

Abstract
Survivin is a member of the inhibitor of apoptosis protein family. Gliomas and many other tumors express survivin at high levels; whereas, normal fully differentiated cells generally do not. Therefore, survivin represents a tumor-specific target for cancer vaccine therapy. It has been shown that it is possible to produce a MHC-I-restricted cellular immunologic response to survivin vaccines. To study differences in immunogenicity between murine and human survivin proteins, we vaccinated C57BL/6 mice with bone marrow dendritic cells (BMDC) transfected with expression vectors containing the murine and human survivin genes. Mice vaccinated with BMDCs expressing a truncated human survivin protein developed cytotoxic T lymphocyte to subcutaneous GL261 glioma cells and exhibited prolonged tumor-free survival compared to mice vaccinated with BMDCs transfected with vector alone (P<0.01). While mice challenged with intracerebral GL261 cells had increased survival, no cures were observed. In contrast, vaccinated mice that fully resisted subcutaneous tumor challenge were rendered resistant to intracerebral GL261 re-challenge. BMDCs transfected with the full-length human survivin molecule were significantly more effective at prolonging survival than BMDCs expressing the full-length murine survivin gene (P=0.0175). Therefore, xenogeneic differences between human and murine sequences might be exploited to develop more immunogenic tumor vaccines.
Keywords: Dendritic cell, Glioma, Survivin, Tumor antigen, Vaccine
Introduction
There is a continuing search for tumor-associated antigens (TAA) that are effective targets for immunotherapy. Among these TAA are mutant forms of common cellular antigens. A number of antitumor vaccine strategies have used cell-surface target molecules such as EGFRvIII [1–3]. Tumor-specific cell surface antigens have been identified, which are accessible to both antibody-mediated and cellular immune attack. Many of these are weakly immunogenic differentiation antigens. A number of these antigens, which were originally described in melanomas, are present in malignant gliomas as well [4]. Epitopes of intracellular proteins also appear on the surface of tumor cells in association with MHC I molecules. If correctly presented by tumor cells and recognizable by specific effector T cells, such intracellular molecules can elicit cytotoxic antitumor responses as well.
Although commonly expressed during fetal development, survivin is one TAA that is rarely detectable in the normal tissues of adult organisms [5]. Malignant gliomas express the intracellular protein survivin at high levels; whereas, low grade gliomas and normal glial cells do not [6–8]. In addition to malignant gliomas, survivin is expressed at high levels in many different tumor types. High-level survivin expression is associated with a poor prognosis [8, 9], a high rate of tumor recurrence, resistance to therapy and a reduced apoptotic index in vivo [10]. Consequently, survivin appears to confer growth and survival advantages to tumor cells that express it. Its tumor specificity makes it an attractive target for immunological based cancer therapy. Also, because survivin is expressed by many different tumor types, its use as a vaccine target has broad therapeutic potential. In fact, survivin has been identified as one of the most specific cancer genes [11].
While peptide vaccines may enter the MHC class I pathway by way of cross-presentation, antigens that are synthesized intracellularly and processed by the proteosome can directly enter this pathway to evoke a cellular antitumor immune response. Peptide epitopes complex with MHC I molecules and are presented at the cell surface leading to a CD8+ T cell-mediated immune response. DNA vaccines can encode antigens that directly enter the MHC I presentation pathway following intracellular synthesis. They may stimulate more robust immune responses than peptide vaccines because the former can present many more antigenic epitopes. Expression of these vectors in dendritic cells ex vivo can provide an effective delivery vehicle for DNA vaccines.
Self proteins like survivin may not stimulate potent antitumor immune responses due to central immunologic tolerance. However, small variations in protein sequence that may exist between homologous proteins of different species can break tolerance to the native antigen. Certain peptides derived from the human melanoma gp100 protein have been shown to act as molecular mimics, binding to murine H-2Db better than the native murine sequence, leading to a more potent cellular antitumor immune response [12]. To study the immunogenicity of a xenogeneic survivin protein, we explored an immunization strategy using the human survivin gene expressed in H2-Kb positive DC to induce resistance of C57BL/6 mice to subcutaneous and intracerebral challenge with GL261 glioma cells.
Materials and methods
Cell lines and culture conditions
GL261 murine glioma cells were grown on 100-mm tissue culture plates in complete Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal calf serum, 5,000 U penicillin/streptomycin, 50 μM 2-mercaptoethanol, 25 mM HEPES, and 1× non-essential amino acids at 37°C in 5% CO2 with media changes two to three times per week.
Western blotting
GL261 cells and normal mouse brain tissue were used for western blotting assays. Assays were preformed as previously described with modifications via chemiluminscence utilizing an Amersham ECL Advance system (Amersham) with semi-dry transfer. Detection was performed with an anti-survivin (FL-172) antibody (Santa Cruz Biotechnologies, Santa Cruz, CA, USA).
Construction of vaccine vectors
Oligodeoxyribonucleotide primers homologous to the coding region ends of the human and murine survivin genes were used in reverse transcription polymerase chain reaction (RT-PCR) to create full-length survivin cDNAs. Fragments I through V of the human survivin molecule (Fig. 1) were also created by RT-PCR with incorporation of an in-frame ATG translation start signal in each vector construct. Primers also contained synthetic NotI (upstream primers) and XhoI (downstream primers) restriction sites for subsequent cloning. Polyadenylated RNA from a human glioblastoma cell line was used as the template for RT-PCR. All PCR products were introduced into the pCR2.1-Topo-TA vector (Invitrogen) and automated sequencing was used to confirm the nucleotide sequence of each cDNA fragment. Each fragment was then cloned into the mammalian expression vector pVAX1 (Invitrogen). The pVAX1 parent vector was designed for use in the development of DNA vaccines. Full-length human survivin cDNA was digested with MbiI to remove the 5′-end of the molecule encoding amino acids 1 through 37. This truncated human survivin gene was introduced into pVAX1 creating pVAX.hSVN(II–V). Amino acid residue 38 of the human survivin protein is methionine, which provides a translation initiation signal for expression of the truncated human survivin gene.
Fig. 1.
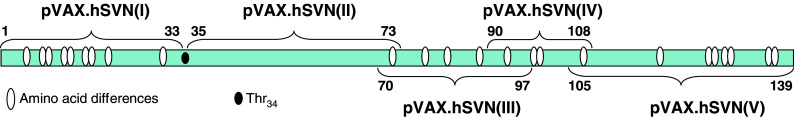
Structure of the survivin gene. Sites of sequence divergence between human and murine survivin molecules are indicated by white ovals. The active phosphorylation site of survivin (Thr34) is indicated by the black oval. Survivin cDNA fragments I through V were inserted into the vector pVAX1 for expression in dendritic cells
Dendritic cell isolation, transfection and vaccination
Dendritic cells were harvested from bone marrow as described [13, 14]. Briefly, bone marrow was harvested from the long bones of euthanized 6-week-old C57BL/6 mice and cells were cultured at a density of 2×106 cells/ml in RPMI containing 10% fetal calf serum, 5,000 U penicillin/streptomycin, 50 μM 2-mercaptoethanol, 25 mM HEPES, 1× non-essential amino acids and 20 ng/ml GM-CSF (PeproTech, Rocky Hill, NJ, USA). After 48 h, non-adherent cells were removed and medium containing GM-CSF was replenished. On day 4, an additional volume of fresh medium containing GM-CSF (20 ng/ml) was added to the culture dishes. After 6 days, non-adherent dendritic cells were harvested for transfection. jetPEI (Q-BIOgene) a linear polyethylenimine, HO–(CH2)2–(CH2–CH2–NH)n–CH2)2–OH was used as a DNA carrier for transfection. We found a nitrogen-to-phosphate ratio of 5:1 to be optimal for DC transfection. The pVAX1 DNA vector (25 μg) was diluted in 1,250 μl of saline and combined with 50 μl of 7.5 mM jetPEI stock solution diluted with 1,250 μl of 0.9% NaCl. The mixture was vortexed and then incubated at 27°C for 15 min. DCs were re-plated in six-well plates at a density of 1.5×106 cells per well in 5 ml of medium containing GM-CSF. A volume of 500 μl of the DNA-jetPEI complex solution was added to each well and incubated overnight. Transfected DCs were then harvested and washed in PBS prior to injection into mice. Flow cytometry was performed on DCs to assess CD11c, CD40, CD86, IA/IE, H2-Kb markers. Delivery of jetPEI complexes into DC was >90% as judged by transfection with FITC-labeled jetPEI and observation by both confocal microscopy and FACS. Vaccinations consisted of three subcutaneous injections of 5×105 DCs, each spaced 1 week apart. All vaccinations were administered into the right flank.
Splenocyte and T cell isolation
Splenic tissue of vaccinated C57BL/6 mice from indicated experimental groups was harvested at endpoint sacrifice. Cells were teased from mouse spleens with sterile forceps and passed through a 70 μm filter. (BD Falcon). Splenocytes were cultured in DMEM containing 10% fetal bovine serum, IL-2 (10 U/ml) and IL-7 (1 ng/ml). Medium was replenished on days 2 and 7. To isolate T cells, splenocytes were centrifuged for 5 min and the pellet was re-suspended in RBC lysis buffer (R&D Systems). Cells were washed twice and re-suspended in complete RPMI culture medium. CD3+ T cells were isolated by negative selection using T cell enrichment columns (R&D Systems) according to the manufacturer’s protocol. Effluent cells were collected and purity was measured to be greater than 90% CD3+ by flow cytometry.
Adoptive transfer and T cell depletion
Naïve C57BL/6 mice underwent adoptive transfer with 106 CD3+ T cells obtained from vaccinated mouse spleens 24 h prior to tumor implantation. CD3+ T lymphocytes were administered in a volume of 1 ml of PBS by i.p. injection. Mice were then implanted intracranially with 5×104 GL261 cells and followed for neurological deficits indicative of tumor growth as described below. Vaccinated mice that had previously rejected both subcutaneous and intracranial tumors were used for CD8+ T cell depletion studies. Mice received i.p. injections of 100 μg of anti-CD8 (2.43) monoclonal antibody in 200 μl PBS 2 days prior to tumor implantation, 3 days post-implantation and every 7 days thereafter, achieving selective depletion (>90%) of CD8+ T cell subsets. These mice were implanted subcutaneously with 1.5×105 GL261 cells into flank opposite that used for the previous tumor implantation.
Tumor cell lysis by cytotoxic T lymphocyte (CTL)
Cellular assays for specific T cell lysis of target were performed using the DELFIA EuTDA cytotoxicity method [15]. GL261 cells were trypsinized and suspended at 1×106 cells/ml followed by addition of 5 μl BATDA label to each 4 ml of cells for 20 min at 37°C. After labeling, cells were centrifuged, washed in PBS, and resuspended at 5×104 cells/ml of DMEM. Next, 5×103 GL261 BATDA-labeled target cells in 100 μl of medium were plated into each well of 96 well plates. Splenocytes from vaccinated animals were added to the target cells in ratios ranging from 1:25 to 1:100 for 2 h at 37°C. Conditions were also set up to assay background levels, spontaneous release and maximal lysis. Plates were centrifuged and 20 μl of supernatant containing released BATDA was transferred to a flat-bottom plate, to which 200 μl of europium solution was added and incubated for 15 min at 27°C. Plates were analyzed on a time-resolved DELFIA fluorometer (Perkin–Elmer). Percentage cytotoxicity was calculated using the formula: (release in assay − spontaneous release)/(maximum release − spontaneous release) × 100. Maximum release was determined by the lysis of 5×103 labeled target cells in triplicate wells with DELFIA lysis buffer (Perkin–Elmer). Spontaneous release was measured by incubating 5×103 target cells in triplicate wells in the absence of effector cells. Results are reported as mean values of triplicate wells.
Tumor challenge
For subcutaneous (s.c.) tumors, a suspension of 1.5×105 GL261 cells in 100 μl DMEM was injected into the shaved left flank of male C57BL/6 mice (Harlan-Sprague-Dawley, Indianapolis, IN, USA). The first appearance of tumor (tumor-free survival) was detected by direct palpation of a 2–3 mm mass. Tumor growth was measured with calipers, and volumes were calculated according to the formula V=(a 2×b 2×c 2)/6, where V represents tumor volume and a, b, and c are perpendicular diameters of the tumor. Statistical significance was assessed using the student’s paired t-test method. For intracerebral (i.c.) tumor challenge, male C57BL/6 mice were anesthetized with an intraperitoneal (i.p.) injection of ketamine (80 mg/kg) and xylazine (20 mg/kg) and fixed in a stereotactic head frame (David Kopf Instruments, Tujunga, CA, USA). A midline scalp incision was made and the bregma was identified. Stereotactic coordinates were measured (2.0 mm lateral to the bregma) for implantation of cells into the deep frontal white matter. A burr hole was drilled at this locus and a 25-gauge needle was passed using a micrometer to a depth of 3.0 mm relative to the dura mater. A suspension of 5×104 GL261 cells in 5 μl of DMEM was injected via a Hamilton syringe at a rate of 2.5 μl/min and the needle was then withdrawn at a rate of 1 mm/min and the incision was sutured. Kaplan–Meier survival plots were prepared and median survival times were determined for all groups. Survival differences were assessed using the logrank Mantel–Cox method.
Detection of CD4+ and CD8+ cells in GL261 tumors
Tumor-containing tissues were removed and immediately frozen in isopentane at −70°C. Tissues were sectioned at 12 μm and sections were air-dried and fixed in acetone/chloroform for 10 min. Non-specific binding was blocked with sera specific for secondary antibodies and stained for tumor-infiltrating lymphocytes using antibodies (BioLegend) to CD4 (GK1.5) and CD8α (2.43) overnight at 4°C. Sections were washed three times with PBS. FITC-labeled secondary antibodies (BioLegend) were incubated with sections for 1 h at 27°C followed by three washes with PBS. Detection was performed utilizing a Nikon Eclipse fluorescence microscope.
Apoptosis in GL261 tumors
TUNEL assays were carried out using an In Situ Cell Death Detection Kit (Roche). Tissue sections containing subcutaneous or intracerebral tumors were fixed with 4% paraformaldehyde in PBS, pH 7.4 for 20 min at 27°C, then washed for 30 min in 1× PBS. Sections were permeabilized in 0.1% Triton X-100, 0.1% sodium citrate for 2 min on ice. Labeling was performed by adding 50 μl of TUNEL reaction mixture (5 μl enzyme solution to 45 μl labeling solution) to each section for 1 h at 37°C in a dark humidified chamber. Tissue sections were washed three times with PBS, mounted in Vectashield mounting media for fluorescence with DAPI and imaged by fluorescence microscopy.
Results
Syngeneic and xenogeneic DNA vaccine vectors
We investigated whether vaccination with bone marrow dendritic cells (BMDC) transfected with an expression vector containing full-length copies of the murine and human survivin genes could induce effective immune responses in C57BL/6 mice leading to the rejection of syngeneic GL261 gliomas. We also explored the possibility that a truncated form of the human survivin gene pVAX.hSVN(II–V) could affect the growth of survivin-expressing murine GL261 gliomas in vivo. This vector was designed to produce a shortened form of the human survivin molecule without a Thr34 residue which is necessary for phosphorylation and activity, yet retains most of the epitopes that might be essential for an effective antitumor immune response (Fig. 1). In addition, individual fragments of the human survivin molecule were expressed in BMDCs to determine whether individual regions of the human survivin molecule were more immunogenic than others and whether any cryptic human survivin epitopes could be unmasked by such an approach.
Survivin expression in tumor cells lines
Endogenous survivin expression was assessed in a range of murine, racine and human tumor cell lines, including murine GL261 glioma cells (Fig. 2). No survivin expression was detected in C57BL/6 murine brain tissue. GL261 murine glioma cells were chosen for these experiments because they serve as a well-characterized syngeneic glioma model in H2-Kb-positive, C57BL/6 mice.
Fig. 2.

Western blot of survivin protein expression in tumor cell lines. Cellular lysates were prepared from F98 rat glioma, Eo771 breast carcinoma, B16 melanoma, K130 immortalized murine astrocytes, CCRF T cell lymphoblast-like cells, GL261 mouse glioma and C57BL/6 brain tissue. Proteins were resolved on 20% SDS-PAGE under reducing conditions and then transferred to PVDF membranes. Anti-survivin antibody detected a 16.5 kDa protein band corresponding to the expected molecular weight of murine, racine, and human survivin
Bone marrow dendritic cells
Bone marrow dendritic cells were isolated and cultured in GM-CSF for 6 days, after which non-adherent BMDCs were harvested for transfection. Following transfection, maturation of BMDCs was observed with expression of CD11c, CD40, CD86, IA/IE, and H2-Kb, as determined by flow cytometry (data not shown). Vaccinations consisted of subcutaneous injections of 5×105 pooled transfected BMDCs, followed by two booster vaccinations of 5x105 cells 7 and 14 days later. Analysis of pVAX.hSVN(II–V) transfected BMDCs by flow cytometry and immunofluorescence revealed robust expression of the truncated survivin protein and accumulation of survivin within cytoplasmic vacuoles (Fig. 3). Control (pVAX1) vector-transfected BMDCs did not express the endogenous survivin molecule at detectable levels.
Fig. 3.

Survivin expression in transfected bone marrow dendritic cells. BMDCs were transfected with pVAX.SVN(II–V) cDNA expression vector or with pVAX1. Over 90% of BMDC took up jetPEI-DNA complexes, as measured by co-transfection with FITC-labeled jetPEI using confocal microscopy and FACS (not shown). Immunofluorescence detection of survivin with anti-survivin antibody reveals a lack of detectable endogenous survivin expression in BMDC’s transfected with pVAX1, and b survivin expression and vacuolar accumulation in CD11c+ BMDCs following transfection with pVAX.hSVN(II–V)
Cytotoxic T lymphocyte responses to BMDC vaccines
We tested splenocytes from mice vaccinated with pVAX1 and pVAX.hSVN(II–V), as described in Materials and methods, for their ability to kill GL261 cells in culture (Fig. 4). CTL assays revealed specific cytotoxicity of lymphocytes from mice vaccinated with pVAX.hSVN(II–V) BMDCs, but not from pVAX1-transfected, BMDC-vaccinated mice.
Fig. 4.

Cellular assays for specific T cell lysis of GL261 target. Cytotoxic T lymphocyte (CTL) assays were performed using the DELFIA EuTDA cytotoxicity method. Splenocyte effector cells from vaccinated animals were added to GL261 target cells at the indicated ratios and were incubated for 2 h at 37°C. Percentage specific release was calculated using the formula: % Specific release = [(Experimental release – Spontaneous Release)/(Maximal release – Spontaneous release)] × 100%. Specific CTL activity against GL261 glioma cells was induced in response to immunization with BMDC transfected with pVAX.hSVN(II–V), but not pVAX1. Data points represent mean values of triplicate wells ± SEM
Subcutaneous tumor growth inhibition
C57BL/5 mice were vaccinated with BMDCs transfected with a vector designated pVAX.hSVN(II–V) encoding a truncated human survivin gene. This vector was constructed specifically to remove the Thr34 phosphorylation site. Four days after initial BMDC vaccination, mice were challenged with subcutaneous (flank) GL261 glioma cell injections. Additional doses of the transfected BMDC’s were given as boosters 7 and 14 days after the first vaccination. Vaccination with pVAX.hSVN(II–V) BMDCs attenuated tumor growth leading to complete tumor-free survival in 12 of 15 mice; whereas, BMDCs transfected with pVAX1 provided no protective effect (Fig. 5a, P<0.001). These observations, combined with the presence of survivin-specific CTL, suggested the existence of a cytotoxic antitumor immune response evoked by the truncated human pVAX.hSVN(II–V) vaccine that is highly effective against a flank GL261 tumor model.
Fig. 5.

Growth of syngeneic GL261 glioma cells in vivo following BMDC vaccination. a Growth of GL261 cells in C57BL/6 mice following immunization with BMDCs transfected with pVAX.hSVN(II–V) (n=7) and BMDCs transfected with pVAX1 control vector (n=15). Vaccinations with BMDCs were performed and then suspensions of 1.5×105 GL261 glioma cells were injected into the flanks of C57BL/6 mice. Tumor-free survival was defined as the point of first appearance of a 2–3 mm subcutaneous mass as detected by direct palpation (P<0.001). b Survival curves of C57BL/6 mice implanted with intracerebral GL261 cells after immunization with BMDC vaccines. Male C57BL/6 mice were anesthetized and fixed in a stereotactic head frame. 5×104 GL261 cells were implanted in the deep white matter of the right frontal cerebral cortex. Mice were injected subcutaneously with 5×105 BMDCs on days −4, +3, and +10 relative to tumor implantation. Survival was defined as the point at which mice died or displayed signs of neurologic deficit, plus 1 day. c Mice vaccinated with pVAX.hSVN(II–V)-transfected BMDC that survived tumor-free for over 180 days, as shown in Fig. 5a, were re-challenged with intracerebral injection of 5×104 GL261 cells. Mice vaccinated with pVAX.hSVN(II–V) and re-challenged with tumor cells survived significantly longer than those vaccinated with pVAX1 (P<0.001). Kaplan–Meier survival plots were prepared significance was determined using logrank Mantel–Cox methods
Survival prolongation in an intracerebral tumor model
C57BL/6 mice were vaccinated with BMDCs transfected with one of four vectors: (1) pVAX1 parent vector, (2) full-length murine survivin pVAX.mSVN(I–V), (3) full-length human survivin pVAX.hSVN(I–V), or (4) truncated human survivin pVAX.hSVN(II–V). Although the human survivin protein was significantly more effective (P=0.0175), both murine and human full-length survivin vaccines prolonged survival (Fig. 5b). The delayed, but progressive growth of intracerebral tumors suggests that site-specific factors, such as the blood brain barrier, could play an important role in modulating the immunologic antitumor response to GL261 cells. Fragments I through V of the human survivin cDNA were also introduced into pVAX1 to test whether individual fragments were either more or less immunogenic than the full-length survivin molecules (Table 1). While each of the individual human survivin fragments (I–V) produced some prolongation of survival, none was superior to either pVAX.hSVN(I–V) or pVAX.hSVN(II–V). Thus, while human survivin is a better immunogen against intracerebral GL261 tumors in C57BL/6 mice than murine survivin, the best response occurs when a full, or nearly full, complement of survivin epitopes is present.
Table 1.
Survival of animals vaccinated with BMDC transfected with full-length human and murine survivin cDNA expression vectors and with vectors expressing human survivin gene fragments I–V and the truncated human survivin gene hSVN(II–V)
| Vaccine vector | Median survival | Range | Mean survival (±SEM) |
|---|---|---|---|
| pVAX1 | 19 | 17–22 | 19.3±0.6 |
| pVAX.hSVN(I–V)* | 27 | 24–39 | 30.2±2.9 |
| pVAX.mSVN(I–V) | 24.5 | 21–26 | 24±0.9 |
| pVAX.hSVN(II–V)** | 26 | 21–34 | 27±1.4 |
| Human SVN fragments | |||
| pVAX.hSVN(I) | 25.5 | 18–29 | 25±1.7 |
| pVAX.hSVN(II) | 23 | 18–28 | 23.5±1.5 |
| pVAX.hSVN(III) | 22 | 21–29 | 25.2±0.7 |
| pVAX.hSVN(IV) | 22.5 | 22–31 | 24.2±1.4 |
| pVAX.hSVN(V) | 21 | 19–21 | 22.3±1.4 |
Vaccinated mice (n=9 per group) were challenged with intracerebral GL261 cells as described. The full-length human survivin {pVAX.hSVN(I–V)} vaccine was significantly more effective than the full-length murine {pVAX.mSVN(I–V)} vaccine (*P=0.0175). In addition, the truncated human survivin {pVAX.hSVN(II–V)} vaccine was significantly more effective than the full-length murine {pVAX.mSVN(I–V)} vaccine (**P=0.041)
Tumor re-challenge of previously cured animals
Twelve of 15 mice (80%) that were vaccinated with pVAX.hSVN(II–V), and that were initially challenged with subcutaneous GL261 implants, had tumor-free survival for greater than 200 days (Fig. 5a). Six of these mice were selected at random and subjected to re-challenge with 5×104 GL261 cells by intracerebral injection. This was done to determine if there was a memory response capable of preventing the subsequent development of a tumor within the brain. In contrast to mice challenged with intracerebral tumor after BMDC vaccination alone, all of the cured, re-challenged animals survived an additional 60 days (Fig. 5c), at which time they were sacrificed. None of these cured, re-challenged mice had detectable intracerebral tumors (data not shown).
Tumors from survivin-vaccinated mice contain CD4+ and CD8+ T cells
Immunofluorescence studies were performed on intracerebral and subcutaneous GL261 tumors from mice vaccinated with three doses of either pVAX.hSVN(II–V) or pVAX1. CD8+ cells were identified in large numbers in flank tumors of mice vaccinated with pVAX.hSVN(II–V) (Fig. 6). Qualitatively fewer numbers of CD8+ cells were observed in the intracerebral tumors of mice undergoing vaccination with pVAX.hSVN(II–V) BMDCs. Very few CD8+ cells were identified in the tumors of mice injected with pVAX1-transfected BMDCs, whether tumor challenge was subcutaneous or intracerebral. Tumors in flank and brain were negative for CD94+ NK cells (data not shown). These findings are consistent with tumor cell apoptosis studies and the results of survival and tumor-free survival experiments. They suggest the existence of site-specific differences, such as those posed by an intact blood brain barrier, affecting the potency of the immune effector response to GL261 tumors.
Fig. 6.

GL261 tumor infiltration by CD4+ and CD8+ T-cells. Frozen sections from intracerebral (c, d, g, h) and subcutaneous (a, b, e, f) GL261 tumors were analyzed for tumor infiltrating lymphocytes (TIL) by direct immunofluorescence. Mice immunized with BMDC transfected with pVAX.hSVN(II–V) (e–h), and pVAX1 (a–d) are shown. All images are shown at ×40 magnification
CD8+ T cell depletion and adoptive immunotherapy
Mice that had rejected both subcutaneous tumor challenge and subsequent intracerebral re-challenge were used for CD8+ T cell depletion and adoptive transfer studies. A group of these mice (n=5) were depleted of CD8+ T cells and challenged a third time with subcutaneous GL261 gliomas implanted into the flank. Following CD8+ T cell depletion, tumor-free survival was significantly reduced compared to non-depleted mice (Fig. 7a, P<0.001). The remaining mice that had rejected both subcutaneous and subsequent intracerebral GL261 gliomas were used as a source of CD3+ T cells for adoptive transfer to naïve mice. Following adoptive transfer, recipient mice were given intracerebral injections of GL261 glioma cells (Fig. 7b). Mice that received CD3+ T cells from pVAX.hSVN(II–V) vaccinated donors survived significantly longer than pVAX1 controls (P<0.01). These studies indicate the presence of a T cell dependent transferable immune response following vaccination with survivin-expressing BMDCs.
Fig. 7.

CD8+ T cell depletion and CD3+ adoptive immunotherapy. a C57BL/6 mice underwent CD8+ T cell depletion and re-challenge with subcutaneous GL261 glioma cells implanted into the flank. Tumor-free survival of CD8+ depleted mice (n=5) was significantly less than non-depleted mice (n=5, P<0.01). b CD3+ T cells were from vaccinated animals that had rejected tumor implantation were transferred adoptively to naïve C57BL/6 mice. CD3+ recipient mice then underwent intracerebral implantation of 5×104 GL261 glioma cells. Mice that received CD3+ cells from survivin-vaccinated donors (n=9) survived significantly longer than pVAX1-vaccinated (n=5) controls (P<0.01)
Tumor apoptosis in response to vaccination
Tumor cell apoptosis was measured in i.c. and s.c. GL261 tumors following vaccination with pVAX1 and pVAX.hSVN(II–V). Very few apoptotic cells were found in the tumors of unvaccinated mice, or in those vaccinated with pVAX1-BMDC (Fig. 8). In contrast, many apoptotic cells were evident in the tumors of mice vaccinated with pVAX.hSVN(II–V). The majority of these apoptotic cells were found in tumors near their interface with surrounding normal tissue. Overall, subcutaneous GL261 gliomas contained greater numbers of apoptotic cells than did intracerebral gliomas, consistent with the results of survival and tumor-free survival studies and the presence and relative numbers of CD8+ cells in tumors of pVAX.hSVN(II–V) vaccinated mice. Since high levels of apoptotic cells are associated with tumor growth inhibition, it is most likely that these apoptotic cells are predominantly glioma cells, rather than tumor infiltrating lymphocytes.
Fig. 8.

Tumor cell apoptosis in vaccinated mice. Frozen sections from intracranial (b, d) and subcutaneous (a, c) GL261 tumors were analyzed for apoptotic cells by TUNEL assays as described. Mice receiving BMDC transfected with pVAX.hSVN(II–V) (c, d) and pVAX1 (a, b) are shown. All images are shown at ×40 magnification
Autoimmune toxicity
A group of seven naïve C57BL/6 mice was treated with a series of three vaccinations with BMDCs transfected with pVAX.hSVN(II–V). No behavioral changes were observed in vaccinated mice from any of the treatment groups. After 180 days, necropsies were performed and histologic sections of the major organs were evaluated by a pathologist. No evidence of toxicity or autoimmune pathology was detected.
Discussion
Immunotherapy has emerged as a potential treatment modality for gliomas because it is: (1) specific to tumor cells, (2) has the potential to eradicate infiltrating tumor cells after surgical debulking, and (3) may be capable of eliciting a long-lasting memory response to prevent tumor recurrence. Both immunological [16–18] and clinical responses [18, 19] have been observed in glioma patients immunized with (1) dendritic cells loaded with peptides eluted from the surface of allogeneic tumor cells [16]; (2) DCs loaded with peptides eluted from autologous tumor cells [17]; (3) DCs pulsed with tumor lysate from autologous tumor cells [18]; and (4) hybrids generated by fusing DCs with autologous tumor cells [19]. In one study [18], patients with recurrent glioblastoma multiforme, who were immunized with tumor lysate-pulsed DCs, had significantly increased survival (median 133 weeks). Thus, DC-based tumor vaccines are of interest for the treatment of malignant gliomas.
Survivin is a 16.5 kDa intracellular protein that belongs to the inhibitor of apoptosis protein (IAP) family. Survivin inhibits apoptosis, acting in concert with the mitotic spindle apparatus to regulate cell division. It is expressed in cells during the G2/M phase of the cell cycle and localizes to the spindle microtubule organizing center during this phase of cell cycle progression [20–22]. Survivin has also been shown to modulate the function of certain caspases, directly inhibiting apoptosis [23–25]. In addition, survivin appears to inhibit the cyclin D/cdk4 complex [26], permitting cell cycle progression and inhibiting apoptosis. Survivin also interacts with the cdk4/p16INK4a complex at the point of cell cycle entry [27]. Thus, survivin appears to function in critical roles at a number of different loci to regulate the cell cycle and inhibit apoptosis. Anti-survivin vaccines or dominant negative survivin gene therapy (i.e., phosphorylation-defective survivin mutants) could be particularly effective in conjunction with apoptosis-inducing agents [28].
We evaluated a truncated non-functional survivin molecule in the context of a transfected DC vaccine to prevent possible immortalization or tumor formation in DC or other cells that might take up and express the pVAX.hSVN(II–V) vaccine plasmid in a stable fashion. A similar approach was used by Mesri et al. [29] to produce a non-dominant survivin mutant by substituting alanine for Thr34. Our studies indicate that vaccination with a DNA vector containing a truncated survivin gene lacking Thr34 is associated with tumor regression. Although this result occurs at the expense of some potentially immunogenic epitopes from the N-terminal portion of the molecule, no significant difference in survival could be detected regardless of whether the truncated or full length human survivin proteins were used as vaccine.
Xiang et al. [30] previously demonstrated that a DNA-based survivin vaccine could evoke a cytotoxic T cell response against tumor cells. This DNA vaccine induced a significant antitumor response against Lewis lung carcinoma, which led to the regression of primary tumor growth and disseminated pulmonary metastases in C57BL/6J mice. The mechanism of the antitumor immunity induced by this DNA vaccine involved MHC class I antigen-restricted CD8+ T cells. The survivin vaccine triggered tumor cell apoptosis, together with release of interferon gamma (IFN-γ). This particular DNA vaccine, however, encoded both survivin and other immune modulators. Similarly, Zeis et al. [31] have shown that transfection of dendritic cells with survivin mRNA leads to resistance to tumor challenge.
The survivin protein contains HLA-A*0201 (HLA-A2) binding motifs. Andersen et al. [32] used peptides with such binding motifs to test for specific T-cell reactivity in leukemia and melanoma patients. CTL responses against two survivin epitopes were detected in both patient groups, but no T-cell reactivity was found in healthy control patients. This suggests that survivin is a TAA that is recognizable by autologous T cells. However, this experiment failed to demonstrate that survivin-reactive T cells had the capacity to lyse tumor cells. Schmitz et al. [33] showed that survivin can induce CD8+ T cell immune responses when presented by dendritic cells. Recombinant survivin protein incubated with DCs can induce specific MHC-I-restricted CTL. Moreover, two survivin-derived peptides elicited a peptide-specific CTL response. One of these peptides was shown to result from the natural intracellular processing of survivin [34].
Currently, immunotherapeutic targeting of glioma-associated antigens is limited by the fact that gliomas are known to be heterogeneous in their antigen-expression profiles and histologically similar gliomas may express different antigens [35, 36]. Therefore, the immune targeting of a single antigen is likely to be only partially effective, due to the possibility of immune escape through the generation of antigen-loss variants [37]. To date, few, if any, common glioma antigens have been identified. Consequently, the development of a strategy that targets a specific antigen may not be suitable for implementation across a large glioma patient population. All of these limitations may be circumvented by the development of an immunotherapeutic approach targeting a single uniformly expressed common tumor antigen or multiple antigens expressed in a heterogeneous fashion.
We have selected a DC-based immunization strategy, since (1) DC are unique among antigen-presenting cells in their ability to stimulate naïve CD4+ and CD8+ T cells, due to their constitutive expression of MHC class I and II antigens and costimulatory molecules [38] and (2) antigen-loaded DC-based immunization strategies can overcome self-tolerance and elicit robust immune responses to self-antigens [39]. Many published reports on immunotherapy of gliomas have focused on immunization strategies utilizing DC that are pulsed with glioma-derived antigens. Ni et al. [40] showed that DC pulsed with GL261 tumor extract significantly increased the survival of mice bearing intracerebral GL261 glioma. In a second study, DC pulsed with GL261 tumor extract-cationic liposome complex was able to elicit GL261-specific cytotoxic responses, and significantly reduced tumor masses in all mice, including several complete remissions [41]. A third study revealed that DC pulsed with gp100- and TRP-2-derived peptides were able to prolong survival in intracerebral GL261 glioma-bearing mice [4]. The DC-based immunization strategies reported to date provide a strong rationale to continue to investigate DC-based immunotherapeutic approaches for gliomas.
Survivin is one of the most commonly expressed TAA [11, 42, 43]. It is an important regulatory molecule in tumor cells and in tumor vascular endothelium [44]. Taken together, these characteristics would appear to make survivin a good candidate for antitumor vaccines. Moreover, survivin and other anti-apoptotic proteins of the IAP family may represent good targets for vaccination because cells that downregulate survivin as an immune escape mechanism might be more susceptible to other apoptotic signals. MHC class I-restricted cytotoxic T cells directed against survivin peptides have been identified in patients with breast cancer, leukemia, and melanoma [45]. Hence, immunologically mediated attack on cells that express survivin epitopes in association with MHC-I antigens might help to prevent immune escape. Moreover, the identification of HLA-A1, HLA-A2 and HLA-A3, and HLA-A11-restricted survivin epitopes [46, 47] suggests that the targeting of multiple survivin epitopes with DNA- or DC-based vaccines, or with multiple HLA-restricted peptides, might be expected to decrease the chance of immune escape by way of HLA allele loss from tumor cell targets.
The mouse, rat, and human survivin genes are highly homologous, but they do contain differences in amino acid sequence. The current study was based on our preliminary observation that a human xenogeneic DNA vaccine against the anti-apoptotic protein survivin inhibits the growth of F98 glioblastoma in syngeneic Fischer rats [48]. We hypothesized that the use of certain human survivin epitopes could elicit an effective immune response to self antigen on tumor cells producing greater antitumor immunity than the murine homolog itself. Thus, xenogeneic differences between human and murine survivin sequences might be exploited to develop more immunogenic glioma vaccines.
Proteosomal processing of the melanoma-associated human gp100 sequence yields a peptide epitope that binds to H-Db with higher affinity and a slower off rate than its murine homolog, thus inducing a more potent antitumor immune response than the homologous murine gp100 peptide. Consequently, the human gp100 sequence acts as a heteroclitic molecule, binding to murine MHC class I better than the native murine sequence, leading to the production of a potent cellular immune response [49]. Alternatively, T cell clones with TCRs recognizing the native peptide may have been deleted during immunological development. Consequently, other T cell clones that recognize similar epitopes (peptide mimics) could mount an immunologic response to the cross-reactive native peptide. As with the melanoma gp100 antigen, xenogeneic differences between the human and murine survivin proteins might be exploited in vaccines to help break tolerance to the endogenous murine survivin protein. Similarly, processed peptides derived from an altered survivin gene might be employed to develop a clinically useful vaccine for humans.
Statistically significant survival differences were observed between vaccine groups; however, all mice that were vaccinated with survivin-expressing DCs died from tumor progression following primary challenge with intracerebral GL261 tumor implants. In contrast, mice that had resisted initial subcutaneous tumor challenge (>180 day tumor-free survival) following pVAX.hSVN(II–V) DC vaccination were also able to resist subsequent re-challenge with intracerebral GL261 tumor implants and to exhibit extended (>60 day) survival. This suggests the possibility that priming with a xenogenic survivin mimic, followed by boosting with syngeneic murine survivin via a cellular vehicle, enhanced the antitumor response to the extent that growth of an intracerebral tumor target could be completely prevented. Similar prime-boost strategies have proven effective for vaccination against high molecular weight melanoma-associated antigen (HMW-MAA) in which vaccination with a mimic, followed by boosting with killed tumor cells produced a greater immune response than with either individually [50]. In particular, this type of prime-boost immunization strategy has been utilized with a number of TAA mimics in several carbohydrate-bearing TAA systems [51–53]. Our data indicate that a prime-boost strategy with a survivin DC vaccine may be useful to elicit a stronger CD4+ response in support of CTL directed against malignant glioma cells.
Acknowledgements
This work was supported by NIH 5R21NS049309–02, NIH 5P30CA16056-29, the Roswell Park Alliance Foundation and the Linda Scime Fund.
References
- 1.Moscatello DK, Ramirez G, Wong AJ. A naturally occurring mutant epidermal growth factor receptor as a target for peptide vaccine immunotherapy of tumors. Cancer Res. 1997;57:1419–1424. [PubMed] [Google Scholar]
- 2.Heimberger AB, Crotty LE, Archer GE, et al. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin Cancer Res. 2003;9:4247–4254. [PubMed] [Google Scholar]
- 3.Ciesielski MJ, Kazim L, Barth RF, Fenstermaker RA. Cellular antitumor immune response to a branched lysine multiple antigenic peptide containing epitopes of a common tumor-specific antigen in a rat glioma model. Cancer Immunol Immunother. 2005;54:107–119. doi: 10.1007/s00262-004-0576-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prins RM, Odesa SK, Liau LM. Immunotherapeutic targeting of shared melanoma-associated antigens in a murine glioma model. Cancer Res. 2003;63:8487–8491. [PubMed] [Google Scholar]
- 5.Adida C, Crotty PL, McGrath J, et al. Developmentally regulated expression of the novel cancer anti-apoptosis gene survivin in human and mouse differentiation. Am J Pathol. 1998;152:43–49. [PMC free article] [PubMed] [Google Scholar]
- 6.Kajiwara Y, Yamasaki F, Hama S, et al. Expression of survivin in astrocytic tumors. Cancer. 2003;97:1077–1083. doi: 10.1002/cncr.11122. [DOI] [PubMed] [Google Scholar]
- 7.Sasaki T, Lopes MB, Hankins GR, Helm GA. Expression of survivin an inhibitor of apoptosis protein in tumors of the nervous system. Acta Neuropathol (Berl) 2002;104:105–109. doi: 10.1007/s00401-002-0532-x. [DOI] [PubMed] [Google Scholar]
- 8.Chakravarti A, Noll E, Black P, et al. Quantitatively determined survivin expression levels are of prognostic value in human gliomas. J Clin Oncol. 2002;20:1063–1068. doi: 10.1200/JCO.20.4.1063. [DOI] [PubMed] [Google Scholar]
- 9.Islam A, Kageyama H, Takada N, et al. A high expression of survivin mapped to 17q25 is significantly associated with poor prognostic factors and promotes cell survival in human neuroblastoma. Oncogene. 2000;19:617–623. doi: 10.1038/sj.onc.1203358. [DOI] [PubMed] [Google Scholar]
- 10.Satoh K, Kaneko K, Hirota M, et al. T expression of survivin is correlated with cancer cell apoptosis and is involved in the development of human pancreatic duct cell tumors. Cancer. 2001;92:271–278. doi: 10.1002/1097-0142(20010715)92:2<271::AID-CNCR1319>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 11.Altieri DC. Validating survivin as a cancer therapeutic target. Nat Rev Cancer. 2003;3:46–54. doi: 10.1038/nrc968. [DOI] [PubMed] [Google Scholar]
- 12.Overwijk WW, Tsung A, Irvine KR, et al. gp100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive tumoricidal T cells using high-affinity altered peptide ligand. J Exp Med. 1998;188:277–286. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:2445–2452. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 14.Steinman RM, Dhodapkar M. Active immunization against cancer with dendritic cells: the near future. Int J Cancer. 2001;4:459–473. doi: 10.1002/ijc.1503. [DOI] [PubMed] [Google Scholar]
- 15.Patel AK, Boyd PN. An improved assay for antibody dependent cellular cytotoxicity based on time resolved fluorometry. J Immunol Methods. 1995;184(1):29–38. doi: 10.1016/0022-1759(95)00071-H. [DOI] [PubMed] [Google Scholar]
- 16.Liau LM, Black KL, Martin NA, et al. Treatment of a glioblastoma patient by vaccination with autologous dendritic cells pulsed with allogeneic major histocompatibility complex class I-matched tumor peptides. Neurosurg Focus. 2000;9:8. doi: 10.3171/foc.2000.9.6.9. [DOI] [PubMed] [Google Scholar]
- 17.Yu JS, Wheeler CJ, Zeltzer PM, et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001;61:842–847. [PubMed] [Google Scholar]
- 18.Yu JS, Liu G, Ying H, et al. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004;64:4973–4999. doi: 10.1158/0008-5472.CAN-03-3505. [DOI] [PubMed] [Google Scholar]
- 19.Kikuchi T, Akasaki Y, Irie M, et al. Results of a phase I clinical trial of vaccination of glioma patients with fusions of dendritic and glioma cells. Cancer Immunol Immunother. 2001;50:337–344. doi: 10.1007/s002620100205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao J, Tenev T, Martins LM, et al. The ubiquitin-proteosome pathway regulates survivin degradation in a cell-cycle dependent manner. J Cell Sci. 2000;113:4363–4371. doi: 10.1242/jcs.113.23.4363. [DOI] [PubMed] [Google Scholar]
- 21.Li F, Ambrosini G, Chu EY, et al. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:580–584. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 22.Fortugno P, Wall NR, Giodini A, et al. Survivin exists in immunochemically distinct subcellular pools and is involved in spindle microtubule function. J Cell Sci. 2002;115:575–585. doi: 10.1242/jcs.115.3.575. [DOI] [PubMed] [Google Scholar]
- 23.Tamm I, Wang Y, Sausville E, et al. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95) Bax caspases and anticancer drugs. Cancer Res. 1998;58:5215–5220. [PubMed] [Google Scholar]
- 24.Conway EM, Pollefeyt S, Cornelissen J, et al. Three differentially expressed survivin cDNA variants encode proteins with distinct antiapoptotic functions. Blood. 2000;95:1435–1442. [PubMed] [Google Scholar]
- 25.Shin S, Sung BJ, Cho Y, et al. An anti-apoptotic protein human survivin is a direct inhibitor of caspase-3 and -7. Biochemistry. 2001;40:1117–1123. doi: 10.1021/bi001603q. [DOI] [PubMed] [Google Scholar]
- 26.Fududa S, Mantel CR, Pelus LM. Survivin regulates hematopoietic progenitor cell proliferation through p21WAF1/Cip1 dependent and independent pathways. Blood. 2004;103:120–127. doi: 10.1182/blood-2003-05-1756. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki A, Hayashida M, Ito T, et al. Survivin initiates cell cycle entry by the competitive interaction with Cdk4/p16INK4a and Cdk2/Cyclin E complex activation. Oncogene. 2000;19:3225–3334. doi: 10.1038/sj.onc.1203665. [DOI] [PubMed] [Google Scholar]
- 28.Grossman D, Kim PJ, Schechner JS, Altieri DC. Inhibition of melanoma tumor growth in vivo by survivin targeting. Proc Natl Acad Sci USA. 2001;98:635–640. doi: 10.1073/pnas.230450097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mesri M, Wall NR, Li J, et al. Cancer gene therapy using a survivin mutant adenovirus. J Clin Invest. 2001;108:981–990. doi: 10.1172/JCI200112983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiang R, Mizutani N, Luo Y, et al. A DNA vaccine targeting survivin combines apoptosis with suppression of angiogenesis in lung tumor eradication. Cancer Res. 2005;65:553–561. [PubMed] [Google Scholar]
- 31.Zeis M, Siegel S, Wagner A, et al. A generation of cytotoxic responses in mice and human individually against hematological malignancies using survivin-RNA-transfected dendritic cells. J Immunol. 2003;170:5391–5397. doi: 10.4049/jimmunol.170.11.5391. [DOI] [PubMed] [Google Scholar]
- 32.Andersen MH, Pedersen LO, Becker JC, Straten P. Identification of a cytotoxic T lymphocyte response to the apoptosis inhibitor protein survivin in cancer patients. Cancer Res. 2001;61:869–872. [PubMed] [Google Scholar]
- 33.Schmitz M, Diestelkoetter P, Weigle B, et al. Generation of survivin-specific CD8+ T effector cells by dendritic cells pulsed with protein or selected peptides. Cancer Res. 2000;60:4845–4849. [PubMed] [Google Scholar]
- 34.Yamanaka R, Yajima N, Abe T, et al. Dendritic cell-based glioma immunotherapy. Int J Oncol. 2003;23:5–15. [PubMed] [Google Scholar]
- 35.Yang L, Ng KY, Lillehei KO. Cell-mediated immunotherapy: a new approach to the treatment of malignant glioma. Cancer Control. 2003;10:138–147. doi: 10.1177/107327480301000205. [DOI] [PubMed] [Google Scholar]
- 36.Ehtesham M, Black KL, Yu JS. Recent progress in immunotherapy for malignant glioma: treatment strategies and results from clinical trials. Cancer Control. 2004;11:192–207. doi: 10.1177/107327480401100307. [DOI] [PubMed] [Google Scholar]
- 37.Dunn GP, Bruce AT, Ikeda H. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 38.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:2445–2452. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 39.Steinman RM, Dhodapkar M. Active immunization against cancer with dendritic cells: the near future. Int J Cancer. 2001;4:459–473. doi: 10.1002/ijc.1503. [DOI] [PubMed] [Google Scholar]
- 40.Ni HT, Spellman SR, Jean WC, et al. Immunization with dendritic cells pulsed with tumor extract increases survival of mice bearing intracranial gliomas. J Neurooncol. 2001;51:1–9. doi: 10.1023/A:1006452726391. [DOI] [PubMed] [Google Scholar]
- 41.Aoki H, Mizuno M, Natsume A, et al. Dendritic cells pulsed with tumor extract-cationic liposome complex increase the induction of cytotoxic T lymphocytes in mouse brain tumor. Cancer Immunol Immunother. 2001;50:463–468. doi: 10.1007/s002620100220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gordan JD, Vonderheide RH. Universal tumor antigens as targets for immunotherapy. Cytotherapy. 2002;4:317–327. doi: 10.1080/146532402760271091. [DOI] [PubMed] [Google Scholar]
- 43.Altieri DC. Survivin and apoptosis control. Adv Cancer Res. 2003;88:31–52. doi: 10.1016/S0065-230X(03)88303-3. [DOI] [PubMed] [Google Scholar]
- 44.O’Connor DS, Schechner JS, Adida C, et al. Control of apoptosis during angiogenesis by survivin expression in endothelial cells. Am J Pathol. 2000;156:393–398. doi: 10.1016/S0002-9440(10)64742-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Altieri DC. The molecular basis and potential role of survivin in cancer diagnosis and therapy. Trends Mol Med. 2001;7:542–547. doi: 10.1016/S1471-4914(01)02243-2. [DOI] [PubMed] [Google Scholar]
- 46.Andersen MH, Pedersen LO, Capeller B, et al. Spontaneous cytotoxic T-cell responses against survivin-derived MHC class I-restricted T-cell epitopes in situ as well as ex vivo in cancer patients. Cancer Res. 2001;61:5964–5968. [PubMed] [Google Scholar]
- 47.Reker S, Meier A, Holten-Andersen L, Svane IM, Becker JC, thor Straten P, Andersen MH. Identification of novel survivin-derived CTL epitopes. Cancer Biol Ther. 2004;3(2):173–179. doi: 10.4161/cbt.3.2.611. [DOI] [PubMed] [Google Scholar]
- 48.Fenstermaker RA, Ciesielski MJ. Immunotherapeutic strategies for malignant gliomas. Cancer Control. 2004;11:181–191. doi: 10.1177/107327480401100306. [DOI] [PubMed] [Google Scholar]
- 49.Hawkins WG, Gold JS, Dyall R. Immunization with DNA coding for gp100 results in CD4 T-cell independent antitumor immunity. Surgery. 2000;128:273–280. doi: 10.1067/msy.2000.107421. [DOI] [PubMed] [Google Scholar]
- 50.Luo W, Hsu JC, Tsao CY, Ko E, Wang X, Ferrone S. Differential immunogenicity of two peptides isolated by high molecular weight-melanoma-associated antigen-specific monoclonal antibodies with different affinities. J Immunol. 2005;174:7104–7110. doi: 10.4049/jimmunol.174.11.7104. [DOI] [PubMed] [Google Scholar]
- 51.Kieber-Emmons TB, Monzavi-Karbassi B, Wang P, et al. Cutting edge: DNA immunization with minigenes of carbohydrate mimotopes induce functional anti-carbohydrate antibody response. J Immunol. 2000;165:623. doi: 10.4049/jimmunol.165.2.623. [DOI] [PubMed] [Google Scholar]
- 52.Cunto-Amesty GP, Luo B, Monzavi-Karbassi A, et al. Peptide mimotopes as prototypic templates of broad-spectrum surrogates of carbohydrate antigens. Cell Mol Biol. 2003;49:245. [PubMed] [Google Scholar]
- 53.Chapman PBD, Wu G, Ragupathi S, et al. Sequential immunization of melanoma patients with GD3 ganglioside vaccine and anti-idiotypic monoclonal antibody that mimics GD3 ganglioside. Clin Cancer Res. 2004;10:4717. doi: 10.1158/1078-0432.CCR-04-0345. [DOI] [PubMed] [Google Scholar]