

Abstract
It is generally believed that priming of efficient T-cell responses takes place in peripheral lymphoid tissues. Although this notion has been rigidly proven for infectious diseases, direct evidence for lymph node priming of in vivo T-cell responses against tumors is still lacking. In the present study, we conducted a full and nonbiased comparison of T-cell clonotypes in melanoma lesions and corresponding sentinel lymph nodes. Whereas most tumor lesions comprised a high number of T-cell clonotypes, only a small number of clonally expanded T cells were detected in the draining lymph nodes. Comparative clonotype mapping demonstrated the presence of identical T-cell clonotypes in the tumors and the respective sentinel lymph nodes, only when tumor cells were present in the latter. However, taking advantage of clonotype specific PCR amplification, TCR sequences representing clonally expanded T cells at the tumor site could be detected in the lymph nodes draining the tumors even in the absence of tumor cells. Evidence for the tumor-specific characteristics of these cells was obtained by in situ staining with peptide/HLA class I complexes demonstrating the presence of MART-1/HLA-A2- and MAGE-3/HLA-A2-reactive T cells at the tumor site, as well as in the draining lymph node. Our data indicate that T-cell responses to melanoma are primed in the sentinel lymph node by cross presentation of tumor antigens by dendritic cells.
Keywords: Human, Tumor immunity, T-Cell receptors, Sentinel lymph node, Peptide/MHC class I multimers
Introduction
A subset of human cancers are capable of eliciting immune responses in vitro, and several lines of evidence have demonstrated that neoplastic cells may be subjected to an immunological response in vivo [3, 34]. In this respect, tumor infiltrating lymphocytes (TIL) and peripheral blood lymphocytes from melanoma patients have been shown to comprise T cells specific for peptides derived from melanoma associated antigens (MAA) [20]. Nevertheless, the immunological response against cancer cells is in most cases inadequate, and tumor progression occurs.
The induction of T-cell responses is dependent upon proper antigen specific priming of naïve T cells by dendritic cells [13]. The transport, processing and presentation of peripheral antigen in the lymph node by the DC, and the subsequent priming of antigen specific T-cell responses represent one of the cornerstones in cellular immunology. The crucial role of the draining lymph node for the induction of localized T-cell reponses has been firmly established for viral infections [9, 14, 23], and has led to the notion that antitumor T-cell responses are initiated similarly. However, other ways of initiating tumor specific responses have been suggested. In murine models, it has been shown that tumor cells may posses the capacity to migrate through the lymphoid system and directly prime tumor specific T cells in the lymph node [35]. Additionally, under certain circumstances, DCs may prime T cells at the site of antigen capture, implying that T cell activation may actually occur at the tumor site provided that DCs are present [17, 29]. To scrutinize cellular immune responses to solid tumors in humans, we have analyzed for clonally expanded T cells in primary melanomas and the corresponding draining lymph nodes.
Materials and methods
Patients and tumor samples
A total of 12 patients were included in the present study. Fresh tissue samples from primary cutaneous melanomas and corresponding sentinel lymph nodes (SLN) were obtained by surgical excision. To this end, SLN is defined as the first lymph node draining from the tumor, detected, excised and divided as described by Gershenwald et al. [11]. Tumors were divided into two parts. One of these was snap frozen in liquid nitrogen immediately after surgery and stored at −80°C. The remaining part, which included the thickest portion of the tumor, was used for routine histology and diagnosis (Table 1). The SLN biopsy material was used for routine histology, immunohistochemistry and RT-PCR based detection of tyrosinase, Melan-A/MART-1, and Mage-3 mRNAs. Informed consent was obtained from all patients prior to any of these measures.
Table 1.
All tumors were superficial spreading melanoma (SSM)
| No | Birth | Localization | Level | Thickness | Ulceration | MAA exp./SLN | Metastasis | Recurrent TCR | Remarks |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1928 | Upper back | IV | 3.8 | + | − | − | + (PCR) | CR |
| 2 | 1934 | Lower leg, left | IV | 3.0 | + | + | − | + (DGGE) | CR |
| 3 | 1941 | Back | IV | 3.75 | − | + | − | + (DGGE) | CR |
| 4 | 1923 | Left flank | III | 1.1 | − | + | − | ND | CR |
| 5 | 1933 | Right shoulder | IV | 1.1 | − | + | − | − | CR |
| 6 | 1941 | Upper leg, left | IV | 1.3 | − | + | − | − | No record |
| 7 | 1950 | Chest | III | 1.15 | − | − | − | − | CR |
| 8 | 1969 | Back | III | 1.6 | + | ± | LN | − | No record |
| 9 | 1968 | Upper arm, left | IV | 1.1 | − | ± | − | − | CR |
| 10 | 1939 | Upper arm, right | IV | 1.6 | − | + | − | − | CR |
| 11 | 1953 | Abdomen, right | III | 1.25 | − | − | − | ND | No record |
| 12 | 1927 | Lower leg, right | III | 0.45 | − | − | LN | + (PCR) | No record |
A single SLN were removed in most patients, however, regarding patients 8, 9, 11, and 12, two SLNs removed. MAA expression in sentinel lymph node is given as positive when the sample were positive for at least one of the markers analyzed by RT-PCR (see Materials and methods). In some patients, two SLN were resected for which MAA expression is given separately (patients 8, 9, and 12). Patients with evidence of metastatic disease (patients 8 and 12), are both dead from disease (year 2000). In the remarks column some patients are denoted, “No record”. These patients never appeared for scheduled clinical evaluation. CR Complete remission
RT-PCR for the detection of MAA
RNA was extracted using the Purescript Isolation Kit (Gentra Systems Inc. MN). 2–5 μg of RNA were used in the synthesis of cDNA using the SuperScript II reverse transcriptase (Gibco-BRL, Life Technologies Inc., Gaithersburg, MD, USA) in a total volume of 50 μl 1× buffer (Gibco-BRL, Life Technologies Inc.) containing 10 mM dithiothreitol (DTT). The RT reactions were primed with a mixture of oligo-dT and random hexamers, and incubations were performed at 37°C for 30 min, 42°C for 30 min, and 72°C for 5 min. Amplification of MAA cDNA was carried out using primers specific for the MAAs tyrosinase, MART-1, and MAGE-3, as described [32].
RT-PCR for TCRBV analysis and denaturing gradient gel electrophoresis (DGGE)
RNA was isolated and cDNA prepared as described above. Quantitation of TCR cDNA in the each sample was carried out as described [31] and enabled the use of equal amounts of TCR template in all reactions. cDNA was amplified using a primer panel amplifying the 24 BV region families of the TCR in DNA fragments suitable for TCR clonotype mapping by DGGE [4]. Amplifications were carried out in a total volume of 25 μl containing 1× PCR buffer [50 mM KCl, 20 mM Tris pH 8.4, 2.0 mM MgCl2, 0.2 mM cresol red, 12% sucrose, 0.005% (wt/v) bovine serum albumin (BSA) (Boehringer-Mannheim)], 2.5 pmol of each primer, 40 mM dNTPs (Pharmacia LKB, Uppsala, Sweden) and 1.25 units of AmpliTaq polymerase (Perkin Elmer Cetus Corporation, Emeryville, CA, USA). Parameters used for amplification were 94°C for 60 s, 60°C for 60 s and 72°C for 60 s for 32 (quantitative PCR) or 40 cycles (DGGE). Taq polymerase and dNTPs were added to the reaction tube at an 80°C step between the denaturation and annealing steps of the first cycle (Hot start).
TCR clonotype mapping
For DGGE analysis, 10 μl aliquots at PCR product were loaded onto a denaturing gradient gel containing 6% polyacrylamide and a gradient of urea and formamide from 20% to 80%. Gels were run at 160 V for 4.5 h in 1× Tris acetate EDTA (TAE) buffer kept at a constant temperature of 58°C. After electrophoresis, the gel was stained with ethidium bromide and photographed under UV transillumination.
Sequencing
DNA bands that resolved at identical positions in the denaturing gradient gel were subjected to sequence analysis using the Thermo Sequenase cycle sequencing kit (Amersham, Life Science, Cleveland, USA). In brief, bands were excised from the denaturing gradient gel, and DNA was eluted in H2O and reamplified. An aliquot (0.2 μl) of the PCR product was used as template in a 40-cycle sequencing reaction with 33P end-labeled BC-region sequencing primer. Gels were dried under vacuum and exposed to a Storage Phosphor Screen.
Clonotype specific PCR and single-strand conformational polymorphism (SSCP)
Sequences revealed from expanded T-cell clonotypes detected in the tumor lesions were used to construct primers for subsequent PCR amplification of these specific transcripts in the sentinel lymph node. The primers were constructed such that the 3′-end of the primers was positioned in the region of N-addition specific for the particular TCR sequence. Sense primers were used together with the constant region antisense primer BC4 (5′-GACCGCGGGTGGGAACAC-3), antisense primers were used in conjunction with the upstream BV-primer [33]. Amplifications were carried out using cDNA from the tumor lesion as positive control, whereas PBL cDNA from several donors served as negative controls. Tumors and lymph nodes from two patients were analyzed by clonotype specific PCR; lymph nodes in both patients were shown to be negative for the expression of MAA (patients 1 and 12). Positive amplicons were analyzed by SSCP to verify sequence identity of sequences amplified in the tumor and the sentinel lymph node. For SSCP analyses, we used the NOVEX Xcell II Mini Cell (NOVEX, San Diego, CA, USA). Ten microliter aliquots were loaded onto the gel and electrophoresed for 4 h at 200 V at room temperature. The gel was stained in ethidium bromide and DNA visualized under ultraviolet light.
Construction of HLA-Peptide complexes for T-cell staining
A recognition site for enzymatic biotinylation using biotin protein ligase (BirA) in fusion with the 5′ end of the extracellular domains of HLA A*0201 (residues 1-275) was expressed in E. coli BL21 (DE3). The recombinant protein was purified by size- (Sephadex G25, Pharmacia) and ion-exchange (mono-Q, Pharmacia) chromatography from inclusion bodies solubilized in 8 M urea. The HLA A*0201 was folded in vitro by dilution in presence of the MAA peptides MART-126-35(ELAGIGILTV) or MAGE-3271-279(FLWGPRALV) and subsequently biotinylated as described previously [1, 3, 27]. After gel filtration on a Pharmacia Sephadex G25 column to remove unbound biotin, the protein was multimerized with streptavidin-FITC conjugated to dextran molecules (DAKO a/s, Denmark) to generate multi-valent HLA-dextran compounds for immunohistochemistry. The HLA A*0201 construct was a kind gift of Dr. Mark M. Davis (Department of Microbiology and Immunology, Stanford University, Palo Alto, CA, USA)
Immunohistochemistry stainings
Frozen sections were fixed in cold acetone for 10 min followed by removal of endogenous peroxidase with 0.03% H2O2 and blocking of collagenous elements with 10% bovine serum albumin. Serial sections were incubated for 30 min with biotinylated antibodies at predetermined dilutions (usually 20 mg per ml). Subsequently, the streptavidin peroxidase complex (DAKO, Hamburg,Germany) was applied for 30 min, followed by a 15 min incubation with the chromogen AEC (DAKO).
The staining procedure for multimeric peptide/MHC complexes has recently been described [3]. Briefly, sections were dried over-night and subsequently fixed in cold acetone for 5 min. All incubation steps were performed at room temperature and in the dark as follows: (1) 45 min of the primary antibody (1:100 diluted), (2) Cy3-conjugated goat antimouse (1:500 diluted; code 115-165-100, Dianova, Hamburg, Germany) for 45 min and finally (3) the multimers for 75 min. Between each step the slides were washed twice for 10 min in PBS/BSA 0.1% and then mounted in vectashield and observed under a Leica Confocal Microscope (TCS 4D, Leica, Mannheim, Germany).
Results
Patients and samples
The project was approved by the local ethical review board. Patients provided informed consent to participate and to donate tumor and lymph node biopsies for analyses. Patient characteristics are given in Table 1.
Antigen mapping of sentinel lymph nodes
Twelve primary melanoma lesions and corresponding sentinel lymph nodes were examined for the presence of identical T-cell clonotypes. To establish whether the sentinel lymph nodes were infiltrated by tumor cells, all lymph nodes were analyzed by RT-PCR using primers specific for the MAAs tyrosinase, MART-1, and MAGE-3 mRNAs. Data from these analyses demonstrated that the lymph nodes from seven of the patients were positive for at least two MAAs. The presence of melanoma cells was confirmed by immunochemistry using antibodies to gp100 and MART-1 (Table 1).
TCR clonotype mapping
Tumor lesions and draining lymph nodes were analyzed for the presence of T-cell clonotypes by RT-PCR/DGGE based TCR clonotype mapping. This method is based on the RT-PCR amplification of T-cell receptor CDR3 regions using primers for the variable region in conjunction with a common primer for the constant part of the receptor, followed by detection of increased numbers of TCR transcripts of a specific T cell. Using DGGE, the method is based on detection of increased numbers of TCR transcripts having distinct identical melting properties of the CDR3 region of the TCR—thus, the detection of clonally expanded T cells via this approach relies on the fact that clonotypic transcripts have no junctional diversity and therefore resolve at a fixed position in the denaturing gradient gel [31]. Importantly, the clonotype mapping reveals a “molecular fingerprint” of the T cell in question, implying that any such T cell may be tracked in time and space.
Applying this approach to tumor lesions revealed the presence of multiple clonotypic TCR transcripts covering the majority of the BV families 1–24 (Fig. 1a). In contrast, the analysis of the corresponding lymph nodes revealed a very limited number of T-cell clonotypes, in the range of 0–6 (Fig. 1b). Furthermore, these bands were faint and detected on a high level of background, indicating that the vast majority of the T cells in the lymph nodes are not clonally expanded.
Fig. 1.
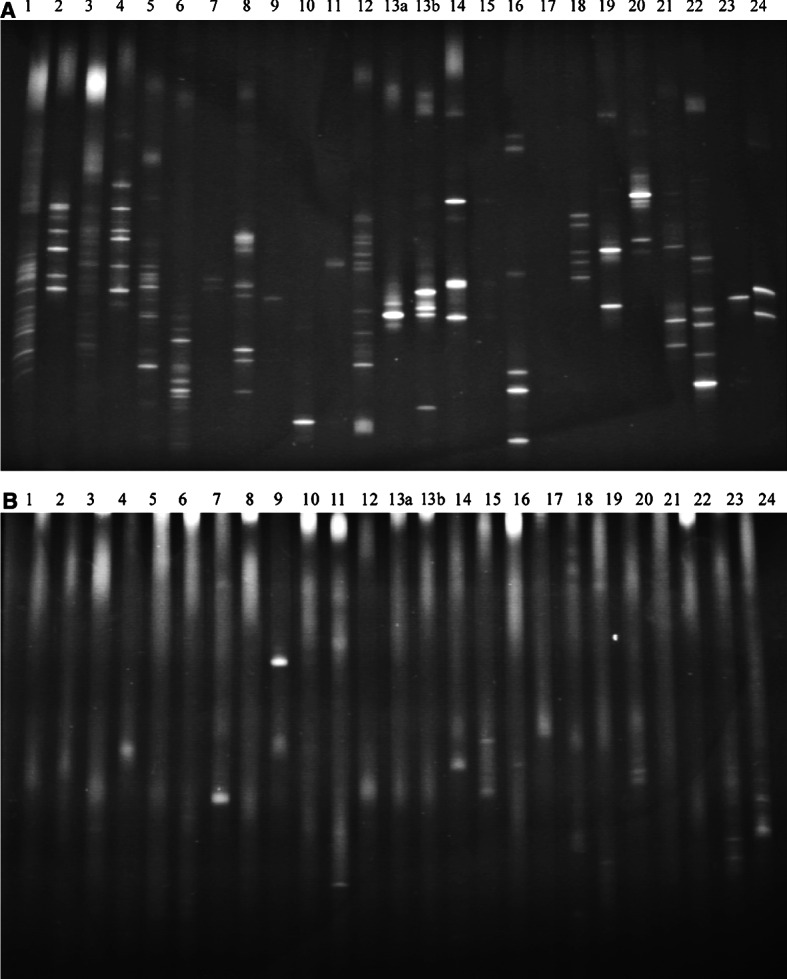
TCR clonotype mapping of the T-cells infiltrate in a primary melanoma (a) and the corresponding SLN (b) covering the BV families 1–24. PCR products were loaded onto a 20–80% denaturing gradient gel and run for 4.5 h at 160 V at a constant temperature of 58°C. DNA was stained with ethidium bromide and photographed under UV light
Comparative DGGE
To distinguish whether identical T cells were present in the tumor and the draining lymph node, clonotypic TCR transcripts detected in the lymph nodes were compared to corresponding transcripts detected in the tumor lesions. To this end, more than 50 clonotypic TCR transcripts detected in the lymph nodes were compared by DGGE to transcripts detected in the tumor lesions. In general, most of the transcripts in the SLN could not be detected in the tumor. However, five recurrent transcripts present in the SLN were detected in the tumor (Fig. 2). These transcripts were subjected to sequence analysis, in all cases verifying identity. These five identical TCR clonotypes, however, were all derived from lymph nodes that were invaded by tumor cells (Table 1).
Fig. 2.
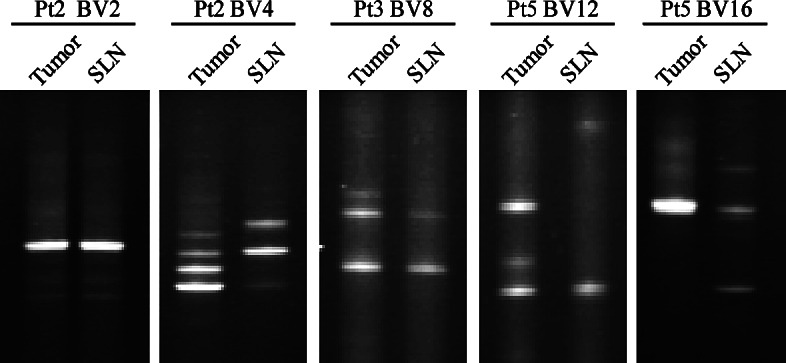
TCR clonotype mapping used for a comparative analysis of clonotypes detected in the sentinel lymph node and the primary tumor from patients 2, 3, and 5. Identity of the transcripts was verified by sequence analysis
Clonotype specific PCR and SSCP
Although the TCR clonotype mapping technology is a highly sensitive technique, the presence of high numbers of polyclonal T cells in the lymph nodes hampers detection of clonotypic transcripts. With the aim of resolving whether TCR transcripts found to be expanded in the tumor were also present in the SLN in numbers below the detection threshold of DGGE-based clonotype mapping, we constructed clonotype specific PCR primers matching TCR transcripts clonally expanded in the tumor lesion. Data from this highly sensitive PCR analysis revealed identical TCR transcripts in the SLN and the tumor in the two patients analyzed. To verify that only the specific transcript was amplified using the constructed primers, SSCP analyses was conducted which confirmed that most amplicons were indeed the result of an amplification of the specific TCR transcript (Fig. 3). SSCP separates DNA molecules based on the sequence dependent conformation of the molecule. Although this technique is not as stringent as DGGE, in app. ninety percent of cases, identical conformation will correspond to identical sequence. In the present case, the importance of the SSCP step was evident from the fact that some amplicons were derived from an unspecific amplification of nonrelevant TCR transcripts. Interestingly, in one patient (patient 12) two MAA-negative sentinel lymph nodes were analyzed revealing that TCR transcripts present in the tumor were recurrently detected in both lymph nodes (Table 1 and Fig. 3).
Fig. 3.

Comparative analyses of TCR clonotypic transcripts using SSCP. Five μl aliquots were loaded onto the gel and electrophoresed for 4 h at 200 V at room temperature. The gel was stained in ethidium bromide and DNA visualized under ultraviolet light. Identical TCR transcripts are detected in SLN for TCRBV2, 16, 20, 24 (patient 1). In patient 12, two SLNs were analyzed for the presence of TCR clonotypes also detected at the tumor site. For two TCR clonotypes; TCRBV11 and TCRBV17—identical clonotypes were detected in both SLNs, whereas for TCRBV19 this clonotype was only detected in one SLN
Staining of melanoma specific T cells using HLA-dextramer compounds
To verify that MAA-specific CTL are indeed present in the SLN, we performed in situ staining for MAA/HLA-A2 reactive TCRs. This technique has been thoroughly validated and is well suited for in-situ detection of antigen specific T cells [3, 25, 28]. The MAA derived peptides MART-126–35(ELAGIGILTV) and MAGE-3271–279(FLWGPRALV), were used for the construction of stable HLA-A2/peptide complexes. These complexes were multimerized using dextran molecules and used to stain snap-frozen samples for the presence of MAA-reactive CTL in a primary tumor and SLN. Using a confocal laser microscope, MART-126–35 as well as MAGE-3271–279/HLA-A2-dextran stained cells were observed both in the tumor and the SLN albeit at low frequencies never exceeding 1% of the CD8+ T-cell population. Figure 4a, b depicts MART-126-35/HLA-A2 reactive T cells in a primary tumor and the corresponding, tumor-infiltrated SLN, whereas Fig. 4c demonstrates the presence of MAGE-3271–279/HLA-A2 reactive T cells in close proximity of tumor cells. Figure 4d gives negative control-sections of a tumor from a HLA-A2 negative patient.
Fig. 4.

Detection of Mart-1/MelanA and MAGE-3 reactive T cells in primary tumors and SLN. Sections of primary tumors of either a HLA-A2+ (a) or a HLA-A2− (d) patient and the SLN of the first patient (b and c) were stained with Cy3-conjugated anti-CD8 (A, B, and D) or anti-Mart-1 antibodies (c) together with either FITC-conjugated Mart-1/MelanA/HLA-A2 (a, b, d) or MAGE-3/HLA-A2 multimers (c). The red channel is depicted in the first row, the green channel in the second, and the digital merge from the two individual images in the third row
Discussion
The notion that cancer cells are subjected to immunological surveillance goes back several decades. Over the past few years, key information has been disclosed in relation to the cells and molecules involved in immunological recognition of antigen, thus providing the means to investigate the interactions between cancer cells and cells of the immune system, in more detail. Importantly, it has been demonstrated that cancer cells express antigens that are recognized by the immune system, and a high number of peptide epitopes recognized by T cells have been characterized [26]. In this respect, significant methodological advances, e.g., the ELISPOT and HLA-tetramer technology, offered the tools to detect the presence of spontaneous T-cell responses against cancer cells in blood and TIL from cancer patients [2, 15, 19, 22]. Thus, it has become clear that cellular tumor-specific immune responses co-exist along with tumor progression, a phenomenon that remains an unresolved enigma of tumor immunology. A number of various ways by which tumors may escape an immunological response have been described [21]. Nevertheless, it is clear that many of these mechanisms only become relevant at late stages of disease, and that antigenic tumors may progress in spite of a fully operational immune system.
Sentinel lymphadenectomy provides a unique opportunity for assessing potential immunological interactions between the primary tumor and the regional lymph node basin. In the present study, we used TCR clonotype mapping to establish whether identical expanded T cells are present in both compartments. We and others have previously demonstrated that metastatic melanoma lesions comprise high numbers of clonally expanded T cells [30]. Accordingly, we identified a similarly high number of expanded T cells in primary melanoma lesions. In contrast, only very limited numbers of expanded T cells were detected in the SLN, and comparisons of these with the clonotypic TCR transcripts present in corresponding tumors, demonstrated that identical T-cell clonotypes could only be detected in lymph nodes infiltrated with melanoma cells. Since the presence of identical T cells in two different tumor lesions from the same patient has been reported previously [24, 31], the presence of the identical TCR transcripts may merely reflect a systemic component of the response. Moreover, none of the very few T-cell clonotypes present above detection threshold in the non-invaded lymph nodes was identical to the TCR transcripts detected in the primary tumor. However, it should be kept in mind that analyses of the present character only reveal a momentary glimpse of the ongoing immune response. From the first immunological detection of “danger”, e.g., expression of MICA/B molecules [5, 12], necrosis [10], or extracellular presence of heat shock proteins [6], the initiation of an immune response is not an instant event. The in vivo time scale for priming and subsequent expansion of T cells is not well established. Another possible pitfall relates to the detection threshold of the employed DGGE-based TCR clonotype mapping, which undoubtedly is compromised by the presence of large numbers of polyclonal T cells, as it is the case in a lymph node. To circumvent this technical limitation, we used clonotype specific PCR amplification to analyze the SLN for the presence of TCR transcripts detected in the tumor, which revealed that approximately half of the analyzed clonotypic TCR transcripts present in the tumor could be recurrently detected in the SLN. Considering the absence of tumor cells in these lymph nodes, this observation supports the notion that APC presenting tumor antigens mediate the priming and limited expansion of tumor specific T cells. However, considering the present data it still cannot be ruled out that tumor cells may have been present in the lymph node at a previous time point.
Clearly, the presence of T cells expressing identical TCR in both the tumor and the SLN does not necessarily signify that these cells are indeed involved in antitumor immune responses. To address this issue, we took advantage of the possibility to stain specific TCR specificities with MAGE-3 or MART-1 epitopes coupled to HLA-dextran compounds [3]. The MART-1 protein frequently elicits reactivity in melanoma patients, but reactivity may also be detected in healthy donors [7]. Although it has been shown that the MAGE-3 271 peptide may not be processed and presented on the surface of cancer cells [18]; however, others have presented contradictory data [16]. Conversely to the Mart-1 protein, MAGE-3 is not likely to elicit spontaneous reactivity in normal donors [8]. Using these constructs to stain melanoma reactive T cells, we show that such cells were present in both the primary tumor as well as the tumor involved and noninvolved draining lymph nodes (Fig. 4). Although the in situ MAA/HLA-multimer staining and the TCR clonotype mapping cannot be performed in direct correspondence, we have previously demonstrated a close connection of such analyses for T cells from PBL or isolated tumor infiltrating lymphocytes. Together, our data suggest that lymph nodes draining the primary tumors are essential for the induction of T-cell responses against melanoma either by hosting APC displaying tumor derived antigens or by hindering the metastatic process of tumor cells themselves in an immune competent microenvironment. Although we cannot pin down the exact cell type involved in these events, and also, cannot exclude that tumor cells are capable of direct priming if present in the lymph node, our data indicate that priming of T-cell responses to solid tumor seem to occur according to the dogma deduced from viral infections.
Acknowledgements
We wish to thank the Danish Melanoma Group for continued interest and support. Grant support: the work was supported by grants from the Danish Research Council, The Danish Cancer Society, The NOVO NORDISK FOUNDATION, the Deutsche Krebshilfe (grant 10-1845-Be1), and the Bundesminesterium für Bildung und Forschung (IZKF Würzburg, Projekt B17, grant 01 KS 903).
References
- 1.Altman JD, Moss PA, Goulder PJR, Barouch DH, McHeyzer Williams MG, Bell JI, McMichael AJ, Davis MM. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94. doi: 10.1126/science.274.5284.94. [DOI] [PubMed] [Google Scholar]
- 2.Andersen MH, Ostergaard Pedersen L, Becker JC, thor Straten P. Identification of a cytotoxic T lymphocyte response to the apoptose inhibitor protein Survivin in cancer patients. Cancer Res. 2001;61:869. [PubMed] [Google Scholar]
- 3.Andersen MH, Ostergaard Pedersen L, Capeller B, Bröcker EB, Becker JC, thor Straten P. Spontaneous cytoxic T-cell responses against survivin-derived MHC class I restricted T-cell epitopes in situ as well as ex vivo in cancer patients. Cancer Res. 2001;61:5964. [PubMed] [Google Scholar]
- 4.Barfoed A, Reichert Petersen T, Kirkin AF, thor Straten P, Claesson M-H, Zeuthen J. Cytotoxic T-lymphocyte clones, established by stimulation with the HLA-A2 binding p5365–73 wild type peptide loaded on dendritic cells in vitro, specifically recognise and lyse HLA-A2 tumour cells overexpressing the p53 protein. Scand J Immunol. 2000;51:128. doi: 10.1046/j.1365-3083.2000.00668.x. [DOI] [PubMed] [Google Scholar]
- 5.Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA (see comments) Science. 1999;285:727. doi: 10.1126/science.285.5428.727. [DOI] [PubMed] [Google Scholar]
- 6.Binder RJ, Han DK, Srivastava PK. CD91: a receptor for heat schock protein pg96. Nat Immunol. 2000;1:151. doi: 10.1038/77835. [DOI] [PubMed] [Google Scholar]
- 7.D’Souza S, Rimoldi D, Lienard D, Lejeune F, Cerottini JC, Romero P. Circulating Melan-A/Mart-1 specific cytolytic T lymphocyte precursors in HLA-A2+ melanoma patients have a memory phenotype. Int J Cancer. 1998;78:699. doi: 10.1002/(sici)1097-0215(19981209)78:6<699::aid-ijc6>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 8.Dhodapkar MV, Young JW, Chapman PB, Cox WI, Fonteneau JF, Amigorena S, Houghton AN, Steinman RM, Bhardwaj N. Paucity of functional T-cell memory to melanoma antigens in healthy donors and melanoma patients. Clin Cancer Res. 2000;6:4831. [PubMed] [Google Scholar]
- 9.Flynn KJ, Belz GT, Altman JD, Ahmed R, Woodland DL, Doherty PC. Virus-specific CD8+ T cells in primary and secondary influenza pneumonia. Immunity. 1998;8:683. doi: 10.1016/S1074-7613(00)80573-7. [DOI] [PubMed] [Google Scholar]
- 10.Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med. 1999;5:1249. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- 11.Gershenwald JE, Thompson W, Mansfield PF, Lee JE, Colome MI, Tseng CH, Lee JJ, Balch CM, Reintgen DS, Ross MI. Multi-institutional melanoma lymphatic mapping experience: the prognostic value of sentinel lymph node status in 612 stage I or II melanoma patients. J Clin Oncol. 1999;17:976. doi: 10.1200/JCO.1999.17.3.976. [DOI] [PubMed] [Google Scholar]
- 12.Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, Spies T. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc Natl Acad Sci USA. 1999;96:6879. doi: 10.1073/pnas.96.12.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol. 2001;19:47–64. doi: 10.1146/annurev.immunol.19.1.47. [DOI] [PubMed] [Google Scholar]
- 14.Jones CM, Cose SC, Coles RM, Winterhalter AC, Brooks AG, Heath WR, Carbone FR. Herpes simplex virus type 1-specific cytotoxic T-lymphocyte arming occurs within lymph nodes draining the site of cutaneous infection. J Virol. 2000;74:2414. doi: 10.1128/JVI.74.5.2414-2419.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawakami Y, Rosenberg SA. T-cell recognition of self peptides as tumor rejection antigens. Immunol Res. 1996;15:179. doi: 10.1007/BF02918248. [DOI] [PubMed] [Google Scholar]
- 16.Keogh E, Fikes J, Southwood S, Celis E, Chesnut R, Sette A. Identification of new epitopes from four different tumor-associated antigens: recognition of naturally processed epitopes correlates with HLA-A*0201-binding affinity. J Immunol. 2001;167:787. doi: 10.4049/jimmunol.167.2.787. [DOI] [PubMed] [Google Scholar]
- 17.Kirk CJ, Hartigan-O’Connor D, Mule JJ. The dynamics of the T-cell antitumor response: chemokine-secreting dendritic cells can prime tumor-reactive T cells extranodally. Cancer Res. 2001;61:8794. [PubMed] [Google Scholar]
- 18.Miconnet I, Servis C, Cerottini JC, Romero P, Levy F. Amino acid identity and/or position determines the proteasomal cleavage of the HLA-A*0201-restricted peptide tumor antigen MAGE-3271-279. J Biol Chem. 2000;275:26892. doi: 10.1074/jbc.M000701200. [DOI] [PubMed] [Google Scholar]
- 19.Nielsen MB, Monsurro V, Migueles SA, Wang E, Perez-Diez A, Lee KH, Kammula U, Rosenberg SA, Marincola FM. Status of activation of circulating vaccine-elicited CD8+ T cells. J Immunol. 2000;165:2287. doi: 10.4049/jimmunol.165.4.2287. [DOI] [PubMed] [Google Scholar]
- 20.Novellino L, Castelli C, Parmiani G. A listing of human tumor antigens recognized by T cells. Cancer Immunol Immunother. 2004;5(3):187–207. doi: 10.1007/s00262-004-0560-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pawelec G, Heinzel S, Kiessling R, Muller L, Ouyang Q, Zeuthen J. Escape mechanisms in tumor immunity: a year 2000 update. Crit Rev Oncog. 2000;11:97. [PubMed] [Google Scholar]
- 22.Pittet MJ, Valmori D, Dunbar PR, Speiser DE, Lienard D, Lejeune F, Fleischhauer K, Cerundolo V, Cerottini JC, Romero P. High frequencies of naive Melan-A/MART-1-specific CD8(+) T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J Exp Med. 1999;190:705. doi: 10.1084/jem.190.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prymowicz D, Moore RN, Rouse BT. Frequency of herpes simplex virus-specific helper T lymphocyte precursors in the lymph node cells of infected mice. J Immunol. 1985;134:2683. [PubMed] [Google Scholar]
- 24.Puisieux I, Even J, Pannetier C, Jotereau F, Favrot M, Kourilsky P. Oligoclonality of tumor-infiltrating lymphocytes from human melanomas. J Immunol. 1994;153:2807. [PubMed] [Google Scholar]
- 25.Reker S, Becker JC, Svane IM, Ralfkiaer E, thor Straten P, Andersen MH. HLA-B35-restricted immune responses against survivin in cancer patients. Int J Cancer. 2004;108:937. doi: 10.1002/ijc.11634. [DOI] [PubMed] [Google Scholar]
- 26.Renkvist N, Castelli C, Robbins PF, Parmiani G. A listing of human tumor antigens recognized by T cells. Cancer Immunol Immunother. 2001;50:3. doi: 10.1007/s002620000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schrama D, Andersen MH, Terheyden P, Schrøder L, Ostergaard Pedersen L, thor Straten P, Becker JC. Oligoclonal TCR usage of melanocyte differentiation antigen-reactive T cells. Cancer Res. 2001;61:493. [PubMed] [Google Scholar]
- 28.Schrama D, Ostergaard Pedersen L, Keikavoussi P, Andersen MH, thor Straten P, Brocker EB, Kampgen E, Becker JC. Aggregation of antigen-specific T cells at the inoculation site of mature dendritic cells. J Invest Dermatol. 2002;119:1443. doi: 10.1046/j.1523-1747.2002.19604.x. [DOI] [PubMed] [Google Scholar]
- 29.Schrama D, thor Straten P, Fischer WH, Merkel A, Bröcker EB, Reisfeld RA, Becker JC. Targeting lymphotoxin alpha to the tumor microenvironment elicits an efficient immune response by induction of peripheral lymphoid tissue. Immunity. 2001;14:111. doi: 10.1016/S1074-7613(01)00094-2. [DOI] [PubMed] [Google Scholar]
- 30.thor Straten P, Becker JC, Guldberg P, Zeuthen J. In situ T cells in melanoma. Cancer Immunol Immunother. 1999;48:386. doi: 10.1007/s002620050591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.thor Straten P, Guldberg P, Grønbæk K, Zeuthen J, Becker JC. In situ T-cell responses against melanoma comprise high numbers of locally expanded T-cell clonotypes. J Immunol. 1999;163:443. [PubMed] [Google Scholar]
- 32.thor Straten P, Kirkin AF, Seremet T, Zeuthen J. Expression of transporter associated with antigen processing 1 and 2 (TAP1/2) in malignant melanoma cell lines. Int J Cancer. 1997;70:582. doi: 10.1002/(SICI)1097-0215(19970304)70:5<582::AID-IJC15>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 33.thor Straten P, Ralfkiaer E, Hendriks J, Seremet T, Vejlsgaard GL, Zeuthen J. T-cell receptor variable region genes in cutaneous T-cell lymphomas. Br J Dermatol. 1998;138:3. doi: 10.1046/j.1365-2133.1998.02022.x. [DOI] [PubMed] [Google Scholar]
- 34.Valmori D, Dutoit V, Rubio-Godoy V, Chambaz C, Lienard D, Guillaume P, Romero P, Cerottini JC, Rimoldi D. Frequent cytolytic T-cell responses to peptide MAGE-A10(254–262) in melanoma. Cancer Res. 2001;61:509. [PubMed] [Google Scholar]
- 35.Wolkers MC, Stoetter G, Vyth-Dreese FA, Schumacher TN. Redundancy of direct priming and cross-priming in tumor-specific CD8+ T cell responses. J Immunol. 2001;167:3577. doi: 10.4049/jimmunol.167.7.3577. [DOI] [PubMed] [Google Scholar]