

Abstract
Bee venom secretory phospholipase A2 (bv-sPLA2) and phosphatidylinositol-(3,4)-bisphosphate (PtdIns(3,4)P2) act synergistically to induce cell death in tumour cells of various origins with concomitant stimulation of the immune system. Here, we investigated the mechanisms involved in such actions and examined structural requirements of PtdIns-homologues to inhibit tumour cells in combination with bv-sPLA2. Renal cancer cells were treated with bv-sPLA2 alone or in combination with PtdIns-homologues. Inhibitory effects on [3H] thymidine incorporation and intracellular signal transduction pathways were tested. Reaction products generated by bv-sPLA2 interaction with PtdIns(3,4)P2 were identified by mass spectrometry. Among the tested PtdIns-homologues those with a phosphate esterified to position 3 of the inositol head group, were most efficient in cooperating with bv-sPLA2 to block tumour cell proliferation. Growth inhibition induced by the combined action of bv-sPLA2 with either PtdIns(3,4)bisphosphate or PtdIns(3,4,5)trisphosphate were synergistic and accompanied by potent cell lysis. In contrast, PtdIns, which lacked the phosphate group at position 3, failed to promote synergistic growth inhibition. The combined administration of PtdIns(3,4)P2 and bv-sPLA2 abrogated signal transduction mediated by extracellular signal regulated kinase 1 and 2 and prevented transduction of survival signals mediated by protein kinase B. Surface expression of the epidermal growth factor (EGF)-receptor was reduced after PtdIns(3,4)P2-bv-sPLA2 administration and associated with a blockade of EGF-induced signalling. In addition, mass spectroscopy revealed that bv-sPLA2 cleaves PtdIns(3,4)P2 to generate lyso-PtdIns(3,4)P2. In conclusion, we suggest that the cytotoxic activity mediated by PtdIns(3,4)P2 and bv-sPLA2 is due to cell death that results from disruption of membrane integrity, abrogation of signal transduction and the generation of cytotoxic lyso-PtdIns(3,4)P2.
Keywords: Bee venom PLA2, Phosphatidylinositol, Signal transduction, Cancer immunotherapy, ESI-MS/MS
Introduction
We have previously shown a strong synergy between bee venom secretory phospholipase A2 (bv-sPLA2) and phosphatidylinositol-(3,4)-bisphosphate (PtdIns(3,4)P2) that mediates inhibition and lysis of a variety of tumour cells from different tissues [35]. The resulting tumour lysates enhanced maturation and immunostimulatory capacities of monocyte derived dendritic cells (moDCs). Such lysates represent a source of tumour-associated antigens in conjunction with adjuvant properties. Thus, these lysates fulfil the basic requirements of a cancer vaccine and can be applied in the context of moDC based cancer immunotherapy. In addition, such lysates can themselves be considered as multivalent vaccines for active immunisations. Alternatively, the observation of tumour cell inhibition by bv-sPLA2 and PtdIns(3,4)P2 implicates a potential of these compounds for the treatment of cancer.
Bee venom secretory phospholipase A2 group III represents a member of the phospholipases A2 (PLA2) family of enzymes that catalyse the hydrolysis of the sn-2 fatty acyl ester bond of membrane glycero-3-phospholipids to generate free fatty acids and lysophospholipids [9, 41, 44]. In principle, sPLA2 enzymes can influence immunogenicity and proliferative capacities of tumour cells by various mechanisms. sPLA2-activity catalytically hydrolyses and digests cell membrane components [26] and consequently disrupts integrity of the lipid bilayers thus making cells susceptible to further degradation. Direct protein interaction of PLA2 enzymes with cell surface receptors regulates a variety of biological activities including proliferation [12, 28, 29]. Immunostimulatory or cytotoxic action of sPLA2 reaction products like lysophosphatidylcholine and lipid mediators has been observed frequently [1, 15, 19]. Although these modes of sPLA2 action differ fundamentally, they all affect intracellular signal transduction, which mediates proliferation and survival. In general, the mitogen-activated protein kinases (MAPK) cascade is associated with proliferative effects. Among the subgroups of MAPK, the extracellular signal regulated kinases (ERK) function as key mediators of mitogenic agonist stimulation [8, 25, 34]. Survival signals are transduced via a pathway that involves phosphatidylinositol 3-OH kinase (PI3-kinase) and protein kinase B (PKB/Akt). PKB/Akt activation is considered both necessary and sufficient for cell survival [6]. sPLA2 isoenzymes investigated so far demonstrated rather a stimulatory effect on MAPK and PKB/Akt, whereas no conclusive data on bv-sPLA2 effects on these kinases in cancer cells are available [4, 16, 22, 27, 31]. However, synthetic analoga of lysolipids, which structurally resemble reaction products of sPLA2 mediate anti-proliferative effects and cytotoxicity via blockades of both, ERKs and PKB/Akt and thus have been suggested for cancer therapies [3, 37-39]. PtdIns(3,4)P2 and other PtdIns-homologues affect signal transduction as second messengers or precursors thereof, or interact directly with signal-transducing proteins [33, 43]. PI3-kinases generate 3-phosphorylated PtdIns which recruit PKB/Akt to the cell membrane where it gets phosphorylated [20]. Once activated, PKB/Akt phosphorylates several signalling proteins, which, in turn, leads to the choice of cellular proliferation or apoptosis [13, 21].
As bv-sPLA2 cooperates with PtdIns(3,4)P2 we hypothesised that this interaction affects signal transduction on the level of PKB/Akt and is related to agonist mediated MAPK signalling. It was also reasonable to speculate that PtdIns(3,4)P2 can act as a substrate for hydrolysis by bv-sPLA2 which would generate a lyso-PtdIns(3,4)P2 with putative anti-cancer properties. Thus, the aim of the current study was to investigate mechanistic aspects of the combined inhibitory action of bv-sPLA2 and PtdIns(3,4)P2 in renal carcinoma cells. Mitogenic signal transduction and survival signalling in response to bv-sPLA2 and PtdIns(3,4)P2 treatment was analysed and reaction products of the catalytical interaction were identified. In addition, we defined further PtdIns-homologues that cooperated with bv-sPLA2 and delineated structural requirements necessary for this interaction.
Materials and Methods
Reagents
Secretory phospholipase A2 (Type III) from bee venom sPLA2 was purchased from Cayman Chemical (Ann Arbor, MI, USA). sPLA2 activity was routinely monitored using the colorimetric sPLA2 assay kit from Cayman Chemical which is based on the synthetic substrate diheptanoyl thiophosphorylcholine. L-α-Phosphatidylinositol [PtdIns] was purchased from Biomol (Hamburg, Germany). Phosphatidylinositol-3,4-bisphosphate (1,2-dipalmitoyl, ammonium salt) [PtdIns(3,4)P2]; phosphatidylinositol-3,4,5-trisphosphate (1,2-dipalmitoyl, ammonium salt) [PtdIns(3,4,5)P3]; phosphatidylinositol-3,4,5-trisphosphate (1-stearyl, 2-arachidonoyl,sodium salt) [PtdIns(3,4,5)P3-AA]; phosphatidylinositol-4,5-bisphosphate (1,2-dipalmitoyl, ammonium salt) [PtdIns(4,5)P2]; D-myo-inositol-1,3,4-trisphosphate (sodium salt) [Ins(1,3,4)P3]; phosphatidylinositol-3-monophosphate (1,2-dipalmitoyl, ammonium salt) [PtdIns(3)P]; phosphatidylinositol-4-monophosphate (1,2-dipalmitoyl, ammonium salt) [PtdIns(4)P] and phosphatidylinositol-5-monophosphate (1,2-dipalmitoyl, ammonium salt) [PtdIns(5)P] were purchased from Cayman Chemical and were prepared according to the manufacturer’s instructions. Concentrations used in this study were established based on a compound to cell ratio. For instance 20,000 cells treated with 2 μg bv-sPLA2 and 2 μg PtdIns(3,4)P2, require 10 μg bv-sPLA2 and 10 μg PtdIns(3,4)P2 in a scaled up experiment with 100,000 cells to yield the indicated effect in the respective bioassay (e.g. proliferation, phosphorylation and flow cytometry). Methanol HPLC grade and chloroform p.a were purchased from Merck (Darmstadt, Germany). 1,2 dipalmitoylphosphatidylinositol (C32:0 PtdIns-ISTD), piperidine and ammonium acetate (MS grade) were purchased from Sigma-Aldrich (Vienna, Austria).
Cell culture
The human kidney carcinoma cell line A498 [10] was propagated at 37°C and 5% CO2 in the presence of 10% fetal calf serum (FCS, heat-inactivated, 30 min, 56°C) in medium consisting of RPMI 1640 supplemented with 10 mM Hepes, non-essential amino acids (1×), 1 mM Na-pyruvate (all from Cambrex Bio Science, Verviers, Belgium), Glutamax (1×) (Invitrogen, Paisley, Scotland, UK), 100 U/ml penicillin and 100 μg/ml streptomycin (PAA Laboratories, Linz, Austria).
Measurement of proliferation
Before each experiment cells were replated and cultured in medium containing 10% FCS for 24 h. An amount of 20,000 cells were then plated in 200 μl medium without serum in 96-well flat-bottomed tissue culture plates (Falcon, BD, Franklin Lakes, NJ, USA). Cells were incubated for 32 h in the presence or absence of PtdIns-homologues and bv-sPLA2. During the last 16 h cells were pulsed with 50 μl fresh medium containing 10% FCS and 1 μCi/well (37 kBq/well) [3H] thymidine (ICN Biomedicals, Eschwege, Germany). Cells were harvested with a Tomtec harvester (Hamden, CT, USA), liquid scintillation counting was performed with a Chameleon multilabel reader (HVD-Life Science, Vienna, Austria). Results are mean cpm ± SEM of triplicate wells. Values were normalised by setting controls to 100%. Typical values for A498 cells (untreated controls) in this set-up ranged from 16,331 to 25,543 cpm.
Measurement of Akt and MAPK phosphorylation
Cells were replated and cultured in medium containing 10% FCS for 24 h. An amount of 600,000 cells were then plated in 300 μl medium without serum in 24-well flat-bottomed tissue culture plates (Costar, Corning Inc., NY, USA). Cells were incubated for 90 min in the presence or absence of PtdIns-homologues (200 μM), epidermal growth factor (EGF) (50 ng/ml) and bv-sPLA2 (200 μg/ml). Subsequently, 200 μl of lysis buffer (Biosource, CA, USA) were added. Lysates were harvested and frozen at 80°C. Analysis of Akt phosphorylation was performed with ELISA kits that detect Akt protein phosphorylated at serine 473 (Biosource). In addition, ELISA kits (Biosource), which detect total Akt independently of phosphorylation status were used for normalising the phospho-serine 473 content of the samples. Untreated controls of normalised phospho-serine 473 levels were set to 100%. For quantification of Erk1/2 phosphorylation ELISA kits that detect both, Erk1 protein phosphorylated at threonine 202 and tyrosine 204, as well as Erk2 protein phosphorylated at threonine 185 and tyrosine 187 (Biosource) were performed. In addition, ELISA kits (Biosource), which detect total Erk1/2 independently of phosphorylation status were used for normalising the phospho-Erk1/2 content of the samples. Untreated controls of normalised dual phosphorylated Erk1/2 levels were set to 100%. Results are mean values ± SEM of at least duplicate measurements.
EGF-receptor expression
Cells were replated and cultured with medium containing 10% FCS for 24 h. Cells were harvested with PBS buffer (Cambrex) supplemented with 1 mM EDTA (Versen; Biochrom, Berlin, Germany). An amount of 100,000 cells were then plated in 100 μl medium without serum in 96-well round-bottomed tissue culture plates (Costar, Corning Inc.). Cells were incubated for 45 min in the presence or absence of PtdIns(3,4)P2 (100 μM) and bv-sPLA2 (100 μg/ml). Subsequently, FCS was added to a final concentration of 10% to attenuate bv-sPLA2 action. Cells were then washed in PBS buffer (Cambrex), resuspended in medium and subjected to immunofluorescence staining for flow cytometric analysis using PE-labelled anti-EGF-receptor (EGFR) antibodies and PE-labelled anti-mouse IgG2b,k antibodies (isotype control), a FACSCalibur and CellQuest software (all from BD, Mountain View, CA, USA).
7-AAD Staining
A498 cells were replated and cultured in medium containing 10% FCS for 24 h. An amount of 100,000 cells were then plated in 100 μl medium without serum in 96-well round-bottomed tissue culture plates (Costar, Corning Inc.). Cells were incubated for 45 min in the presence or absence of PtdIns(3,4)P2 (100 μM) and bv-sPLA2 (100 μg/ml). Subsequently, NaN3 was added to a final concentration of 1%, and FCS was added to a final concentration of 10% to attenuate bv-sPLA2 action. Cells were then incubated with 10 μg/ml of the fluorescent DNA stain 7-amino-actinomycin D (7-AAD; Sigma-Aldrich) for 3 min and were washed in PBS buffer (Cambrex). The percentage of 7-AAD+ cells was determined with a FACSCalibur flow cytometer (BD) and CellQuest software (BD).
Mass spectrometry
Lipids were extracted from cell culture and media control samples using a modified procedure of Folch et al. [11] omitting the salt addition. Mass spectrometric analyses were performed on a triple-quadrupole mass spectrometer, the API4000 Qtrap (Applied Biosystems/MDS Sciex Toronto, ON, Canada) operated in negative ionisation modes with a turbo-ionspray source and Analyst Version 1.4.1 data system (Applied Biosystems/MDS Sciex). Samples (50 μl) were injected via an CTC-HTC-PAL autosampler (Zwingen, Switzerland) or infused (100 μl) with a syringe into the electrospray source at a flow rate of 25 μl/min by a binary Agilent HP 1100 HPLC pump (Böblingen, Germany) or 10 μl/min with a syringe pump (Harvard, MA, USA), respectively. For HPLC a carrier solvent of 4:1 CH3OH/CHCl3 was used. Nitrogen was used for the collision and sheath gas. Precursor scans (PS in negative ion mode) of lipid head groups included PS 255, 241, 321 and 401.
Sample preparation for mass spectrometric lipid analysis of A498 cells
A498 cells (105/ml) were incubated in serum-free medium (6 ml) in the presence or absence of bv-sPLA2 (10 μg/ml), PtdIns(3,4)P2 (10 μM) or a combination thereof for 2 h at 37°C and 5% CO2. Supernatants and cells were harvested, 4.5 ml of this suspension were concentrated by lyophilisation. Samples were then reconstituted in 300 μl H2O and transferred into a 10 ml glass tube. One millilitre of methanol was used to wash residual material followed by a further wash with 2 ml of chloroform and 3 ml of chloroform/methanol (2:1 v/v). Finally 250 pmol of 1,2 dipalmitoylphosphatidylinositol was added to each tube as an internal standard and the mixture was incubated for 1 h in the dark at room temperature whilst shaking. Then, 1.5 ml of distilled H2O was added to the mixture and shaken for further 30 min. The samples were centrifuged for 10 min at 1,500g and the lower organic phase was collected and dried under a gentle stream of nitrogen. Prior to electrospray ionisation mass spectrometry (ESI-MS/MS) analysis the samples were reconstituted in 1 ml of chloroform/methanol/H2O (2:1:01 v:v:v).containing 10 mM ammonium acetate.
Sample preparation for mass spectrometric lipid analysis in cell-free media
Serum-free medium (2 ml) supplemented with bv-sPLA2 (10 μg/ml), PtdIns(3,4)P2 or a combination thereof was incubated for 2 h at 37°C and 5% CO2 and was processed and analysed as previously described but with the following changes to improve recovery of the PtdIns(3,4)P2 and its purported lyso product. After the 1 h incubation in the dark at room temperature whilst shaking, water was substituted with a 20 mM ammonium acetate solution and the upper phase including the interface was dried and reconstituted with methanol (200 μl) for desalting with a (pre-washed) reverse-phase extraction cartridge (Isolut C18 endcapped, 50 mg/1 ml). The samples were desalted on-column with 2 ml of distilled water and then eluted with 300 μl methanol and 500 μl of chloroform/methanol/H2O (2:1:0.1 v:v:v). The combined eluants were dried with a gentle stream of nitrogen gas. Prior to mass spectrometric analyses the samples were reconstituted in 400 μl of chloroform/methanol/H2O (2:1:01 v:v:v) containing 30 mM piperidine.
Statistical analysis
Statistics were performed by analysing data with a two-sided Student’s t-test. Results were considered statistically significant at p ≤0.05.
Results
PtdIns-homologues can act synergistically with bv-sPLA2 to inhibit tumour cell growth
We investigated the effects of various PtdIns-homologues and of bv-sPLA2 on the proliferation of the well-characterised kidney cancer cell line A498. Cells were treated with a PtdIns-homologue (10 μM) and bv-sPLA2 (10 μg/ml) either alone or in combination. After 32 h of incubation A498 cells were pulsed with [3H] thymidine and cell proliferation was measured as [3H] thymidine incorporation. Figure 1 demonstrates that either substance alone had little effect (p > 0.05) on the proliferation of A498 cells. However, when bv-sPLA2 was present in combination with 3-phosphorylated PtdIns a potent reduction of [3H] thymidine incorporation occurred (Fig. 1). The most prominent inhibition could be detected when bv-sPLA2 was co-administered with PtdIns(3,4)P2, PtdIns(3,4,5)P3 and with PtdIns(3,4,5)P3-AA (all p < 0.05). These compounds clearly synergised with bv-sPLA2. All other compounds failed to act synergistically with bv-sPLA2 in blocking cell growth. These data pinpoint a structural hierarchy in which the 3-phosphorylation of the PtdIns appears to be critically important for the combined action with bv-sPLA2, since the three most potent substances were all 3-phosphorylated. In contrast, PtdIns lacking the phosphorylation at position 3 of the inositol head group did not inhibit in a synergistic manner. Ins(1,3,4)P3 alone exhibited the weakest activity indicating that the phosphatidyl is important as well.
Fig. 1.

Treatment of A498 kidney cancer cells with PtdIns-homologues in combination with bv-sPLA2 inhibits proliferation. Cells were left untreated (C control), were treated with bv-sPLA2 (10 μg/ml), with the indicated PtdIns-homologue (each at 10 μM) or a combination of bv-sPLA2 plus the indicated PtdIns-homologue. Light bars demonstrate the effects of single compounds on the proliferation, whereas dark bars depict the combined action of bv-sPLA2 plus the indicated PtdIns-homologue. Proliferation was determined by assessing [3H] thymidine incorporation. Shown are normalised mean values ± SEM of at least three independent experiments
Bv-sPLA2 and PtdIns(3,4)P2 abrogate signal transduction mediated by PKB/Akt
As PtdIns(3,4)P2 was the most potent in synergising with bv-sPLA2 to inhibit 3[H] thymidine incorporation, these compounds were used for the further analysis of the combinatory effects on intracellular signal transduction. 3-phosphorylated PtdIns-homologues affect subcellular organisation of signalling components of the PI3-kinase/PKB/Akt pathway that mediates cytoprotective events [6, 45]. Thus, tumour cell inhibition induced by bv-sPLA2 and PtdIns(3,4)P2 is likely to be related to the activation status of PKB/Akt. In order to test this hypothesis, A498 cells were treated with bv-sPLA2 and PtdIns(3,4)P2 for 90 min. The activation of PKB/Akt was then monitored by quantitative ELISA that determine PKB/Akt phosphorylation at serine 473. Treatment of A498 cells with bv-sPLA2 halved basal activity (p < 0.05) of the multifunctional survival signal mediator PKB/Akt, whereas PtdIns(3,4)P2 alone only moderately (p > 0.05) affected phosphorylation at serine 473 (Fig. 2). However, the combination of bv-sPLA2 and PtdIns(3,4)P2 completely abrogated basal PKB/Akt phosphorylation (p < 0.05). In the presence of the survival-promoting factor EGF, PKB/Akt signalling in A498 cells was intact as demonstrated by a 2.6-fold increase in EGF-induced serine 473 phosphorylation (p < 0.05). However, when EGF was co-administered with bv-sPLA2 PKB/Akt phosphorylation was prevented (p < 0.05). Basically similar effects were obvious in the presence of PtdIns(3,4)P2 alone which inhibited EGF-induced phosphorylation of serine 473 (p < 0.05). Activity of PKB/Akt was completely abrogated when bv-sPLA2, PtdIns(3,4)P2 and EGF were co-administered simultaneously (p < 0.05). Consequently, bv-sPLA2-PtdIns(3,4)P2 treatment resulted in a down-regulation of basal activity of PKB/Akt and completely inhibited the transduction of EGF-induced survival signals of exogenous origin.
Fig. 2.

Combined treatment of A498 kidney cancer cells with bv-sPLA2 and PtdIns(3,4)P2 inhibits phosphorylation of PKB/Akt. Cells were left untreated (C control), were treated with bv-sPLA2 (200 μg/ml), with PtdIns(3,4)P2 (200 μM) with EGF (50 ng/ml) or a combination thereof as indicated. Phosphorylation of serine 473 of PKB/Akt was normalised to total PKB/Akt levels and was quantitatively determined by specific ELISA. Shown are normalised mean values ± SEM of four independent experiments
Bv-sPLA2 and PtdIns(3,4)P2 abrogate signal transduction mediated by ERK1/2
Exogenous growth factor stimuli like EGF signals are mediated by the MAPK pathway that integrates extracellular signals to converge at the level of ERK1 and 2. Thus, the inhibitory combination of bv-sPLA2 and PtdIns(3,4)P2 possibly targets also this growth-related signal transduction pathway. We evaluated whether bv-sPLA2 and PtdIns(3,4)P2 affected the phosphorylation and activation status of ERK1/2 in A498 cells by performing ELISA that quantify phosphorylated ERK 1 at threonine 202 and tyrosine 204 and ERK2 at threonine 185 and tyrosine 187. Figure 3 indicates that bv-sPLA2 alone moderately reduced basal activation of ERK1/2 (p > 0.05), whereas single treatment with PtdIns(3,4)P2 displayed a weak-promoting effect on activation of ERK1/2 (p < 0.05). In contrast, the combined treatment of A498 cells with bv-sPLA2 and PtdIns(3,4)P2 fully abrogated basal activity of ERK1/2 (p < 0.05). Stimulation with exogenous EGF increased the extent of phosphorylation of ERK1/2 indicative of a functional, intact MAPK pathway in A498 cells. Simultaneous administration of EGF and bv-sPLA2 prevented EGF-induced ERK1/2 phosphorylation (p < 0.05). In the presence of PtdIns(3,4)P2, ERK1/2 activation still was inducible by EGF (p < 0.05). However, when EGF, bv-sPLA2 and PtdIns(3,4)P2 were co-administered simultaneously, the EGF signal could not be transduced to activate ERK1/2 (p < 0.05). In conclusion, the combined treatment with bv-sPLA2 and PtdIns(3,4)P2 blocked basal ERK1/2 activity and inhibited phosphorylation induced by exogenous EGF.
Fig. 3.

Combined treatment of A498 kidney cancer cells with bv-sPLA2 and PtdIns(3,4)P2 inhibits phosphorylation of ERK1/2. Cells were left untreated (C control), were treated with bv-sPLA2 (200 μg/ml), with PtdIns(3,4)P2 (200 μM) with EGF (50 ng/ml) or a combination thereof as indicated. Phosphorylation of ERK1/2 was normalised to total ERK1/2 levels and was quantitatively determined by specific ELISA. Shown are normalised mean values ± SEM of four independent experiments
Bv-sPLA2 and PtdIns(3,4)P2 reduce EGF-receptor cell surface expression
Epidermal growth factor has been shown to promote proliferation of A498 cells [40] and the EGF-receptor mediates autocrine growth stimulatory pathways in renal cancer [5]. The fact that PI3-kinase/PKB/Akt and MAPK signalling functions downstream of the EGFR prompted us to investigate the influence of bv-sPLA2 and PtdIns(3,4)P2 on the cell surface expression of this growth factor receptor. A498 cells were treated with bv-sPLA2 and PtdIns(3,4)P2 for 45 min and were then subjected to flow cytometric analysis. EGFR expression detected by a specific antibody in untreated cells was >90% (Fig. 4). Treatment of A498 cells with bv-sPLA2 alone did not significantly alter the number of EGFR+ cells. However, the overall mean fluorescence intensity (MFI) of EGFR staining in the presence of bv-sPLA2 was reduced from 153 MFI (control) to 73 MFI. Administration of PtdIns(3,4)P2 alone decreased the MFI to 125. The combination of bv-sPLA2 and PtdIns(3,4)P2 resulted in an MFI of 19 and reduced the total number of EGFR+ cells to 60%. This data clearly indicate a diminished EGFR cell surface expression and a reduction of EGFR+ cells. Figure 5 demonstrates that the reduction of the EGFR staining was a dose-dependent consequence of bv-sPLA2 and PtdIns(3,4)P2 treatment.
Fig. 4.

Combined treatment of A498 kidney cancer cells with bv-sPLA2 and PtdIns(3,4)P2 reduces cell surface expression of the EGFR. Cells were left untreated (C control), were treated with bv-sPLA2 (100 μg/ml), with PtdIns(3,4)P2 (100 μM) or a combination thereof as indicated. Expression of the EGFR was determined by flow cytometry. Shown is a representative of three independently performed experiments
Fig. 5.

Dose-dependent reduction of EGFR staining by bv-sPLA2 and PtdIns(3,4)P2 in A498 cells. Cells were left untreated (C control), were treated with bv-sPLA2, with PtdIns(3,4)P2 or a combination thereof as indicated. Expression of the EGFR was determined by flow cytometry [mean fluorescence intensity (MFI)]. Mean values ± SEM are presented
Bv-sPLA2 and PtdIns(3,4)P2 disrupt membrane integrity
Flow cytometry revealed the presence of two distinct cell populations even after 45 min of short-term treatment with bv-sPLA2 and PtdIns(3,4)P2 (Fig. 6, left panel) The main A498 cell population (91.4%) showed high forward/side scatter characteristics (Fig. 6, FSC/SSC-hi, blue), whereas a small proportion (8.6%) demonstrated low scattering (Fig. 6, FSC/SSC-low, orange). Incubation with bv-sPLA2 or PtdIns(3,4)P2 alone subjected A498 cells to a change in morphology as demonstrated by a shift towards the FSC/SSC-low population (Fig. 6, orange). However, when cells were treated with the combination of bv-sPLA2 and PtdIns(3,4)P2 the number of FSC/SSC-hi cells was decreased to 3.3%, whereas the FSC/SSC-low population increased to 96.7% (Fig. 6). A putative mechanism how bv-sPLA2 may act on cells is disruption of cell membranes. Thus we investigated the two distinct A498 populations for their susceptibility to staining with 7-AAD, which binds double-stranded DNA of disrupted cells but is excluded from intact cells. Treatment of A498 cells with single substances enhanced the overall number of 7-AAD+ cells from 2.6 (untreated control) to 18.9% (bv-sPLA2) and 71.2% (PtdIns(3,4)P2), respectively (Fig. 6, right panel). The combination of bv-sPLA2 with PtdIns(3,4)P2 resulted in 98.5% 7-AAD+ cells. Bv-sPLA2 administration rapidly induced intense 7-AAD staining located predominantly in the FSC/SSC-low population. PtdIns(3,4)P2 alone affected all cell populations and resulted in 7-AAD staining with lower MFI particularly in FSC/SSC-hi cells. Possibly this is due to a 7-AAD influx as a consequence of membrane disturbance induced by incorporation of PtdIns(3,4)P2 via its C16 fatty acid side chains into the lipid layer. This population (79.8% of all A498 cells) displays morphology of viable cells in the forward/side scatter diagram, whereas the FSC/SSC-low population has intense 7-AAD staining and rather reflects cells on the verge of cell death. Taken together, the combination of bv-sPLA2 and PtdIns(3,4)P2 resulted in a homogenous FCS/SSC-low population with high 7-AAD staining. Thus, basically all cells treated with the bv-sPLA2-PtdIns(3,4)P2 combination had been disrupted.
Fig. 6.
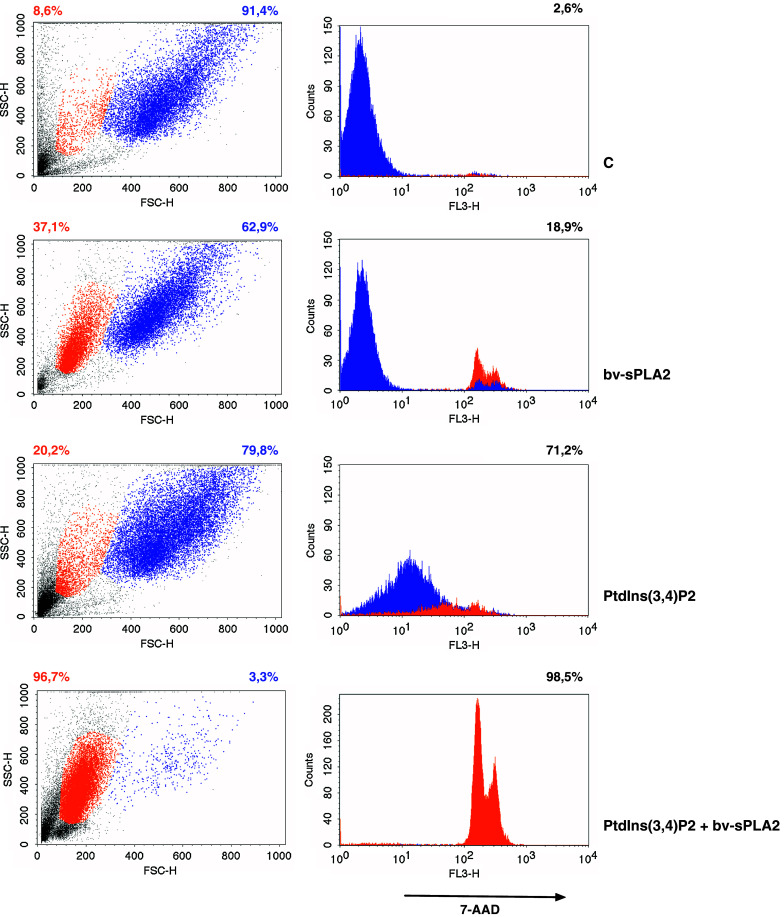
Combined treatment with PtdIns(3,4)P2 and bv-sPLA2 enhances the number of 7-AAD+ cells. Cells were left untreated (C control), were treated with bv-sPLA2 (100 μg/ml), with PtdIns(3,4)P2 (100 μM) or a combination thereof as indicated. Distribution of FSC/SSC-hi (blue) or FSC/SSC-low (orange) populations are displayed in scatters diagrams and corresponding percentages are shown in appropriate colours (left panel). Overall 7-AAD stainings are presented in histograms and percentages (right panel). 7-AAD staining of the FSC/SSC-hi population is depicted in blue, whereas the 7-AAD staining of the FSC/SSC-low population is presented in orange
Bv-sPLA2 hydrolyses PtdIns(3,4)P2 to generate lyso-PtdIns(3,4)P2
A forementioned data collectively point towards combined effects of bv-sPLA2 and PtdIns(3,4)P2 which include abrogation of signal transduction and membrane damage. One further possibility for a cooperative effect is that PtdIns(3,4)P2 serves as a substrate of bv-sPLA2 which cleaves the fatty acid from the sn-2 position to generate the corresponding lyso-PtdIns(3,4)P2 with putative cytotoxic properties. This hypothesis was tested in a set of experiments, where bv-sPLA2 (10 μg/ml) was incubated with PtdIns(3,4)P2 (10 μM) for 2 h in the absence of A498 cells. Commercially available 1,2-dipalmitoylphosphatidylinositol (C32:0 PtdIns-ISTD) was added to samples prior to extraction as an ISTD for quantification of all samples including lyso-PtdIns(3,4)P2. The reaction products were extracted and subjected to analysis by mass spectroscopy. Lipids other than PtdIns in general were monitored in this system requiring ammonium acetate. These are not the ideal conditions for observing highly phosphorylated PtdIns as they undergo in-source defragmentation giving rise to product ions including losses of fatty acid and phosphate causing artefactual ions correspondingly. Hence analysis of the bv-sPLA2 digest of the PtdIns(3,4)P2 gives rise to a prominent peak corresponding to a monoacyl-PtdIns at m/z 571. Supporting evidence of this is shown by the addition of piperidine to the cell-free lysis mixture prior to ESI-MS/MS analysis. Piperidine is reported to stabilise the phosphate groups minimising the in-source fragmentation [46]. The mass spectrometry analysis of the PtdIns(3,4)P2 (denoted C32:0 PtdInsP2) by the InsP head group specific ion scan PS 241, clearly shows this (Fig. 8a, b) and to a lesser extent in PS 321 and PS 401 scans (data not shown), albeit with severely diminished ionisation responses and an ever present in-source fragmentation series of product ions. The lyso product is clearly observed in Fig. 8b at m/z 731.3 supporting the original premise that PtdIns(3,4)P2 is a substrate for bv-sPLA2 in the original A498 tumour cell lysis experiments (Table 1). A small background of m/z 571 is also present in the ISTD + media only (Fig. 7a) due to a minor in-source fragmentation of the ITSD itself. This small amount of the m/z 571 is due to the same palmitoyl moiety and InsP head group and hence requires subtraction from other analyses. Future ISTD choices when available will avoid this complication. In the cell lysis experiments summarised in Table 1, lyso-PtdIns(3,4)P2 was increased from 0.08 to 0.98 μM, a 12-fold increase, when bv-sPLA2 and PtdIns(3,4)P2 were incubated together with cells. However, the administration of bv-sPLA2 or PtdIns(3,4)P2 alone did not increase lyso-PtdIns(3,4)P2 levels significantly above background (0.2 μM). Taken together these data suggest that bv-sPLA2 hydrolyses PtdIns(3,4)P2 to generate lyso-PtdIns(3,4)P2. This hydrolysis occurs in the presence and absence of cells but is somewhat reduced in the presence of A498 cells. Most likely this is due to other cellular enzymes (PtdIns-Phospholipase C and/or Phospholipase D) competing with the bv-sPLA2 for the PtdIns(3,4)P2 as a substrate and giving other products.
Fig. 8.
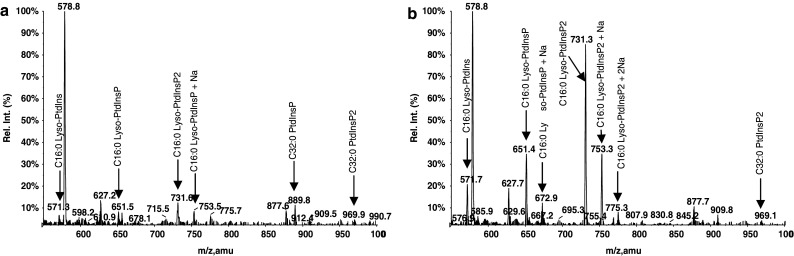
Precursor ion scan 241 of bv-sPLA2 products of PtdIns(3,4)P2 in a cell free system to detect lyso-PtdIns(3,4)P2 by attenuated in source fragmentation using piperidine. a Cell culture medium containing PtdIns(3,4)P2 only, b cell culture medium containing a mixture of bv-sPLA2 (10 μg/ml) and PtdIns(3,4)P2 (10 μM). Lyso-PtdIns(3,4)P2 observable at m/z 731, was detected by the precursor ion scans 241 with the corresponding loss of InsP head group. The lyso-PtdIns observable at m/z 571, was produced from an in-source fragmentation of the PtdIns(3,4)P2 and also from the C32:0 PtdIns used as ISTD (C32:0 PtdIns-ISTD). Other in source fragments are also observable, e.g. product ions seen at m/z 651 and 673 and products thereof are from in-source fragmentation of (M − H)- parent ion m/z 969 and at (M −0H + Na)- m/z 991, respectively
Table 1.
Generation of lyso-PtdIns(3,4)P2
| Treatment | Cell-free lyso-PtdIns(3,4)P2 (μM) | A498 cells lyso-PtdIns(3,4)P2 (μM) |
|---|---|---|
| Control (medium only) | 0.43 | - |
| Control (cells + medium) | - | 0.08 |
| bv-sPLA2 | 0.50 | 0.1 |
| PtdIns(3,4)P2 | 0.11 | 0.2 |
| bv-sPLA2 + PtdIns(3,4)P2 | 2.06 | 0.98 |
Quantification of lyso-PtdIns(3,4)P2 (m/z 571) was based on comparing intensities of the lyso species with known concentration of the ISTD using a precursor ion scan 241. Relative response factors were not considered in these calculations
Fig. 7.
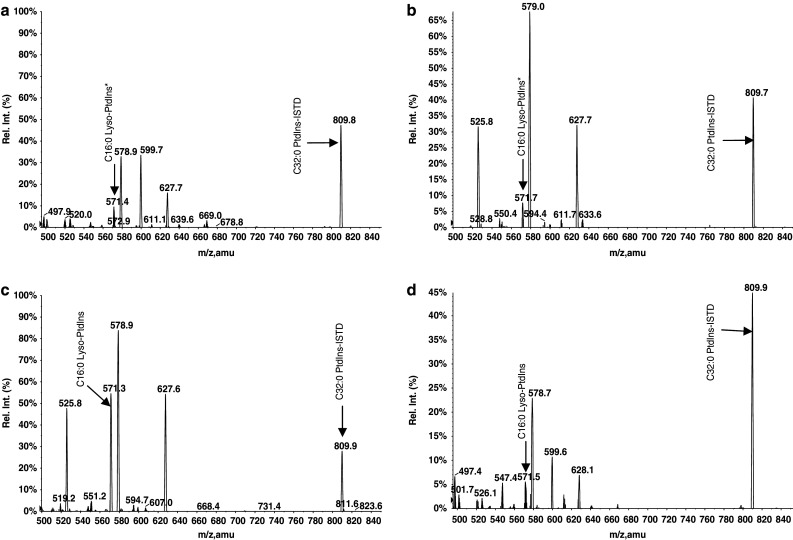
Precursor ion scans of 241 showing bv-sPLA2 products of the PtdIns(3,4)P2 substrate in a cell-free system. a Cell culture medium as a control containing, b bv-sPLA2 (10 μg/ml), c a mixture of bv-sPLA2 (10 μg/ml) and PtdIns(3,4)P2 (10 μM) and d PtdIns(3,4)P2 only. The lyso-PtdIns(3,4)P2 product of PtdIns(3,4)P2 is observable at m/z 571, an in-source bis-dephosphorylated fragment, in the presence of 10 mM ammonium acetate. The precursor ion scan 241 in negative ion mode corresponds to the loss of the InsP head group. The asterisk indicates the lyso-PtdIns derived from an in-source fragmentation of the C32:0 PtdIns used as internal standard (ISTD)
Discussion
In the present study we analysed cytotoxic actions of bv-sPLA2 in combination with PtdIns-homologues. We show, that 3-phosphorylated PtdIns, i.e. a phosphate group esterified to position 3 of the inositol head group, and in particular PtdIns(3,4)P2 were the most effective in synergising with bv-sPLA2 to inhibit the growth of renal cancer cells (Fig. 1). PtdIns or its homologues have fatty acids attached to positions sn-1 and sn-2. In our studies, 3-phosphorylated PtdIns with the saturated fatty acid palmitic acid (C16) were the most potent. However, increased length of the fatty acids did not seem to correlate with an increase in toxicity. If one acyl group was a polyunsaturated fatty acid such as arachidonic acid (C20), the ability to synergise with bv-sPLA was slightly reduced. Of note, PtdIns(3,4)P2 and PtdIns(4,5)P2 strongly varied in their inhibitory potential. These compounds differed only in the phosphorylation of the inositol head group suggesting, that the observed growth inhibition is not restricted to a cytotoxic action of the C16 acyl groups, which were present in both compounds. Figure 1 depicts the hierarchy of the structural requirements of PtdIns-homologues with regard to the ability to synergise with bv-sPLA2. Taken together, administration of bv-sPLA2 in combination with 3-phosphorylated PtdIns-homologues efficiently blocked proliferation of renal carcinoma cells. In a previous work we have shown that bv-sPLA2 in combination with 3-phosphorylated PtdIns-homologues can also inhibit the proliferation of cancer cells from prostate, breast and lung tissue [35].
We further provide evidence that the potent reduction of tumour cell survival was associated with a complete down-regulation of PKB/Akt phosphorylation (Fig. 2). The PI3-kinase/PKB/Akt pathway represents a central survival-related signal transduction pathway. In various studies it has been demonstrated that PKB/Akt activation enhances cell survival and promotes growth and invasiveness of tumours [6, 7]. PKB/Akt is expressed in A498 cells and it has been shown previously that phosphorylation at serine 473 correlated well with PKB/Akt kinase activity in renal cell carcinoma cells [2]. In this study, we demonstrate that bv-sPLA2 prevents activation of PKB/Akt in A498 cells. This result was surprising, since other PLA2 isoenzymes like groups IB and IIA have been shown to promote activation of PKB/Akt in other cell types [4, 31]. Parts of these effects were transduced by cell surface sPLA2 receptors, whereas bv-sPA2-mediated PKB/Akt inhibition in our exprimental setup can be attributed to the catalytic activity of bv-sPLA2 [35]. PtdIns(3,4)P2 alone reduced EGF-induced PKB/Akt phosphorylation but was less effective than bv-sPLA2 in abrogating activity of PKB/Akt. 3-Phosphorylated PtdIns-homologues are products of the PI3-kinase that can act as membrane anchor or modulator of PKB/Akt activity [6]. The inhibitory effect of PtdIns(3,4)P2 in our study is not in line with studies which demonstrated that PtdIns(3,4)P2 can activate PKB/Akt [14, 24]. However, the PI3-kinase/PKB/Akt pathway has been reported to be serum regulated and over activation of PI3-kinase in the absence of serum results in apoptosis [23, 45]. This suggests, that PtdIns(3,4)P2 stimulation in the absence of serum could provoke the induction of cell death. Indeed, PKB/Akt signalling in A498 was serum dependent, since addition of FCS induced PKB/Akt phosphorylation (data not shown). We did not further investigate the action of PtdIns(3,4)P2 in the presence of serum because under such conditions no inhibition of tumour cell survival would be achieved together with bv-sPLA2 [35]. The combined administration of bv-sPLA2 and PtdIns(3,4)P2 completely prevented the activation of PKB/Akt. This treatment also affected the MAPK signalling mediated by ERK1 and 2 [8, 25]. Activation of ERKs has been reported to be essential for cell growth [30]. In A498 cells ERKs are constitutively activated [17] and this pathway is a potential target for the treatment of renal cell carcinoma [32, 42]. In general, the action of sPLA2 isoenzymes on the MAPK pathway is not clear yet. In rat renal mesangial cells naja-sPLA2 group II stimulated a signalling cascade that involved the activation of ERK2 [18]. Effects of other sPLA2 isoenzymes on MAPK activation seem to be rather stimulatory and were transduced by the corresponding cell surface receptors [16, 22, 27]. Here we demonstrate a clear inhibition by bv-sPLA2 that affected basal ERK1/2 activity and fully prevented EGF-induced ERK1/2 phosphorylation (Fig. 3). This inhibition was further potentiated by PtdIns(3,4)P2 which even stimulated basal ERK1/2 activity on its own.
As EGF-induced signals were abrogated in the presence of bv-sPLA2 and PtdIns(3,4)P2 we considered the possibility that the EGFR which acts upstream of the PI3-kinase and MAPK-pathways is targeted by this treatment. Indeed we found a reduced cell surface staining of the EGFR when cells were treated with bv-sPLA2 as demonstrated by a diminished MFI in flow cytometric analysis (Figs. 4, 5). Effects of PtdIns(3,4)P2 on EGFR expression were less pronounced, but it clearly supported bv-sPLA2 when administered together. Under such conditions, the overall number of EGFR+ cells, as well as the corresponding MFI were reduced which indicates diminished EGFR cell surface expression. The disruption of membrane integrity by bv-sPLA2 and PtdIns(3,4)P2 (Fig. 6) obviously interfered with the localisation of the transmembraneous EGFR. Although it remains unclear whether the EGFR was internalised or whether its function was impaired, EGF-induced signalling was unequivocally prevented in the presence of bv-sPLA2 and PtdIns(3,4)P2.
In addition, the observed induction of a 7-AAD+ population with low scatter characteristics (Fig. 6) after administration of bv-sPLA2-PtdIns(3,4)P2 provides evidence for altered cell morphology. The massive staining with 7-AAD provides indirect evidence for increased membrane permeability of this treatment and suggests that disruption of membrane integrity is a crucial step in bv-sPLA2-PtdIns(3,4)P2 action. Thus, data presented here reveal a mode of combined bv-sPLA2-PtdIns(3,4)P2 action on renal cancer cells that targets cell membranes as well as associated signal transduction modules. Furthermore, our data suggest that lyso-PtdIns(3,4)P2 which is generated by bv-sPLA2-mediated hydrolysis of PtdIns(3,4)P2 (Figs. 7, 8; Table 1) may be involved in the observed cytotoxicity. Of note, structurally resembling anti-cancer drugs like alkyl-lysophospholipids have been reported to induce rapid internalisation but no phosphorylation of the EGFR, which is in line with our results [36]. In addition, such synthetic lysolipids affected survival and apoptotic pathways. For future studies, it will be interesting to delineate putative anti-tumour characteristics of lyso-PtdIns(3,4)P2. Presently, this compound is commercially not available and needs to be synthesised.
In conclusion, we demonstrate that bv-sPLA2 acts in combination with 3-phosphorylated PtdIns to inhibit proliferation and survival of renal cancer cells. Of note, at least the effects of bv-sPLA2 and PtdIns(3,4)P2 are not simply specific in regard to the growth of A498 cells but have been shown previously to affect also cancer cells from various origins [35]. The observed cytotoxic effect in renal cancer cells is mediated by membrane disruption that interferes with the expression of growth modulatory cell surface proteins and prevents the activation of key regulatory signal transduction modules. This further suggests that bv-sPLA2 and 3-phosphorylated PtdIns display defined inhibitory effects on renal cancer cells which may further support the previously suggested application in cancer immunotherapy [35].
Footnotes
This work was supported by a grant to MT of the kompetenzzentrum medizin tirol (kmt), a centre of excellence.
References
- 1.Andresen TL, Jensen SS, Madsen R, Jorgensen K. Synthesis and biological activity of anticancer ether lipids that are specifically released by phospholipase A2 in tumor tissue. J Med Chem. 2005;48:7305–7314. doi: 10.1021/jm049006f. [DOI] [PubMed] [Google Scholar]
- 2.Asakuma J, Sumitomo M, Asano T, Hayakawa M. Selective Akt inactivation and tumor necrosis actor-related apoptosis-inducing ligand sensitization of renal cancer cells by low concentrations of paclitaxel. Cancer Res. 2003;63:1365–1370. [PubMed] [Google Scholar]
- 3.Ashagbley A, Samadder P, Bittman R, Erukulla RK, Byun HS, Arthur G. Synthesis of ether-linked analogues of lysophosphatidate and their effect on the proliferation of human epithelial cancer cells in vitro. Anticancer Res. 1996;16:1813–1818. [PubMed] [Google Scholar]
- 4.Choi YA, Lim HK, Kim JR, Lee CH, Kim YJ, Kang SS, Baek SH. Group IB secretory phospholipase A2 promotes matrix metalloproteinase-2-mediated cell migration via the phosphatidylinositol 3-kinase and Akt pathway. J Biol Chem. 2004;279:36579–36585. doi: 10.1074/jbc.M314235200. [DOI] [PubMed] [Google Scholar]
- 5.Ciardiello F, Caputo R, Bianco R, Damiano V, Pomatico G, Pepe S, Bianco AR, Agrawal S, Mendelsohn J, Tortora G. Cooperative inhibition of renal cancer growth by anti-epidermal growth factor receptor antibody and protein kinase A antisense oligonucleotide. J Natl Cancer Inst. 1998;90:1087–1094. doi: 10.1093/jnci/90.14.1087. [DOI] [PubMed] [Google Scholar]
- 6.Coffer PJ, Jin J, Woodgett JR. Protein kinase B (c-Akt): a multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem J. 1998;335(Pt 1):1–13. doi: 10.1042/bj3350001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 8.Davis RJ. The mitogen-activated protein kinase signal transduction pathway. J Biol Chem. 1993;268:14553–14556. [PubMed] [Google Scholar]
- 9.Dennis EA. Diversity of group types, regulation, and function of phospholipase A2. J Biol Chem. 1994;269:13057–13060. [PubMed] [Google Scholar]
- 10.Fogh J, Fogh JM, Orfeo T. One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J Natl Cancer Inst. 1977;59:221–226. doi: 10.1093/jnci/59.1.221. [DOI] [PubMed] [Google Scholar]
- 11.Folch J, Lees M, Sloane Stanley G. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226(1):497–509. [PubMed] [Google Scholar]
- 12.Fonteh AN, Atsumi G, LaPorte T, Chilton FH. Secretory phospholipase A2 receptor-mediated activation of cytosolic phospholipase A2 in murine bone marrow-derived mast cells. J Immunol. 2000;165:2773–2782. doi: 10.4049/jimmunol.165.5.2773. [DOI] [PubMed] [Google Scholar]
- 13.Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/S0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 14.Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 15.Graler MH, Goetzl EJ. Lysophospholipids and their G protein-coupled receptors in inflammation and immunity. Biochim Biophys Acta. 2002;1582:168–174. doi: 10.1016/s1388-1981(02)00152-x. [DOI] [PubMed] [Google Scholar]
- 16.Hernandez M, Burillo SL, Crespo MS, Nieto ML. Secretory phospholipase A2 activates the cascade of mitogen-activated protein kinases and cytosolic phospholipase A2 in the human astrocytoma cell line 1321N1. J Biol Chem. 1998;273:606–612. doi: 10.1074/jbc.273.1.606. [DOI] [PubMed] [Google Scholar]
- 17.Horiguchi A, Oya M, Marumo K, Murai M. STAT3, but not ERKs, mediates the IL-6-induced proliferation of renal cancer cells, ACHN and 769P. Kidney Int. 2002;61:926–938. doi: 10.1046/j.1523-1755.2002.00206.x. [DOI] [PubMed] [Google Scholar]
- 18.Huwiler A, Staudt G, Kramer RM, Pfeilschifter J. Cross-talk between secretory phospholipase A2 and cytosolic phospholipase A2 in rat renal mesangial cells. Biochim Biophys Acta. 1997;1348:257–272. doi: 10.1016/s0005-2760(97)00073-8. [DOI] [PubMed] [Google Scholar]
- 19.Kabarowski JH, Xu Y, Witte ON. Lysophosphatidylcholine as a ligand for immunoregulation. Biochem Pharmacol. 2002;64:161–167. doi: 10.1016/S0006-2952(02)01179-6. [DOI] [PubMed] [Google Scholar]
- 20.Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615–675. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- 21.Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, Hay N. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997;11:701–713. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- 22.Kinoshita E, Handa N, Hanada K, Kajiyama G, Sugiyama M. Activation of MAP kinase cascade induced by human pancreatic phospholipase A2 in a human pancreatic cancer cell line. FEBS Lett. 1997;407:343–346. doi: 10.1016/S0014-5793(97)00373-6. [DOI] [PubMed] [Google Scholar]
- 23.Klippel A, Escobedo MA, Wachowicz MS, Apell G, Brown TW, Giedlin MA, Kavanaugh WM, Williams LT. Activation of phosphatidylinositol 3-kinase is sufficient for cell cycle entry and promotes cellular changes characteristic of oncogenic transformation. Mol Cell Biol. 1998;18:5699–5711. doi: 10.1128/mcb.18.10.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klippel A, Kavanaugh WM, Pot D, Williams LT. A specific product of phosphatidylinositol 3-kinase directly activates the protein kinase Akt through its pleckstrin homology domain. Mol Cell Biol. 1997;17:338–344. doi: 10.1128/mcb.17.1.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- 26.Kudo I, Murakami M. Phospholipase A2 enzymes. Prostaglandins Other Lipid Mediat. 2002;68-9:3–58. doi: 10.1016/S0090-6980(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 27.Kundu GC, Mukherjee AB. Evidence that porcine pancreatic phospholipase A2 via its high affinity receptor stimulates extracellular matrix invasion by normal and cancer cells. J Biol Chem. 1997;272:2346–2353. doi: 10.1074/jbc.272.47.29468. [DOI] [PubMed] [Google Scholar]
- 28.Lambeau G, Lazdunski M. Receptors for a growing family of secreted phospholipases A2. Trends Pharmacol Sci. 1999;20:162–170. doi: 10.1016/S0165-6147(99)01300-0. [DOI] [PubMed] [Google Scholar]
- 29.Lambeau G, Schmid-Alliana A, Lazdunski M, Barhanin J. Identification and purification of a very high affinity binding protein for toxic phospholipases A2 in skeletal muscle. J Biol Chem. 1990;265:9526–9532. [PubMed] [Google Scholar]
- 30.Pages G, Lenormand P, L’Allemain G, Chambard JC, Meloche S, Pouyssegur J. Mitogen-activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. Proc Natl Acad Sci USA. 1993;90:8319–8323. doi: 10.1073/pnas.90.18.8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park DW, Kim JR, Kim SY, Sonn JK, Bang OS, Kang SS, Kim JH, Baek SH. Akt as a mediator of secretory phospholipase A2 receptor-involved inducible nitric oxide synthase expression. J Immunol. 2003;170:2093–2099. doi: 10.4049/jimmunol.170.4.2093. [DOI] [PubMed] [Google Scholar]
- 32.Patel PH, Chaganti RS, Motzer RJ. Targeted therapy for metastatic renal cell carcinoma. Br J Cancer. 2006;94:614–619. doi: 10.1038/sj.bjc.6602978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Payrastre B, Missy K, Giuriato S, Bodin S, Plantavid M, Gratacap M. Phosphoinositides: key players in cell signalling, in time and space. Cell Signal. 2001;13:377–387. doi: 10.1016/S0898-6568(01)00158-9. [DOI] [PubMed] [Google Scholar]
- 34.Pouyssegur J, Volmat V, Lenormand P. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Biochem Pharmacol. 2002;64:755–763. doi: 10.1016/S0006-2952(02)01135-8. [DOI] [PubMed] [Google Scholar]
- 35.Putz T, Ramoner R, Gander H, Rahm A, Bartsch G, Thurnher M. Antitumor action and immune activation through cooperation of bee venom secretory phospholipase A2 and phosphatidylinositol-(3,4)-bisphosphate. Cancer Immunol Immunother. 2006;55:1347–1388. doi: 10.1007/s00262-006-0143-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruiter GA, Verheij M, Zerp SF, Moolenaar WH, Van Blitterswijk WJ. Submicromolar doses of alkyl-lysophospholipids induce rapid internalization, but not activation, of epidermal growth factor receptor and concomitant MAPK/ERK activation in A431 cells. Int J Cancer. 2002;102:343–350. doi: 10.1002/ijc.10741. [DOI] [PubMed] [Google Scholar]
- 37.Ruiter GA, Verheij M, Zerp SF, van Blitterswijk WJ. Alkyl-lysophospholipids as anticancer agents and enhancers of radiation-induced apoptosis. Int J Radiat Oncol Biol Phys. 2001;49:415–419. doi: 10.1016/S0360-3016(00)01476-0. [DOI] [PubMed] [Google Scholar]
- 38.Ruiter GA, Zerp SF, Bartelink H, van Blitterswijk WJ, Verheij M. Anti-cancer alkyl-lysophospholipids inhibit the phosphatidylinositol 3-kinase-Akt/PKB survival pathway. Anticancer Drugs. 2003;14:167–173. doi: 10.1097/00001813-200302000-00011. [DOI] [PubMed] [Google Scholar]
- 39.Samadder P, Bittman R, Byun HS, Arthur G. Synthesis and use of novel ether phospholipid enantiomers to probe the molecular basis of the antitumor effects of alkyllysophospholipids: correlation of differential activation of c-Jun NH(2)-terminal protein kinase with antiproliferative effects in neuronal tumor cells. J Med Chem. 2004;47:2710–2713. doi: 10.1021/jm0302748. [DOI] [PubMed] [Google Scholar]
- 40.Sion-Vardy N, Vardy D, Rodeck U, Kari C, Levin RM, Malkowicz SB. Antiproliferative effects of tyrosine kinase inhibitors (tyrphostins) on human bladder and renal carcinoma cells. J Surg Res. 1995;59:675–680. doi: 10.1006/jsre.1995.1222. [DOI] [PubMed] [Google Scholar]
- 41.Six DA, Dennis EA. The expanding superfamily of phospholipase A(2) enzymes: classification and characterization. Biochim Biophys Acta. 2000;1488:1–19. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- 42.Staehler M, Rohrmann K, Haseke N, Stief CG, Siebels M. Targeted agents for the treatment of advanced renal cell carcinoma. Curr Drug Targets. 2005;6:835–846. doi: 10.2174/138945005774574498. [DOI] [PubMed] [Google Scholar]
- 43.Toker A. Phosphoinositides and signal transduction. Cell Mol Life Sci. 2002;59:761–779. doi: 10.1007/s00018-002-8465-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valentin E, Lambeau G. What can venom phospholipases A(2) tell us about the functional diversity of mammalian secreted phospholipases A(2)? Biochimie. 2000;82:815–831. doi: 10.1016/S0300-9084(00)01168-8. [DOI] [PubMed] [Google Scholar]
- 45.Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 46.Wenk MR, Lucast L, Di Paolo G, Romanelli AJ, Suchy SF, Nussbaum RL, Cline GW, Shulman GI, McMurray W, De Camilli P. Phosphoinositide profiling in complex lipid mixtures using electrospray ionization mass spectrometry. Nat Biotechnol. 2003;21:813–817. doi: 10.1038/nbt837. [DOI] [PubMed] [Google Scholar]