

Abstract
Linkage of doxorubicin (Dox) to a water-soluble synthetic N-(2-hydroxypropyl)methacrylamide copolymer (PHPMA) eliminates most of the systemic toxicity of the free drug. In EL-4 lymphoma-bearing C57BL/6 mice, a complete regression of pre-established tumours has been achieved upon treatment with Dox–PHPMA–HuIg conjugate. The treatment was effective using a range of regimens and dosages, ranging from 62.5 to 100% cured mice treated with a single dose of 10–20 mg of Dox eq./kg, respectively. Fractionated dosages producing lower levels of the conjugate for a prolonged time period had substantial curative capacity as well. The cured mice developed anti-tumour protection as they rejected subsequently re-transplanted original tumour. The proportion of tumour-protected mice inversely reflected the effectiveness of the primary treatment. The treatment protocol leading to 50% of cured mice produced only protected mice, while no mice treated with early treatment regimen (i.e. starting on day 1 after tumour transplantation) rejected the re-transplanted tumour. Exposure of the host to the cancer cells was a prerequisite for developing protection. The anti-tumour memory was long lasting and specific against the original tumour, as the cured mice did not reject another syngeneic tumour, melanoma B16-F10. The immunity was transferable to naïve recipients in in vivo neutralization assay by spleen cells or CD8+ lymphocytes derived from cured animals. We propose an effective treatment strategy which eradicates tumours without harming the protective immune anti-cancer responses.
Keywords: Targeted tumour therapy, HPMA, Human immunoglobulin, Doxorubicin, Complete tumour regression, Protective anti-tumour response
Introduction
Many drug delivery systems have been developed to improve the therapeutic effect of cytotoxic drugs and to reduce their systemic toxicity. The concept of macromolecular carriers of low-molecular-weight (anti-cancer) drugs has evolved continuously over the last century, being started in 1906 with the Ehrlich’s “magic bullet” phrase [1] and accomplished with the concept of the use of polymers as targeted drug carriers by Ringsdorf in 1975 [2]. Macromolecular drugs based on N-(2-hydroxypropyl)methacrylamide (HPMA) copolymers (PHPMA) possess a higher anticancer efficacy and better pharmacokinetic profile than their low-molecular-weight counterparts. The rationales for the use of HPMA-based conjugates are biocompatibility of the polymeric carrier, solubilization of hydrophobic drugs, targetability, and preferential accumulation in solid tumours due to enhanced permeability and retention (EPR) effect [3, 4]. Passive targeting of the macromolecular compound to the tumour is due to increased permeability of discontinuous vascular endothelium, which allows extravasations of macromolecules into the interstitial space of the tumour tissue, and the poor lymphatic drainage, which hampers elimination of accumulated macromolecules from the site. The accumulation and retention of polymeric drugs are greatly enhanced in tumour tissue compared with those in normal tissue. This EPR effect is applicable to macromolecules and lipid particles, but not to low-molecular-weight drugs [5, 6], to which category most of clinically relevant cytotoxic drugs belong.
Doxorubicin (Dox) possesses a broad spectrum of anti-cancer activities and is used in chemotherapy of various cancers, including adenocarcinomas, melanomas, sarcomas, lymphomas and leukaemia’s. As with other cytostatic drugs, its acute and cumulative dose-related toxicity poses a major obstacle in therapeutic outcomes, with cardiotoxicity and myelotoxicity being the most dangerous side effects [7]. It has been repeatedly documented that Dox and other cytostatic or immunosuppressive drugs conjugated to PHPMA copolymer show reduced non-specific toxicity and better therapeutic profile [8–10]. Different intracellular trafficking of these conjugates in membrane-limited organelles in contrast to free diffusion for low-molecular-weight compounds might partially overcome P-glycoprotein (Pgp)-mediated multidrug resistance [11, 12]. The PHPMA–Dox conjugates have been shown to have anti-tumour effect in vitro and in vivo [13, 14].
Immunotherapeutic strategy of tumour treatment does not rely solely on targeting the tumour cells themselves; by trying to induce anti-cancer mechanisms or to increase immunogenicity of the tumour cells or both it strives to induce the active reaction of the host to the cancer. However, immunotherapy as a single treatment modality is generally effective against small tumours ( < 3 mm in diameter) in animal models of cancer [15]. Larger tumours appear either to impair the anti-tumour immune response or avoid it [16, 17]. Combination of an agent of proven clinical utility such as chemotherapy or radiotherapy together with immunotherapy therefore could form an ideal basis for therapeutic success. Indeed, it may only be effective if the conventional treatment is not overtly suppressive to the host’s immune anti-tumour responses. Therefore, it is emphasized that a compound having its own anti-tumour efficacy together with protective effect on the immune anti-tumour functions could be an optimal tool for effective tumour treatment strategy.
A copolymer conjugate Dox–PHPMA–HuIg has already been evaluated in a pilot study and displayed a good performance in patients with disseminated cancers. All patients (8 so far) showed good tolerance of the conjugate and partial clinical response or stabilization of the disease lasting for months. Five patients were tested for selected immune anti-cancer responses. Three of them displayed periodical activation of natural killer (NK) cells and lymphokine-activated killers (LAK) following the conjugate administration [18, 19], and the other had increased NK or LAK activity after at least one conjugate administration. This finding was suggestive of a possible activation of anti-cancer immune mechanisms.
In order to analyse the anti-cancer effects of the Dox–PHPMA–HuIg conjugate, we performed a series of experiments with C57BL/6 mice bearing syngeneic T cell lymphoma EL-4.
Materials and methods
Cell lines
Mouse T cell lymphoma EL-4 (ATCC TIB-39) and subline EL-4.IL-2 (ATCC TIB-181) were purchased from ATCC and grown in RPMI 1640 medium containing 10% foetal calf serum (RPMI-10), antibiotics (penicillin/streptomycin, Sigma, USA) and supplemented with 4 mM l-glutamine, 0.05 mM 2-mercaptoethanol, and 4.5 g/l glucose. B16-F10 melanoma [20] was kindly donated by Prof. M. Pospisil, Institute of Microbiology ASCR, and was grown in RPMI-10 medium with antibiotics, 2 mM l-glutamine, and 0.05 mM 2-mercaptoethanol.
Animals, models of tumour growth
Male C57BL/6 mice (B/6) were from the Animal Facility of the Institute of Physiology, AS CR, and female nu/nu CD-1 mice from the Institute of Molecular Genetics, ASCR. Mice were 2–3 months old at the start of experiments and were given food and water ad libitum.
The C57BL/6 mice (minimum 8 per group) were injected subcutaneously (s.c.) into the right shaven ridge with 1 × 105 EL-4 cells or 1 × 106 B16-F10 melanoma cells to produce continuously growing solid tumours. Nu/nu mice were s.c. injected with 5 × 105 EL-4. Tumour sizes were measured with callipers in two perpendicular diameters every 2 days and expressed as tumour volume V = a × b 2/2 (a = longer diameter, b = shorter diameter). Survival time was regularly scored.
Conjugate synthesis and characterization
The Dox–PHPMA–HuIg conjugate was prepared from the polymer precursor—a random copolymer of HPMA with methacryloylglycyl-d,l-phenylalanyl-l-leucylglycine 4-nitrophenyl ester (Ma–Gly–Phe(d, l)–Leu–Gly–ONp) by consecutive aminolysis with HuIg (Intraglobin F, human immunoglobulin suitable for intravenous use, Biotest Pharma GmbH, Germany) and Dox hydrochloride (Dox.HCl) (Adriblastina CS, Pharmacia & Upjohn, USA) using a modified procedure originally described in [21]. HPMA was prepared by modified method of methacryloylation of 1-aminopropan-2-ol with methacryloyl chloride [21] carried out in methylene chloride containing sodium bicarbonate, Ma–Gly–Phe(d, l)–Leu–Gly–ONp was prepared from methacryloyl chloride and respective dipeptides using the methods of peptide synthesis [21], and reactive polymer precursor was prepared by radical precipitation copolymerization of HPMA with Ma–Gly–Phe(d, l)–Leu–Gly–ONp in acetone (initiator AIBN, 0.6 wt%; concentration of monomers 12.5 wt%; molar ratio HPMA: reactive esters 12:1; 60°C; 24 h) as described earlier [21].
Aminolytic reaction of the polymer precursor with Dox.HCl and HuIg was performed in water as follows: The polymer precursor (13.5 g) was dissolved in ice-cooled double-distilled water (150 ml) vigorously stirred in Erlenmayer flask on ice bath (approx. 30 min). A total of 75 ml of a solution of HuIg (4.5 g) in double-distilled water (isolated from its dosage form Intraglobin F by ultrafiltration in an Amicon cell using Millipore membrane with cut-off 100,000) was added, the pH of the reaction mixture was adjusted to 7.8 (pH-stat) and the mixture was stirred at 10°C for 30 min. Temperature was raised to 20°C and an original solution of Adriblastina (1.15 g Dox.HCl, 575 ml) was added in small portions within 1 h. pH was kept at 7.8 (pH-stat, sodium tetraborate) for another 18 h, the reaction was stopped by adding 0.5 ml of 1-aminopropan-2-ol and the mixture was left overnight in a refrigerator. The rough product was purified by gel filtration after adjusting pH to 6.5 on a column filled with Sephadex G-25 Medium using double-distilled water as eluent. Purified conjugate was freeze-dried.
As a control, polymer conjugates Dox–PHPMA and PHPMA–HuIg were prepared, Dox–PHPMA using a procedure described in [22] (M w = 24.8 kDa, Dox content 5.1 wt%) and PHPMA–HuIg using a modified method described above for Dox conjugate (no Dox.HCl added) (M w = 1,000 kDa, content of HuIg 18 wt%).
The polymer precursor and polymer conjugate were characterized by composition of the oligopeptide spacer and a content of HuIg in the conjugate using amino acid analysis (Amino Acid Analyser LDC Analytical, precolumn OPA derivatization), weight- and number-average molecular weights (LC Äkta Explorer equipped with RI and multiangle LS DAWN-DSP-F (Wyatt Technology) detectors using Superose 6 (Pharmacia) column), and content of Dox (UV–VIS spectrophotometry, ε = 11,500 l/mol/cm, λ = 488 nm, water).
The conjugate was tested for the contents of free polymer, free drug or free antibody by combination of methods: FPLC, electrophoresis (Pharmacia-LKB Phast System, SDS-PAGE) and extraction combined with HPLC. Neither method showed significant amounts of free antibody or free Dox in the conjugate. Characteristics of the conjugate: composition of the spacer Gly:L-Phe:D-Phe:L-Leu 2.03:0.53:0.47:1.0; molecular weight of the polymer (polymer precursor) M w = 25.0 kDa, molecular weight of the conjugate M w = 900 kDa, M w/M n ∼ 4, Dox content = 4.75 wt%, HuIg content = 16 wt%, content of free Dox < 1% of the total Dox content.
Proliferation assay in vitro
The subline EL-4.IL-2 was chosen for in vitro assays because the original cell line EL-4, forming solid tumours in mice does not incorporate [3H]thymidine at a sufficient quantity. The cells were collected from an exponential growth phase culture, washed, and loaded into 96-well culture plates (Nunc, Denmark). The tested samples were then added (0.05 ml) so as to achieve the desired concentrations and 0.25 ml final volume, 5 × 104 cells per well. The plates were cultivated for 2 days, with 0.5 μCi of [3H]thymidine added in 0.05 ml for the last 4 h. The cell proliferation was evaluated through the use of automatic cell harvestor (Tomtec Mach III, USA), and solid scintillator Meltilex (Wallac, Finland) for counting in 1450 Microbeta Trilux (Wallac, Finland).
Treatment, tumour protection assay
Therapeutic setting
Drugs were administered when the tumours were well established (day 8–9 post-transplantation), either as a single dose administered intravenously (i.v.; 10–20 mg Dox eq./kg) or divided into five or ten doses given intraperitoneally (i.p.). Only mice with developed tumour of appropriate size were selected and randomized for the therapeutic regimen experiments. Early treatment setting: A total dose of 20 mg Dox eq./kg divided into five equivalent doses, given i.p. every other day starting on day 1 after tumour transplantation. Commercial dosage form of Dox was used (Adriblastina CS, Pharmacia & Upjohn, USA), diluted in sterile saline before injection. Detailed dosage and times of administration are given in Results. The doses of polymeric conjugates were determined as equivalents of Dox/kg, based on the mean body weight in every experimental group at the time of the first drug administration.
The treated mice surviving at least 60 days without tumour were considered long-term survivors (LTS). To evidence their tumour resistance, the LTS were injected with 1 × 105 living EL-4 cells or 1 × 106 B16-F10 cells s.c. and left without treatment. Tumour growth and survival were monitored.
In vivo tumour neutralization (Winn’s assay)
To determine whether the LTS-derived lymphocytes were able to destroy living tumour cells, we performed the in vivo neutralization assays (Winn’s assay) [23, 24]. Spleens were removed from the cured mice, washed, counted and 1 × 107 cells mixed with the living 1 × 105 EL-4 cells (ratio 100:1) and injected s.c. to naïve mice. CD8+ cells were isolated from spleens using Spin Sep cell enrichment procedure (StemCell Technologies, USA), in which the CD8+ were negatively selected. FACS analysis revealed that 96% of the resulting population were CD3+CD4−CD8+ lymphocytes. CD8+ cells were mixed with 1 × 105 EL-4 at a ratio lymphocyte:tumour cell = 20:1 and 10:1, respectively, assuming that approximately 10% of normal spleen cell population is CD8+. Mice in the control group received the respective cell population taken from naïve donors together with the tumour cells. All mice in control groups became moribund at the same time as control mice receiving the tumour cells only.
Ag-specific response in vitro
Re-stimulation of LTS-derived spleen cells was performed using mitomycin C-treated EL-4 cells as the specific antigen. A total of 5 × 107 EL-4 cells/ml were incubated in PBS with 0.05 mg/ml fresh mitomycin C (Sigma, USA) in the dark for 20 min at 37°C, and thoroughly washed (four times) with RPMI-10 medium. The spleen cells (4 × 105) were incubated with 4 × 104 mitomycin-treated EL-4 cells in a 2 ml volume in 24-well culture plates (Nunc, Denmark) for 6 days. Mitomycin C-treated B16-F10 melanoma cells were used as a control stimulus and naïve spleen cells were employed for comparison with LTS-derived ones.
The cytotoxicity assay was performed with EL-4.IL-2 cells as the targets, labelled with [3H]thymidine (JAM technique) [25], using several effector:target ratios. The IFNγ detection in supernatants of re-stimulated cells was done by capture ELISA (antibody pair, Pharmingen, USA), protocol as recommended by the producer, and with murine rIFNγ (Peprotech, Asia) as a standard.
Histology
Tumours from treated animals and from untreated animals as a control were dissected at times specified in Results, as well as spleen, liver, lung, and kidney, fixed in 5% formalin and embedded in paraffin. The sections were stained with haematoxylin/eosin.
Statistical analysis
All in vivo experiments were repeated at least once, with similar results obtained. Results were expressed as means and SD, and the Student’s t test was used for evaluating statistical significance. P < 0.05 value was considered significant.
Results
In vitro activity of Dox bound to PHPMA copolymer
The Dox–PHPMA–HuIg (Fig. 1) conjugate was poorly active against EL-4.IL-2 lymphoma cells in vitro. Incubation of EL-4.IL-2 cells with Dox-containing polymeric conjugates and free Dox.HCl for 2 days showed a large difference in the sensitivity of the EL-4.IL-2 cells to polymer-bound and free Dox.HCl, respectively (Fig. 1). The IC50 values of polymeric conjugates with or without HuIg were similar.
Fig. 1.

Structure of the Dox–PHPMA–HuIg conjugate. The Dox–PHPMA–HuIg conjugate consists of the HPMA [poly-N-(2-hydroxypropyl)methacrylamide] backbone, which was modified by biodegradable oligopeptide side chains terminating in the targeting immunoglobulin molecules, and the drug (Dox) randomly distributed along the polymer chain. Structure of the Dox–PHPMA conjugate is identical, except that it does not contain the immunoglobulin molecule, and the side chains end with Dox only (z = 0). Comparison of the IC50 values of the free Dox and HPMA-based conjugates Dox–PHPMA and Dox–PHPMA–HuIg: IC50 values were calculated as the concentration of Dox which inhibits the incorporation of [3H]thymidine to 50% of the control level. The EL-4.IL-2 cells were incubated at 5 × 104 per well with different concentrations of the tested samples for 2 days, and proliferation of cells was measured by [3H]thymidine incorporation. Data are representative of at least three independent experiments
High IC50 values were in accordance with the assumed negligible binding capacity of the Dox–PHPMA–HuIg conjugate to EL-4 and EL-4.IL-2 lymphoma cells. This was also documented by FACS analysis which did not reveal any HuIg-mediated binding of the conjugate to the surface of EL-4 cells (not shown).
Anti-tumour effect of non-targeted polymer-bound Dox and HuIg-targeted Dox in vivo
The EL-4 tumours grew progressively in untreated B/6 mice, generally developing palpable tumours (5–8 mm in diameter) on day 8–9. At this time, percentage of mice with developed tumours generally exceeded 90%. Death of untreated controls occurred between 29 and 38 days (average survival time 32.4 days, SD 1.007, median 32; n = 62). No spontaneous tumour regression was recorded in untreated animals. Therapy schemes were largely designed as therapeutic, so that established tumours were treated, starting usually on day 9 after the transplantation. Dox–PHPMA–HuIg treatment significantly reduced the tumour growth rate in EL-4 lymphoma-bearing B/6 mice (Fig. 2a, c) and, consequently, extended the survival (Fig. 2b, d). The non-targeted conjugate Dox–PHPMA also suppressed the tumour growth, but was less effective than the HuIg-containing conjugate (Fig. 2a, b) in any in vivo assessment. Difference between the efficacy of the Dox–PHPMA and the Dox–PHPMA–HuIg conjugate was significant (Fig. 2a, b). On repeated administration, Dox-free vehicle (conjugate PHPMA–HuIg) did not convey any growth reduction advantage (Fig. 2a, b).
Fig. 2.

Tumour growth and survival of B/6 mice treated with Dox–PHPMA–HuIg. a, b The mice were transplanted s.c. with 1 × 105 EL-4 lymphoma cells (day 0) and treated in the therapeutic setting with Dox–PHPMA–HuIg, Dox–PHPMA, Dox-free vehiculum PHPMA–HuIg, and free Dox (Adriblastina CS, Pharmacia & Upjohn, USA). Total dose of polymeric conjugates was 20 mg Dox eq./kg, divided into five doses injected i.p. on days 12, 14, 16, 19, and 21. Free Dox was injected i.p. in a total dose of 12.5 mg/kg, time schedule as above. Statistical significance: *P < 0.05 and **P < 0.01. c, d The EL-4-bearing B/6 mice were treated with a single dose of the Dox–PHPMA–HuIg conjugate, Dox eq. 15 mg/kg, injected i.v. on day 10
Notably, the curative effects of the Dox–PHPMA–HuIg conjugate were evident at different treatment schedules (Table 1). We used either a single dose of the conjugate, injected i.v. on day 9 or 10, which led to a high conjugate concentration after the administration. The effectiveness of this treatment was proportional to the conjugate dose; with maximum dose equivalent of 20 mg Dox/kg injected i.v. This dose cured all treated mice, and was still tolerated by the recipients. On the other hand, fractionated dosage also proved to be successful. A total of 20 mg Dox eq. given as five partial doses i.p. produced 55.2% of cured mice and prolonged survival in the remaining mice due to treatment-induced tumour growth reduction (Table 1).
Table 1.
Therapeutic dosages and effectiveness of the treatment with Dox–PHPMA–HuIg conjugate in EL-4-lymphoma-bearing B/6 mice
| Total dose (mg Dox/kg) | Number of dosesa | Administration | Cured mice (%) | Mean survival of the treated mice that developed tumour (days) |
|---|---|---|---|---|
| 20 | 1 | i.v. | 100 | – |
| 20 | 5b | i.p. | 55.2 | 52.2c |
| 15 | 1 | i.v. | 81 | 52.8c |
| 15 | 10d | i.p. | 100 | –c |
| 10 | 1 | i.v. | 62.5 | 45.5c |
aSingle doses were injected as therapeutic, on day 9 after tumour transplantation
bAdministration of the conjugate on days 10, 12, 14, 17, and 19
cMean survival of untreated control mice with s.c. transplanted EL-4 lymphoma was 32.4 days (SD 1.007)
dAdministration of the conjugate on days 3, 6, 8, 10, 12, 14, 16, 18, 20, and 22
A lower Dox–PHPMA–HuIg conjugate dose (15 mg Dox/kg) producing 81% cures when given as a single injection i.v. proved fully curative when divided into ten consecutive low doses administered i.p. on days 3, 6, 8, 10, 12, 14, 16, 18, 20, and 22 (Table 1). In the course of the treatment, palpable tumours were manifested in all treated mice between days 10–14. All tumours regressed until day 20. Thus, lower levels of the drug administered over an extended time periods were also sufficient to completely suppress the tumour growth.
For comparison, the non-targeted Dox–PHPMA conjugate produced 12.5% of LTS using the fractionated dosage (20 mg Dox/kg injected in five doses i.p., days 10, 12, 14, 16, and 19), and 69% of LTS, using single dose (15 mg Dox/kg injected i.v., day 9). Lower dose of the Dox–PHPMA conjugate (10 mg Dox/kg injected i.v., day 9) cured only 25% of mice.
Activity of the Dox–PHPMA–HuIg conjugate in other in vivo models
In nude mice bearing the EL-4 lymphoma, the tumour growth reduction resulting from the Dox–PHPMA–HuIg therapy was remarkable until day 30 post-tumour transplantation. At that time, the tumours in the treated group started progressive growth (Fig. 3). Yet the treated mice revealed prolonged survival (mean 49, median 53.5 days vs. mean 37, median 37.5 days in untreated controls). No complete cure was recorded in nude mice after the treatment with the Dox–HPMA–HuIg conjugate.
Fig. 3.

Efficacy of the Dox–PHPMA–HuIg tretatment in other in vivo models. Tumour growth of EL-4 lymphoma in nu/nu mice: CD-1 nu/nu mice were transplanted s.c. on day 0 with 5 × 105 EL-4 and treated with five doses of Dox–PHPMA–HuIg (total 20 mg Dox eq./kg), injected i.p. on days 11, 14, 16, 18, and 21. No complete cure was recorded in nude mice with EL-4 lymphoma. Tumour growth in the B/6 mice with syngeneic melanoma B16-F10: 1 × 106 melanoma cells were transplanted s.c., and the treatment with Dox–PHPMA–HuIg was again therapeutic. Total dose 20 mg Dox eq./kg, injected on days 11, 14, 16, 18, and 21. No complete cure of melanoma was seen. Control—non-treated mice. Results of representative experiments are shown
In B/6 mice with another syngeneic tumour, B16-F10 melanoma, the therapeutic effect of the Dox–PHPMA–HuIg conjugate showed measurable slowing down of tumour progression (mean survival 50.5 days vs. 37.2 days in the untreated group, P < 0.05) (Fig. 3), but survival extension was less pronounced than in syngeneic EL-4 lymphoma model. No tumour regression was recorded in B/6 mice-bearing B16-F10 melanoma treated either with fractionated dosage (as in Fig. 3) or with a single dose eq. 15 mg Dox/kg (not shown).
Tumour resistance in EL-4-cured animals
Importantly, in a significant proportion of the Dox–HPMA–HuIg treated EL-4-bearing B/6 mice the tumours regressed. The regression was due to involution and resorption of the tumour mass. The proportion of the LTS depended on the treatment schedule (Table 2). The regimen using five partial doses given i.p. every other day (total dose 20 mg Dox eq./kg) was far less effective, but still cured more than a half of treated mice (in total, 55.2% of mice cured, n = 29). By contrast, the EL-4-bearing mice could be completely cured with one single dose (20 mg Dox eq./kg) of the conjugate, given i.v. on day 9 or 10. This was the highest single dose used in our experiments. It cured 100% mice without any early toxic death (Table 2), but was near the maximum tolerated dose, because the body weight of the treated mice dropped significantly. The decrease was 20%, peaking 7 days after the conjugate administration.
Table 2.
Effectiveness of the primary treatment of EL-4 lymphomas in C57BL/6 mice and proportion of tumour-resistant long-term survivors
| Treatment with Dox–PHPMA–HuIg | Pa | Tb | Tb | Tb | Tb | Tb |
|---|---|---|---|---|---|---|
| Dose (Dox eq. mg/kg) | 5 ×, total 20 | 5 ×, total 20 | 1 ×, total 10 | 1 ×, total 15 | 2 ×, total 15 | 1 ×, total 20 |
| Route of administration | i.p. | i.p. | i.v. | i.v. | i.v. | i.v. |
| Complete regression of primary tumour (%) | 80 | 55.2 | 62.5 | 81 | 88.9 | 100 |
| Mice resistant to a second tumour (%)c | 0 | 100 | ND | 66.7 | ND | 50 |
Summary data from all experiments are listed
P protective treatment scheme, T therapeutic scheme, ND not determined
aThe therapy started on day 1 after tumour transplantation and was given as five equal doses injected every other day
bThe first or the single dose was given when tumours were well established, usually on day 9–10 post-transplantation
cThe resistance in long-term survivors was verified by the injection of 1 × 105 EL-4 tumour cells s.c. After the tumour transplantation, the mice were left without treatment
In any of our experiments, irrespective of the conjugate dose and therapeutic regimen, more than 50% of the cured mice developed effective protection against the same tumour, as they survived a subsequent challenge by a lethal dose of viable tumour cells (Table 2). The percentage of the mice surviving the tumour re-challenge indirectly reflected the efficacy of the primary therapy (Table 2). Sixty-six per cent of the LTS re-transplanted with the same tumour as late as 6 months after the primary therapy were still resistant to the same tumour.
The presence of a significant load of tumour cells during the treatment course was apparently important for the protection development. This was confirmed in experiments using early treatment regimen of the treatment. EL-4 bearing mice were efficiently cured (80%, n = 10) when the Dox–PHPMA–HuIg conjugate was injected five times every other day (total 20 mg Dox eq./kg) starting on day 1 after the tumour transplantation (Table 2). However, the cured mice revealed no tumour protection, as they did not survive the subsequent tumour challenge and died exactly as control untreated mice.
The protection was specific for the original tumour. Mice that previously rejected the EL-4 tumour due to the Dox–HPMA–HuIg therapy were subsequently challenged either by the original tumour (1 × 105 EL-4 cells s.c.), or by another syngeneic tumour (1 × 106 B16-F10 cells s.c) Fig. 4. The LTS re-challenged by the same tumour were protected and did survive without any tumour growth (Fig. 4a). By contrast, all LTSs cured from EL-4 lymphoma and transplanted with a tumour of different type (syngeneic melanoma B16-F10), finally developed tumours and died (Fig. 4b). The tumour growth was delayed (Fig. 4b, inset) and survival time was longer compared to melanoma-bearing mice with no previous tumour history (Fig. 4b), suggesting that multiple mechanisms were involved in the control of the tumour growth.
Fig. 4.

Survival of EL-4-cured LTS, transplanted with second tumour. a The mice were re-transplanted with the same tumour, i.e. 1 × 105 EL-4 cells s.c. Primary treatment of the mice was as in Fig. 1a, b. Control—naïve mice transplanted with the same number of EL-4 cells. b EL-4-cured LTSs as in (a) were transplanted with non-cross-reacting melanoma B16-F10 to demonstrate the specificity of the anti-tumour memory. Control—naïve mice transplanted with the B16-F10 melanoma, 1 × 106 cells s.c. The difference in the survival between LTS and control mice was not statistically significant. b (inset) Growth of the B16-F10 melanoma tumours in EL-4-cured LTSs and in control mice
Transfer of LTS-derived lymphocytes to naïve mice
Transfers of cells derived from cured mice to naïve recipient were performed to document that the anti-tumour protection was mediated by lymphocytes. The mice that had previously rejected the EL-4 tumour due to the Dox–PHPMA–HuIg therapy were donors of spleen lymphocytes. In in vivo neutralization assay (Winn’s assay), in which the lymphocytes were injected to naïve recipients s.c. together with 1 × 105 live EL-4 cells, lymphocytes of Dox–PHPMA–HuIg-cured mice prevented tumour development in the naïve recipients (Fig. 5). Given that the lymphocytes were taken from donors that proved resistant by repeated challenge with the EL-4 lymphoma, the protection rate was 100%. If the lymphocyte donors were mice cured from the EL-4 lymphoma with a single administration of the Dox–PHPMA–HuIg conjugate, the protection of naïve recipients was less effective, but still significant (Fig. 5).
Fig. 5.

Winn’s assay. Recipients were naïve B/6 mice, injected s.c. with donor-derived cells together with 1 × 105 EL-4 lymphoma cells. The cells were injected in a small volume (0.1 ml), mixed and kept on ice until the injection. Donor’s cells: spleen cells of mice cured from the EL-4 lymphoma (a single dose of Dox–PHPMA–HuIg conjugate eq. 15 mg Dox/kg on day 9 post-tumour transplantation). The spleen cells for the Winn’s test were isolated on day 45 post-transplantation. CD8+ cells were isolated from the above suspension using the Spin Sep procedure (StemCell Technologies, USA). Resistant donors were mice cured with ten doses of the Dox–PHPMA–HuIg conjugate, total dose eq. 15 mg Dox/kg (see Table 1), re-transplanted with the EL-4 lymphoma 70 days after the primary transplantation, and used as donors for Winn’s assay another 94 days after the second tumour. Number of transferred cells: 1 × 107 spleen cells of cured mice or spleen cells of resistant donors were injected together with 1 × 105 EL-4 lymphoma cells (100:1). Number of CD8+ cells was 2 × 106 (20:1), and 1 × 106 (10:1), respectively. For the control, 1 × 107 naïve spleen cells were used as a donor cell population, and 1 × 105 EL-4 alone
We also performed experiments with isolated CD8+ cells. On day 45, post-tumour transplantation, the CD8+ lymphocytes were isolated from spleens of B/6 mice after complete tumour regression following the treatment with Dox–PHPMA–HuIg (15 mg Dox eq./kg). 42.9% (three out of seven) mice injected with 2 × 106 CD8+ lymphocytes and 1 × 105 EL-4 were fully protected by transferred cells, as they did not develop any tumour. The recipient protection depended on the number of transferred CD8+ cells, as animals receiving only 1 × 106 of CD8+ cells revealed almost no protection (Fig. 5). A prolonged lifespan and survival > 50 days after the cell transfer was recorded in just one mouse of this group, albeit with a slowly growing tumour.
In vitro activities of LTS-derived lymphocytes
The cytotoxic response of spleen cells freshly isolated from spleens of EL-4-cured animals 60 days after the tumour transplantation was negligible, and so was the response of naïve cells. When the LTS-derived lymphocytes were re-stimulated in vitro by cultivation with mitomycin C-treated EL-4 cells and/or mitomycin C-treated B16-F10 melanoma cells as a control, an enhancement of the specific cytotoxic response over the naïve splenocytes was evident (Fig. 6a). The LTS-derived lymphocytes killed more EL-4 cells than naïve lymphocytes, while the non-specific response in LTS-derived and naïve cells was comparable. However, the difference between LTS-derived and naïve cells was rather small. IFNγ detection in supernatants of the in vitro re-stimulated cells revealed far stronger differences between immune (LTS-derived) and non-immune (naïve) cells (Fig. 6b). The IFNγ secretion was faster in LTS, as the EL-4-restimulated splenocytes of LTS showed high IFNγ production soon (24 h, Fig. 6b) after in vitro re-stimulation, while naïve splenocytes did not produce substantial levels of IFNγ. The IFNγ production was tested at further intervals until 4 days after the in vitro re-stimulation. At all intervals, the LTS-derived spleen cells produced higher IFNγ levels than their naïve counterparts (not shown).
Fig. 6.

In vitro activity of spleen cells derived from EL-4-cured LTS. Spleen cells were re-stimulated in vitro by cultivation either with mitomycin C-treated EL-4 cells, or with non-cross-reacting mitomycin C-treated melanoma cells B16-F10. The responding spleen cells were incubated at 4 × 105 together with 4 × 104 stimulatory mitomycin-treated EL-4 (B16-F10) cells in a volume of 2 ml in 24-well culture plates. a After 6 days of cultivation, the cells were washed and their cytotoxic capacity was determined using JAM test [25]. The target cells EL-4.IL-2 were labelled by incubation with 5 μCi [3H]thymidine for 4 h at 37°C, 5% CO2, thoroughly washed and used as target cells at 1 × 104 per well. The results were calculated as percentage of target cell killing at a ratio of E:T = 40:1. Naïve spleen cells were used as a control. b IFNγ concentrations in supernatants of spleen cells of the LTS and of naïve mice, (re-)stimulated in vitro as in (a). The supernatant samples were assayed after 24 h of cultivation by capture ELISA, using a pair of monoclonal antibodies and murine rIFNγ as a standard. Statistical significance **P < 0.01
Histological analysis of regressing tumours
Histological analysis of the tumours revealed that in EL-4 lymphoma-bearing mice the cancer cells propagated to organs in the far progressed stages of the tumour development. As late as 3–4 weeks after the tumour cell transplantation, overt micrometastases appeared in spleens of untreated EL-4-bearing mice. Along with the appearance of compact tumour infiltrates in the spleen, the architecture of white pulp follicles was effaced, pointing to probable impairment of immune responses together with the propagation of the tumour. This finding conforms to our previous finding that in EL-4-bearing mice, some antibodies against the tumour cell were found in the sera of animals treated with HPMA-based Dox-containing conjugate, and the titre peaked on day 15 post-tumour transplantation [26]. Since in these mice the subcutaneous tumours after the day 15 formed a mass greater than 1 cm in diameter, it could be emphasized that later in the tumour development the antibodies were absorbed by binding to the tumour.
A continuous collateral layer was found at the periphery of tumours regressing due to treatment with the Dox–PHPMA–HuIg (10 mg Dox/kg, injected i.v. on day 10, see Table 1). The layer was formed by mononuclear cells, probably of inflammatory origin (Fig. 7a). On the contrary, progressively growing tumours of non-treated animals only showed scarce to minimal round-cell demarcation, although some perifocal oedema and scattered patches of haemorrhage were seen (Fig. 7b). The same was found by the analysis of tumour in one Dox–PHPMA–HuIg-treated mouse; the tumour growth was arrested due to the treatment (10 mg Dox/kg, injected i.v. on day 10), but finally resumed its growth (Fig. 7c).
Fig. 7.
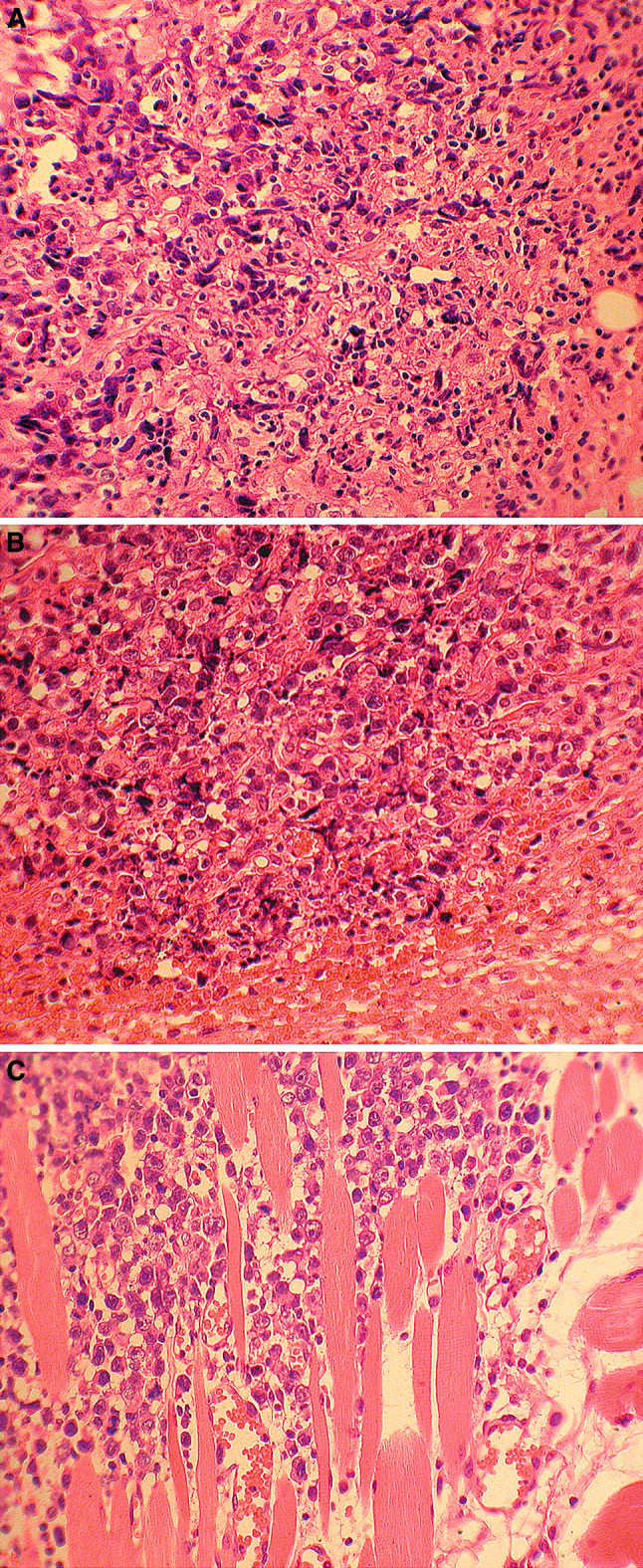
Histological analysis (haematoxylin/eosin staining). a Regressive changes of tumour: prominent cell polymorphism and shrinkage (left). Round cell inflammatory border within and around the periphery of the tumour infiltrate (right centre, b). Treatment: Dox–PHPMA–HuIg conjugate, 10 mg Dox/kg i.v., day 10. The mouse was killed for histological analysis of organs on day 21 post-transplantation. b Periphery of the tumour focus displaying round anaplasic cells with indistinct cytoplasm. Some elements undergo pyknosis and shrinkage, but widespread necrosis and consistent inflammatory border are lacking. Perifocal edema and hemorrhage (bottom and right). Treatment: saline (control, a); histology, day 21. c Detail of tumour periphery invading the dorsal striated muscle (top left), slight perifocal edema (bottom right) without structures of inflammatory demarcation wall. Treatment: as in (a), the tumour growth was arrested and finally resumed its growth. Histology, day 32 post-transplantation
Discussion
The Dox–PHPMA–HuIg conjugate has been designed with the aim of treating patients with solid tumours. In humans, identifying suitable tumour-associated antigens for targeting is still limited for most cancers, and the results so far obtained from clinical trials are not satisfactory to become good Ag-specific therapy [27]. The rationale for using intravenous immunoglobulin (IVIg), an effective therapy of autoimmune conditions, in the treatment of cancer stems from the bi-directional relationship between autoimmunity and cancer. IVIg exerts certain immunomodulatory effect [28]. It is believed that it operates in many different ways, which probably act together in a synergistic manner. Some anti-cancer effect of the human IVIg has already been described in experimental mouse system with tumours of mouse and/or human origin [29]. Enhanced IL-12 production by mononuclear cells in vitro was shown [29], as well as an inhibition of matrix metallopreoteinase-9 production in macrophages [30]. We suggest that a possible effect of the IVIg itself could occur locally rather than systemically. We know that Intraglobin F injected together with the Dox–PHPMA conjugate at a dose equivalent to that injected in the Dox–PHPMA–HuIg conjugate to EL-4-bearing B/6 mice did not alter the anti-cancer effect of the conjugate (unpublished observation). It was published that IVIg induced apoptosis of cells of T cell lymphoma, B cell lymphoma via Fas–Fas ligand and caspase activation and increased Fas protein expression [31], or increased arrest of cells in the G1 phase [32]. The effect of IVIg was ambivalent, as the pro-apoptotic and anti-proliferative effects were more pronounced in transformed cells than in small, non-activated mononuclear cells [31, 32], and could differ in different cell lines. Mechanisms of action of the Dox–PHPMA–HuIg conjugate on the cellular and subcellular level are still poorly understood and remain to be elucidated.
A pilot clinical study was conducted in Czech Republic in patients with generalized cancers refractory to conventional therapy. In the first two patients, we used autologous IgG for passive/active targeting of farmorubicin and Dox [26]. To eliminate the disadvantages of the individualized therapy and allow repeated administrations of the conjugate, the autologous IgG was then replaced with commercially available human IVIg (Intraglobin F). The results revealed that “non-specific” immunoglobulin used as the passive/active targeting moiety of the polymer-bound cytotoxic drug could considerably improve the therapy of solid tumours.
Here, we present data illustrating the efficacy of treatment of murine EL-4 lymphoma with Dox–PHPMA–HuIg conjugate. The conjugate was highly effective in a wide spectrum of therapeutic schemes and dosages. The effect was evidenced by tumour growth reduction, prolonged survival time, and complete regression of tumours in a significant proportion of animals. The conjugate was effective either at a high concentration originating from a single administration of a high dose, or at a lower concentration persisting for an extended time period in the organism. This we consider as one of the most important advantages over the non-targeted conjugate Dox–HPMA and namely free Dox.HCl, as both the Dox-HPMA and free cytotoxic substance Dox.HCl produced cured animals only at a high dosage. Moreover, the free drug administered in a therapeutically efficient dosage elicited in treated animals late life-threatening toxicity [33].
Protocols effective against well established EL-4 lymphoma tumours in B/6 mice have already been reported leading to a proportion of tumour-protected (immune) LTS, as evidenced by rejection of the same re-implanted tumour. All the protocols used a combination of a cytostatic/cytotoxic treatment with immunomodulation by a cytokine: cyclophosphamide plus TNFα [34–36], Dox plus TNFα [37, 38], Dox plus interleukin-2 [39, 40]. Combination of Dox and IL-2 was also effective in murine renal cell carcinoma [41]. In our hands, the therapeutic effect on the EL-4 lymphoma tumours was conferred solely by the Dox–PHPMA–HuIg conjugate. The tumour growth-reducing capacity was substantial and the therapy led to > 50% survivors effectively protected against tumour re-growth.
The conjugate most probably combines the effect of a direct toxic hit against the tumour with an effective stimulation of immunocompetent cells. High molecular weight of the conjugate facilitates its accumulation in the tumour tissue due to EPR effect [3–6]. This is corroborated by the unquestioned stability of the HPMA-based conjugates in the circulation [42–44]. Long persistence of the Dox–PHPMA–HuIg conjugate in the peripheral blood was documented also in our pilot study [18]. The limited side-toxic effect of HPMA-based macromolecular therapeutics against bone marrow, heart, thymus, kidney, and liver has been repeatedly described [8–10, 18, 26]. Tumour destruction provides a burst of tumour-associated antigens to induce the anti-tumour immune responses in individuals with unimpaired immune system. The importance of the antigenic load was confirmed in experiments with the early treatment, where the exposure of the host to the cancer cells was much lower than that in the treatment of pre-established tumours. We have shown a similar phenomenon in another murine tumour treated with HPMA-based Dox conjugate. In BCL1 mouse lymphoma model [45], the lymphoma-bearing mice were treated with a conjugate containing monoclonal antibody B1 specific to the tumour-associated idiotype expressed exclusively on the BCL1 cells. A negative correlation between the efficacy of the treatment and the development of resistance/protection was also seen in the BCL1 model. Systemic resistance was achieved in a proportion of mice cured in the therapeutic setting, after the disease had been well established (between days 7 and 11), but not in mice cured with a single dose of the conjugate given on day 3 [46, 47]. The conjugate administered on day 15 was non-effective, as the anti-tumour capacity of the immune system was already exhausted as a consequence of the tumour growth.
It was repeatedly documented that the PHPMA-bound Dox does not impair immune functions [8, 26, 48]. We have shown that spleen lymphocytes and also isolated CD8+ cells were able to neutralize the tumour cells in vivo. This is in accordance with the generally accepted idea of an important role of CD8+ cytotoxic T cells in eradication of tumours [49]. We demonstrated a small increase of specific cytotoxicity against EL-4 cells in the LTS, as compared to naïve mice. However, reports showing that T cells are non-lytic in culture but can acquire CTL activity in vivo exist [50–52]. The IFNγ production readily induced by the Ag-specific stimulus supports the view of CD8+-mediated immune response as the decisive mechanism in rejecting the tumours and maintaining the memory. The need for the intact specific cell-mediated immunity is also evident, as the Dox–PHPMA–HuIg treatment of EL-4 lymphoma in nu/nu mice did not produce any survivors.
The histological pictures must be interpreted with some caution. The untreated (or relapsing) lymphoma was a solid mass consisting of oval or polysomal poorly demarcated cells with light polymorph nuclei, prominent nucleoli and scattered mitoses. As usual in malignant tumours, dystrophy and necrosis of individual cells or circumscribed areas were seen, mainly in central areas of the tumour. However, most elements were well preserved in the invasive tumour periphery (cf. Fig. 7c). On the other hand, signs of cell dystrophy and disruption prevailed both in the central areas and at the periphery of treated tumour (cf. Fig. 7a, b). As for collateral oedema, haemorrhage and (rather scarce) inflammatory infiltration (Fig. 7b), they seem to represent the non-specific demarcation of degraded or necrotic tumour mass rather than a sign of cytotoxic cellular response. In fact, the cytotoxic reaction against the given tumour detected in situ always remained rather weak.
The B16-F10 melanoma was chosen for assessing the specificity of protection in the disease-free survivors, as the model of subcutaneous melanoma was widely used to evaluate the efficacy of HPMA-based polymeric drugs [53]. However, the B16-F10 melanoma is considered rather less sensitive to these therapeutics, which could be the reason for an incomplete cure seen in Dox–PHPMA–HuIg treated animals.
In summary, we propose a cancer treatment strategy using a single compound containing “non-specific” immunoglobulin, which has its own anti-tumour efficacy together with a protective and, possibly even stimulatory effect on the immune anti-cancer responses. This may bring benefit to patients by suppressing or eradicating tumours without harming potential immune responses protecting the host against a potential disease relapse. Considering potential introduction of the conjugate into clinical use, certain contradictions between experiment and clinical practice become obvious. First, the treatment must be aimed at the highest cure rate, regardless of the potentially less effective resistance in the future. Second, the concept of low immunogenicity of tumours in humans is generally accepted. Speculatively, chances to corroborate the anti-cancer responses after the primary treatment by means of vaccines based on gene-modified tumour cells [54], or tumour antigen/tumour derived peptide-based vaccines [55], exist. Definitely, the treatment with polymer-bound drugs such as with the Dox–PHPMA–HuIg conjugate leaves the immune system with no serious harm, and therefore capable of producing next responses.
Acknowledgements
The project was supported by the Ministry of Education, Youth and Sports (grant 1M 4635608802), the Czech Science Foundation (grant 305/05/2268), and the Institutional Research Concept AV0Z50200510. We thank Prof. I. Lefkovits for helpful suggestions, and Ms. H. Mišurcová and Ms. H. Semorádová for excellent technical assistance.
Abbreviations
- HPMA
N-(2-hydroxypropyl)methacrylamide
- PHPMA
N-(2-hydroxypropyl)methacrylamide copolymer
- Dox
Doxorubicin
- HuIg
Human intravenous immunoglobulin
- IVIg
Intravenous immunoglobulin
- NK
Natural killer cells
- LAK
Lymphokine-activated killers
- LTS
Long-term survivor
- MMC
Mitomycin C
References
- 1.Ehrlich P. Studies in immunity. New York: Plenum; 1906. [Google Scholar]
- 2.Ringsdorf H. Structure and properties of pharmacologically active polymers. J Poly Sci Polym Symp. 1975;101:135–153. [Google Scholar]
- 3.Maeda H, Matsumura Y. Tumoritropic and lymphotropic principles of macromolecular drugs. Crit Rev Ther Drug Carrier Syst. 1989;6:193–210. [PubMed] [Google Scholar]
- 4.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271–284. doi: 10.1016/S0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 5.Noguchi Y, Wu J, Duncan R, Strohalm J, Ulbrich K, Akaike T, Maeda H. Early phase tumor accumulation of macromolecules: a great difference in clearance rate between tumor and normal tissues. Jpn J Cancer Res. 1998;89:307–314. doi: 10.1111/j.1349-7006.1998.tb00563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu J, Akaike T, Hayashida K, Okamoto T, Okuyama A, Maeda H. Enhanced vascular permeability in solid tumor involving peroxynitrite and matrix metalloproteinases. Jpn J Cancer Res. 2001;92:439–451. doi: 10.1111/j.1349-7006.2001.tb01114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hortobagyi GN. Anthracyclines in the treatment of cancer. An overview. Drugs. 1997;54(Suppl 4):1–7. doi: 10.2165/00003495-199700544-00003. [DOI] [PubMed] [Google Scholar]
- 8.Rihova B, Kopeckova P, Strohalm J, Rossmann P, Vetvicka V, Kopecek J. Antibody-directed affinity therapy applied to the immune system: in vivo effectiveness and limited toxicity of daunomycin conjugated to HPMA copolymers and targeting antibody. Clin Immunol Immunopathol. 1988;46:100–114. doi: 10.1016/0090-1229(88)90010-4. [DOI] [PubMed] [Google Scholar]
- 9.Rossmann P, Rihova B, Strohalm J, Ulbrich K. Morphology of rat kidney and thymus after native and antibody-coupled cyclosporin A application (reduced toxicity of targeted drug) Folia Microbiol (Praha) 1997;42:277–287. doi: 10.1007/BF02819003. [DOI] [PubMed] [Google Scholar]
- 10.St’astny M, Ulbrich K, Strohalm J, Rossmann P, Rihova B. Abnormal differentiation of thymocytes induced by free cyclosporine is avoided when cyclosporine bound to N-(2-hydroxypropyl)methacrylamide copolymer carrier is used. Transplantation. 1997;63:1818–1827. doi: 10.1097/00007890-199706270-00020. [DOI] [PubMed] [Google Scholar]
- 11.St’astny M, Strohalm J, Plocova D, Ulbrich K, Rihova B. A possibility to overcome P-glycoprotein (PGP)-mediated multidrug resistance by antibody-targeted drugs conjugated to N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer carrier. Eur J Cancer. 1999;35:459–466. doi: 10.1016/S0959-8049(98)00373-6. [DOI] [PubMed] [Google Scholar]
- 12.Kopecek J, Kopeckova P, Minko T, Lu Z. HPMA copolymer-anticancer drug conjugates: design, activity, and mechanism of action. Eur J Pharm Biopharm. 2000;50:61–81. doi: 10.1016/S0939-6411(00)00075-8. [DOI] [PubMed] [Google Scholar]
- 13.Jelinkova M, Strohalm J, Plocova D, Subr V, St’astny M, Ulbrich K, Rihova B. Targeting of human and mouse T-lymphocytes by monoclonal antibody-HPMA copolymer–doxorubicin conjugates directed against different T-cell surface antigens. J Control Release. 1998;52:253–270. doi: 10.1016/S0168-3659(97)00210-1. [DOI] [PubMed] [Google Scholar]
- 14.Kovar M, Strohalm J, Ulbrich K, Rihova B. In vitro and in vivo effect of HPMA copolymer-bound doxorubicin targeted to transferrin receptor of B-cell lymphoma 38C13. J Drug Target. 2002;10:23–30. doi: 10.1080/10611860290007496. [DOI] [PubMed] [Google Scholar]
- 15.Kanwar J, Berg R, Lehnert K, Krissansen G. Taking lessons from dendritic cells: multiple xenogeneic ligands for leukocyte integrins have the potential to stimulate anti-tumor immunity. Gene Ther. 1999;6:1835–1844. doi: 10.1038/sj.gt.3301016. [DOI] [PubMed] [Google Scholar]
- 16.Hersey P. Impediments to successful immunotherapy. Pharmacol Ther. 1999;81:111–119. doi: 10.1016/S0163-7258(98)00038-2. [DOI] [PubMed] [Google Scholar]
- 17.Griffioen AW, Damen CA., Mayo KH, Barendsz-Janson AF, Martinotti S, Blijham GH, Groenewegen G. Angiogenesis inhibitors overcome tumor induced endothelial cell anergy. Int J Cancer. 1999;80:315–319. doi: 10.1002/(SICI)1097-0215(19990118)80:2<315::AID-IJC23>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 18.Rihova B, Strohalm J, Prausova J, Kubackova K, Jelinkova M, Rozprimova L, Sirova M, Plocova D, Etrych T, Subr V, Mrkvan T, Kovar M, Ulbrich K. Cytostatic and immunomobilizing activities of polymer-bound drugs: experimental and first clinical data. J Control Release. 2003;91:1–16. doi: 10.1016/S0168-3659(03)00235-9. [DOI] [PubMed] [Google Scholar]
- 19.Rihova B, Kubackova K. Clinical implications of N-(2-hydroxypropyl)methacrylamide copolymers. Curr Pharm Biotechnol. 2003;4:311–322. doi: 10.2174/1389201033489711. [DOI] [PubMed] [Google Scholar]
- 20.Fidler IJ. Selections of successive tumour lines for metastasis. Nat New Biol. 1973;242:148–149. doi: 10.1038/newbio242148a0. [DOI] [PubMed] [Google Scholar]
- 21.Ulbrich K, Subr V, Strohalm J, Plocova D, Jelinkova M, Rihova B. Polymeric drugs based on conjugates of synthetic and natural macromolecules. I. Synthesis and physico-chemical characterisation. J Control Release. 2000;64:63–79. doi: 10.1016/S0168-3659(99)00141-8. [DOI] [PubMed] [Google Scholar]
- 22.Rihova B, Bilej M, Vetvicka V, Ulbrich K, Strohalm J, Kopecek J, Duncan R. Biocompatibility of N-(2-hydroxypropyl) methacrylamide copolymers containing adriamycin. Immunogenicity, and effect on haematopoietic stem cells in bone marrow in vivo and mouse splenocytes and human peripheral blood lymphocytes in vitro. Biomaterials. 1989;10:335–342. doi: 10.1016/0142-9612(89)90075-6. [DOI] [PubMed] [Google Scholar]
- 23.Winn HJ. Immune mechanisms in homotransplantation. II. Quantitative assay of the immunologic activity of lymphoid cells stimulated by tumor homografts. J Immunol. 1961;86:228–239. [PubMed] [Google Scholar]
- 24.Parajuli P, Pisarev V, Sublet J, Steffel A, Varney M, Singh R, LaFace D, Talmadge JE. Immunization with wild-type p53 gene sequences coadministered with Flt3 ligand induces an antigen-specific type 1 T-cell response. Cancer Res. 2001;61:8227–8234. [PubMed] [Google Scholar]
- 25.Matzinger P. The JAM test. A simple assay for DNA fragmentation and cell death. J Immunol Methods. 1991;145:185–192. doi: 10.1016/0022-1759(91)90325-A. [DOI] [PubMed] [Google Scholar]
- 26.Rihova B, Strohalm J, Kubackova K, Jelinkova M, Hovorka O, Kovar M, Plocova D, Sirova M, St’astny M, Rozprimova L, Ulbrich K. Acquired and specific immunological mechanisms co-responsible for efficacy of polymer-bound drugs. J Control Release. 2002;78:97–114. doi: 10.1016/S0168-3659(01)00489-8. [DOI] [PubMed] [Google Scholar]
- 27.Kawakami Y, Fujita T, Matsuzaki Y, Sakurai T, Tsukamoto M, Toda M, Tsumimoto H. Identification of human tumor antigens and its implications for diagnosis and treatment of cancer. Cancer Sci. 2004;95:784–791. doi: 10.1111/j.1349-7006.2004.tb02182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sapir T, Shoenfeld Y. Facing the enigma of immunomodulatory effects of intravenous immunoglobulin. Clin Rev Allergy Immunol. 2005;29:185–199. doi: 10.1385/CRIAI:29:3:185. [DOI] [PubMed] [Google Scholar]
- 29.Shoenfeld Y, Fishman P. Gamma-globulin inhibits tumor spread in mice. Int Immunol. 1999;11:1247–1252. doi: 10.1093/intimm/11.8.1247. [DOI] [PubMed] [Google Scholar]
- 30.Shapiro S, Shoenfeld Y, Gilburd B, Sobel E, Lahat N. Intravenous gamma globulin inhibits the production of matrix metalloproteinase-9 in macrophages. Cancer. 2002;95:2032–2037. doi: 10.1002/cncr.10905. [DOI] [PubMed] [Google Scholar]
- 31.Prasad NK, Papoff G, Zeuner A, Bonnin E, Kazatchkine MD, Ruberti G, Kaveri SV. Therapeutic preparations of normal polyspecific IgG (IVIg) induce apoptosis in human lymphocytes and monocytes: a novel mechanism of action of IVIg involving the Fas apoptotic pathway. J Immunol. 1998;161:3781–3790. [PubMed] [Google Scholar]
- 32.Ekberg C, Nordstrom E, Skansen-Saphir U, Mansouri M, Raqib R, Sundqvist VA, Fernandez C. Human polyspecific immunoglobulin for therapeutic use induces p21/WAF-1 and Bcl-2, which may be responsible for G1 arrest and long-term survival. Hum Immunol. 2001;62:215–227. doi: 10.1016/S0198-8859(00)00250-0. [DOI] [PubMed] [Google Scholar]
- 33.Mrkvan T, Sirova M, Etrych T, Chytil P, Strohalm J, Plocova D, Ulbrich K, Rihova B. Chemotherapy based on HPMA copolymer conjugates with pH-controlled release of doxorubicin triggers anti-tumor immunity. J Control Release. 2005;110:119–129. doi: 10.1016/j.jconrel.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 34.Krawczyk CM, Verstovsek S, Ujhazy P, Maccubbin D, Ehrke MJ. Protective specific immunity induced by cyclophosphamide plus tumor necrosis factor alpha combination treatment of EL4-lymphoma-bearing C57BL/6 mice. Cancer Immunol Immunother. 1995;40:347–357. doi: 10.1007/BF01525385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ehrke MJ, Verstovsek S, Pocchiari SK, Krawczyk CM, Ujhazy P, Zaleskis G, Maccubbin D, Meer JM, Mihich E. Thymic anti-tumor effectors in mice cured of lymphoma by cyclophosphamide plus TNF-alpha therapy: phenotypic and functional characterization up to 20 months after initial tumor inoculation. Int J Cancer. 1998;76:579–586. doi: 10.1002/(SICI)1097-0215(19980518)76:4<579::AID-IJC22>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 36.Ehrke MJ, Verstovsek S, Krawczyk CM, Ujhazy P, Zaleskis G, Maccubbin DL, Mihich E. Cyclophosphamide plus tumor necrosis factor-alpha chemoimmunotherapy cured mice: life-long immunity and rejection of re-implanted primary lymphoma. Int J Cancer. 1995;63:463–471. doi: 10.1002/ijc.2910630327. [DOI] [PubMed] [Google Scholar]
- 37.Ehrke MJ, Verstovsek S, Maccubbin DL, Ujhazy P, Zaleskis G, Berleth E, Mihich E. Protective specific immunity induced by doxorubicin plus TNF-alpha combination treatment of EL4 lymphoma-bearing C57BL/6 mice. Int J Cancer. 2000;87:101–109. doi: 10.1002/1097-0215(20000701)87:1<101::AID-IJC15>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 38.Ehrke MJ, Verstovsek S, Ujhazy P, Meer JM, Eppolito C, Maccubbin DL, Mihich E. Doxorubicin plus tumor necrosis factor alpha combination treatments in EL4-lymphoma-bearing C57BL/6 mice. Cancer Immunol Immunother. 1998;45:287–298. doi: 10.1007/s002620050445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ehrke MJ, Verstovsek S, Zaleskis G, Ho RL, Ujhazy P, Maccubbin DL, Mihich E. Specific anti-EL4-lymphoma immunity in mice cured 2 years earlier with doxorubicin and interleukin-2. Cancer Immunol Immunother. 1996;42:221–230. doi: 10.1007/s002620050274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ho RL, Maccubbin D., Ujhazy P, Zaleskis G, Eppolito C, Mihich E, Ehrke MJ. Immunological responses critical to the therapeutic effects of adriamycin plus interleukin 2 in C57BL/6 mice bearing syngeneic EL4 lymphoma. Oncol Res. 1993;5:363–372. [PubMed] [Google Scholar]
- 41.Gautam SC, Chikkala NF, Ganapathi R, Hamilton TA. Combination therapy with adriamycin and interleukin 2 augments immunity against murine renal cell carcinoma. Cancer Res. 1991;51:6133–6137. [PubMed] [Google Scholar]
- 42.Vasey PA, Kaye SB, Morrison R, Wilson P, Twelves C, Duncan R, Thomson AH, Murray LS, Hilditch TE, Murray T, Burtles S, Fraier D, Frigerio E, Cassidy J. Phase I clinical and pharmacokinetic study of PK1 [N-(2-hydroxypropyl)methacrylamide copolymer doxorubicin]: first member of a new class of chemotherapeutic agents–drug–polymer conjugates. Cancer Research Campaign Phase I/II Committee. Clin Cancer Res. 1999;5:83–94. [PubMed] [Google Scholar]
- 43.Julyan PJ, Seymour LW, Ferry DR, Daryani S, Boivin CM, Doran J, David M, Anderson D, Christodoulou C, Young AM, Hesslewood S, Kerr DJ. Preliminary clinical study of the distribution of HPMA copolymers bearing doxorubicin and galactosamine. J Control Release. 1999;57:281–290. doi: 10.1016/S0168-3659(98)00124-2. [DOI] [PubMed] [Google Scholar]
- 44.Jelinkova M, Strohalm J, Etrych T, Ulbrich K, Rihova B. Starlike vs. classic macromolecular prodrugs: two different antibody-targeted HPMA copolymers of doxorubicin studied in vitro and in vivo as potential anticancer drugs. Pharm Res. 2003;20:1558–1564. doi: 10.1023/A:1026170830782. [DOI] [PubMed] [Google Scholar]
- 45.Kovar M, Mrkvan T, Strohalm J, Etrych T, Ulbrich K, St’astny M, Rihova B. HPMA copolymer-bound doxorubicin targeted to tumor-specific antigen of BCL1 mouse B cell leukemia. J Control Release. 2003;92:315–330. doi: 10.1016/S0168-3659(03)00340-7. [DOI] [PubMed] [Google Scholar]
- 46.Kovar M, Mrkvan T, Sirova M, Sťastny M, Etrych T, Strohalm J, Plocova D, Ulbrich K, Rihova B (2004) HPMA copolymer-bound doxorubicin targeted with monoclonal antibody to tumor-specific antigen of BCL1 leukemia cells can completely cure tumor-bearing mice and establish long-term immunological memory to the original tumor. 31st annual meeting and exposition of the Controlled Release Society. Honolulu, HI, USA
- 47.Rihova B, Strohalm J, Kovar M, Mrkvan T, Subr V, Hovorka O, Sirova M, Rozprimova L, Kubackova K, Ulbrich K. Induction of systemic antitumour resistance with targeted polymers. Scand J Immunol. 2005;62(Suppl 1):100–105. doi: 10.1111/j.1365-3083.2005.01617.x. [DOI] [PubMed] [Google Scholar]
- 48.Rihova B, Strohalm J, Hoste K. Immunoprotective therapy with targeted anticancer drugs. Macromol Symp. 2001;172:21–28. doi: 10.1002/1521-3900(200107)172:1<21::AID-MASY21>3.0.CO;2-C. [DOI] [Google Scholar]
- 49.Hanson HL, Donermeyer DL, Ikeda H, White JM, Shankaran V, Old LJ, Shiku H, Schreibe RD, Allen PM. Eradication of established tumors by CD8+ T cell adoptive immunotherapy. Immunity. 2000;13:265–276. doi: 10.1016/S1074-7613(00)00026-1. [DOI] [PubMed] [Google Scholar]
- 50.Miki S, Ksander B, Streilein JW. Studies on the minimum requirements for in vitro “cure” of tumor cells by cytotoxic T lymphocytes. Reg Immunol. 1992;4:352–362. [PubMed] [Google Scholar]
- 51.Ksander BR, Acevedo J, Streilein JW. Local T helper cell signals by lymphocytes infiltrating intraocular tumors. J Immunol. 1992;148:1955–1963. [PubMed] [Google Scholar]
- 52.Guerder S, Carding SR, Flavell RA. B7 costimulation is necessary for the activation of the lytic function in cytotoxic T lymphocyte precursors. J Immunol. 1992;155:5167–5174. [PubMed] [Google Scholar]
- 53.Seymour LW, Ulbrich K, Steyger PS, Brereton M, Subr V, Strohalm J, Duncan R. Tumour tropism and anti-cancer efficacy of polymer-based doxorubicin prodrugs in the treatment of subcutaneous murine B16F10 melanoma. Br J Cancer. 1994;70:636–641. doi: 10.1038/bjc.1994.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eager Nemunaitis R. J. GM-CSF gene-transduced tumor vaccines. Mol Ther. 2005;12:18–27. doi: 10.1016/j.ymthe.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 55.Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54:721–728. doi: 10.1007/s00262-004-0653-2. [DOI] [PMC free article] [PubMed] [Google Scholar]