] [Google Scholar]
- 11.Shigenmori C, Wada H, Matsumoto K, Shioklu H, Nakamura S, Suzuki H. Tissue factor expression and metastatic potential of colorectal cancer. Thromb Haemostas. 1998;80:894–898. [PubMed] [Google Scholar]
- 12.Bierhaus A, Nawroth PP. Coagulation, inflammation and immune response-an evolutionary conserved plan as cause for disseminated intravasal coagulation? Hamostaseologie. 2005;25:23–32. doi: 10.1267/hämo05010023. [DOI] [PubMed] [Google Scholar]
- 13.Dvora HF, Senger DR, Dvorak AM. Fibrin as a component of the tumor stroma: origins and biological significance. Cancer Metastasis Rev. 1983;2:41–73. doi: 10.1007/BF00046905. [DOI] [PubMed] [Google Scholar]
- 14.Bardos H, Molnar P, Csecsei G, Adany R. Fibrin deposition in primary and metastatic human brain tumors. Blood Coagula Fibrinolysis. 1996;7:536–548. doi: 10.1097/00001721-199607000-00005. [DOI] [PubMed] [Google Scholar]
- 15.Wojtukiewicz MZ, Zacharaski LR, Memoli VA. Fibrinogen-fibrin transformation in situ in renal cell carcinoma. Anticancer Res. 1990;10:579–582. [PubMed] [Google Scholar]
- 16.Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, Jirouskova M, Degen JL. Platelets and fibrin (ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood. 2005;105:178–185. doi: 10.1182/blood-2004-06-2272. [DOI] [PubMed] [Google Scholar]
- 17.Gunji Y, Lewis J, Gorelik E. Fibrin formation inhibits the in vitro cytotoxic activity of human natural and lymphokine-activated killer cells. Blood Coagul Fibrinolysis. 1990;1:663–672. [PubMed] [Google Scholar]
- 18.Gorelik E. Protective effect of fibrin on tumour metastasis. Fibrinolysis. 1992;6:35–38. doi: 10.1016/0268-9499(92)90091-U. [DOI] [Google Scholar]
- 19.Carr ME, Jr, Sajer SA, Spaulding A. Fibrin coating of bladder tumor cells (T24) is not protective against LAK cell cytotoxicity. J Lab Clin Med. 1992;119:132–138. [PubMed] [Google Scholar]
- 20.Reijerkerk A, Meijers JC, Havik SR, Bouma BN, Voest EE, Gebbink MF. Tumor growth and metastasis are not affected in thrombin-activatable fibrinolysis inhibitor-deficient mice. J Thromb Haemost. 2004;2:769–779. doi: 10.1111/j.1538-7836.2004.00682.x. [DOI] [PubMed] [Google Scholar]
- 21.Kunzelmann-Marche C, Satta N, Toti F, Zhang Y, Nawroth PP, Morrissey JH, Freyssinet JM. The influence exerted by a restricted phospholipid microenvironment on the expression of tissue factor activity at the cell plasma membrane surface. Thromb Haemost. 2000;83:282–289. [PubMed] [Google Scholar]
- 22.Dietzen DJ, Jack GG, Page KL, Tetzloff TA, Hall CL, Mast AE. Localization of tissue factor pathway inhibitor to lipid rafts is not required for inhibition of factor VIIa/tissue factor activity. Thromb Haemost. 2003;89:65–73. [PubMed] [Google Scholar]
- 23.Mulder AB, Smit JW, Bom VJ, Blom NR, Ruiters MH, Halie MR, van der Meer J. Association of smooth muscle cell tissue factor with caveolae. Blood. 1996;88:1306–1313. [PubMed] [Google Scholar]
- 24.Weiss L. Metastatic inefficiency. Adv Cancer Res. 1990;54:159–211. doi: 10.1016/S0065-230X(08)60811-8. [DOI] [PubMed] [Google Scholar]
- 25.Shoji M, Hancock WW, Abe K, Micko C, Casper KA, Baine RM, Wilcox JN, Danave I, Dillehay DL, Matthews E, Contrino J, Morrissey JH, Gordon S, Edgington TS, Kudryk B, Kreutzer DL, Rickles FR. Activation of coagulation and angiogenesis in cancer: immunohistochemical localization in situ of clotting proteins and vascular endothelial growth factor in human cancer. Am J Pathol. 1998;152:399–411. [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Tissue factor controls the balance of angiogenic and antiangiogenic properties of tumor cells in mice. J Clin Invest. 1994;94:1320–1327. doi: 10.1172/JCI117451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mueller B, Ruf W. Requirement for binding of catalytically active factor VIIa in tissue factor-dependent experimental metastasis. J Clin Invest. 1998;101:1372–1378. doi: 10.1172/JCI930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Contrino J, Hair G, Kreutzer DL, Rickles FR. In situ detection of tissue factor in vascular endothelial cells: correlation with the malignant phenotype of human breast disease. Nat Med. 1996;2:209–215. doi: 10.1038/nm0296-209. [DOI] [PubMed] [Google Scholar]
- 29.Bromberg ME, Sundaram R, Homer RJ, Garen A, Konigsberg WH. Role of tissue factor in metastasis: functions of the cytoplasmic and extracellular domains of the molecule. Thromb Haemost. 1999;82:88–92. [PubMed] [Google Scholar]
- 30.Versteeg HH, Richel DJ, Peppelenbosch MP, Spek CA. Concerted action of coagulation factors on cell survival. J Thromb Haemost. 2004;2:673–674. doi: 10.1111/j.1538-7836.2004.00691.x. [DOI] [PubMed] [Google Scholar]
- 31.Bromberg ME, Konigsberg WH, Madison JF, Pawashe A, Garen A. Tissue factor promotes melanoma metastasis by a pathway independent of blood coagulation. Proc Natl Acad Sci USA. 1995;92:8205–8209. doi: 10.1073/pnas.92.18.8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brunner KT, Mauel J, Cerottini JC, Chapuis B. Quantitative assay of the lytic action of immune lymphoid cells on 51-Cr-labelled allogeneic target cells in vitro; inhibition by isoantibody and by drugs. Immunology. 1968;14:181–196. [PMC free article] [PubMed] [Google Scholar]
- 33.Brown CE, Wright CL, Naranjo A, Vishwanath RP, Chang WC, Olivares S, Wagner JR, Bruins L, Raubitschek A, Cooper LJ, Jensen MC. Biophotonic cytotoxicity assay for high-throughput screening of cytolytic killing. J Immunol Methods. 2005;297:39–52. doi: 10.1016/j.jim.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 34.Sorensen BB, Rao LV, Tornehave D, Gammeltoft S, Petersen LC. Antiapoptotic effect of coagulation factor VIIa. Blood. 2003;102:1708–1715. doi: 10.1182/blood-2003-01-0157. [DOI] [PubMed] [Google Scholar]
- 35.Abe K, Shoji M, Chen J, Bierhaus A, Danave I, Micko C, Casper K, Dillehay DL, Nawroth PP, Rickles FR. Regulation of vascular endothelial growth factor production and angiogenesis by the cytoplasmic tail of tissue factor. Proc Natl Acad Sci USA. 1999;96:8663–8668. doi: 10.1073/pnas.96.15.8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Versteeg HH, Peppelenbosch MP, Spek CA. Tissue factor signal transduction in angiogenesis. Carcinogenesis. 2003;24:1009–1013. doi: 10.1093/carcin/bgg039. [DOI] [PubMed] [Google Scholar]
- 37.Paborsky LR, Tate KM, Harris RJ, Yansura DG, Band L, McCray G, Gorman CM, O’Brien DP, Chang JY, Swartz JR, Fung VP, Thomas JN, Vehar GA. Purification of recombinant human tissue factor. Biochemistry. 1989;28:8072–8077. doi: 10.1021/bi00446a016. [DOI] [PubMed] [Google Scholar]
- 38.Paborsky LR, Caras IW, Fisher KL, Gorman CM. Lipid association, but not the transmembrane domain, is required for tissue factor activity. Substitution of the transmembrane domain with a phosphatidylinositol anchor. J Biol Chem. 1991;266:21911–21916. [PubMed] [Google Scholar]
- 39.Belting M, Dorrell MI, Sandgren S, Aguilar E, Ahamed J, Dorfleutner A, Carmeliet P, Mueller BM, Friedlander M, Ruf W. Regulation of angiogenesis by tissue factor cytoplasmic domain signaling. Nat Med. 2004;10:502–509. doi: 10.1038/nm1037. [DOI] [PubMed] [Google Scholar]
- 40.Pawlinski R, Pedersen B, Erlich J, Mackman N. Role of tissue factor in haemostasis, thrombosis, angiogenesis and inflammation: lessons from low tissue factor mice. Thromb Haemostas. 2004;92:444–450. doi: 10.1160/TH04-05-0309. [DOI] [PubMed] [Google Scholar]
- 41.Watanabe T, Yasuda M, Yamamoto T. Angiogenesis induced by tissue factor in vitro and in vivo. Thrombosis Res. 1999;96:183–189. doi: 10.1016/S0049-3848(99)00101-2. [DOI] [PubMed] [Google Scholar]
- 42.James NJ, Ettelaie C, Bruckdorfer KR. Inhibition of tissue factor activity reduces the density of cellular network formation in an in vitro model of angiogenesis. Biochem Soc Trans. 2002;30:217–221. doi: 10.1042/BST0300217. [DOI] [PubMed] [Google Scholar]
- 43.Camerer E, Gjernes E, Wiiger M, Pringle S, Prydz H. Binding of factor VIIa to tissue factor on keratinocytes induces gene expression. J Biol Chem. 2000;275:6580–6585. doi: 10.1074/jbc.275.9.6580. [DOI] [PubMed] [Google Scholar]
- 44.Poulsen LK, Jacobsen N, Sorensen BB, Bergenhem NC, Kelly JD, Foster DC, Thastrup O, Ezban M, Petersen LC. Signal transduction via the mitogen-activated protein kinase pathway induced by binding of coagulation factor VIIa to tissue factor. J Biol Chem. 1998;273:6228–6232. doi: 10.1074/jbc.273.11.6228. [DOI] [PubMed] [Google Scholar]
- 45.Hartzell S, Ryder K, Lanahan A, Lau LF, Nathan D. A growth factor-responsive gene of murine BALB/c 3T3 cells encodes a protein homologous to human tissue factor. Mol Cell Biol. 1989;9:2567–2573. doi: 10.1128/mcb.9.6.2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Versteeg HH, Spek CA, Peppelenbosch MP, Richel DJ. Tissue factor and cancer metastasis: the role of intracellular and extracellular signaling pathways. Mol Med. 2004;10:6–11. doi: 10.2119/2003-00047.Versteeg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Versteeg HH, Spek CA, Richel DJ, Peppelenbosch MP. Coagulation factors VIIa and Xa inhibit apoptosis and anoikis. Oncogene. 2004;23:410–417. doi: 10.1038/sj.onc.1207066. [DOI] [PubMed] [Google Scholar]
- 48.Versteeg FVIIa: TF induces cell survival via G12/G13-dependent Jak/STAT activation and BclXL production. Circ Res. 2004;94:1032–1040. doi: 10.1161/01.RES.0000125625.18597.AD. [DOI] [PubMed] [Google Scholar]