

Abstract
Agents that enhance T cell co-stimulatory signaling have emerged as promising cancer immunotherapies. Our laboratory has been evaluating the TNF receptor co-stimulatory molecule, OX40, which has the capacity to augment critical aspects of T cell function and induce tumor regression in animal models. Effective stimulation of OX40 expressing T cells was accomplished with agonist antibodies to OX40 that were eventually translated into a clinical trial for cancer patients. A recent attempt to assess the affect of immune senescence on OX40 therapy, revealed a dramatic loss of efficacy of the agonist therapy in older tumor-bearing mice. The deficiency in OX40-enhanced anti-tumor responses in older mice correlated with a decrease in the number of differentiated effector T cells. Further investigation suggests that the underlying age-related decline in the agonist OX40-mediated T cell responses was not inherent to the T cells themselves, but related to the host environment. Thus, effective use of immunotherapies based on T cell co-stimulatory molecules may require additional modifications, such as immune stimulants to increase innate immunity, to address age-related defects that reside outside of the T cell and within the host environment.
Keywords: OX40, Co-stimulation, Aging
Introduction
Efforts to cure cancer with immunotherapy have spanned over a century, from the seminal finding of Coley [7] to our current understanding of how this multi-faceted disease interfaces with cells of the immune system; yet an effective immunotherapy remains elusive. It is widely accepted that the immune system can eradicate tumors that develop, and slow the growth of existing tumors. Unfortunately, escape of tumors from the control of immune-surveillance is common. One strategy to restore effective immune destruction of tumors is to activate the tumor-associated antigen-specific T cells that can mediate tumor destruction. Fundamentally, increases in the number and function of this population of T cells should increase the killing of cancer cells. A promising approach to boost the number of tumor-specific T cells is the engagement of members of the TNF receptor superfamily (TNFR), which have been shown to enhance expansion, function, and persistence of antigen-specific T cells. Targeted stimulation of co-stimulatory TNFRs, including OX40, has been recognized to have great potential for broad usage against multiple types of tumors, with the intent to boost T cell stimulation [5]. In addition, preclinical studies have shown that OX40 engagement may be effective as a stand alone therapy against a number of different tumor types. Our laboratory has used agonist antibodies specific for both human and mouse OX40 to identify the role of OX40 in T cell activation and have applied this form of co-stimulation in both preclinical and clinical tumor studies. An aspect of the OX40-mediated therapeutic strategy that has not been fully addressed is the influence of aging.
OX40 expression, signaling, and T cell co-stimulation
OX40 is a type I transmembrane protein that is expressed primarily on antigen-primed T cells and mediates potent co-stimulatory signals to differentiating effector T cells. The pattern of OX40 expression is unique, as it is for the most part restricted to lymphoid tissue with transient expression on activated CD4 and CD8 T cells [14, 17]. TCR/CD28-activation of naïve T cells initiates the expression of OX40, with peak expression occurring within 24–48 h after TCR engagement, and then returning to undetectable levels a few days later [17]. The presence of OX40 on recently activated T cells can be engagement by its cognate ligand, OX40L. The OX40L has been shown to be expressed on activated and mature antigen-presenting cells (DCs, macrophages and B cells), driven by the activation of Toll-like receptors (TLRs) or by CD40 engagement [1, 4, 33]. Thus, the expression of OX40L is primarily restricted to sites of inflammation in vivo, limiting the extent of OX40 activation [1, 54]. The self-assembled OX40L trimer binds to the trimeric OX40 receptor at a different conformational angle than other TNF–TNFR family members [8], but it is unknown how this conformational difference affects OX40–OX40L signaling. In the end, engagement of OX40 (by OX40L or other exogenous agonists) mediates the recruitment of TNF receptor associated factors (TRAFs) that initiate the activation of various intracellular signaling pathways, including JNK and NF-κB [2, 21, 50].
OX40-mediated signals primarily influence the activation and survival of effector CD4 and CD8 T cells [9, 35, 39]. This is evident in OX40L transgenic mice that displayed a dramatic increase in T cell survival and memory T cell generation following immunization [32]. Furthermore, OX40 deficient T cells are more susceptible to T cell apoptosis compared to their OX40 intact counterparts [16]. While OX40 stimulation has been mainly associated with increased T cell survival, there is evidence that OX40-mediated co-stimulation can also greatly enhance both CD4 and CD8 effector T cell function. CD4 T cells produced tenfold greater levels of IFNγ and IL-2 (on a per cell basis) compared to controls following in vivo stimulation with antigen and agonist anti-OX40 [20, 53] and CD8 effector T cells produced more TNFα, IFNγ, and granzyme B after OX40-stimulation [25, 35]. Taken together, these studies demonstrate that OX40-mediated co-stimulation of effector CD4 and CD8 T cells generated after antigen-priming drives increased accumulation of these cells and enhances their effector function. In addition to the potent effects on differentiated effector T cells, OX40 signaling also affects regulatory T cells (Tregs), where OX40 signaling was shown to prevent the generation of Tregs and abrogate their suppressive activity [42, 44, 47, 48].
OX40 co-stimulation as a cancer immunotherapy
The ability of OX40 co-stimulation to enhance T cell differentiation and function led to therapeutic studies in mice. An OX40 antibody has been used, by our laboratory and others, to describe increases in antigen-specific T cell numbers, enhanced cytokine production, and promotion of increased survival [12, 16, 30, 35, 37]. In addition, anti-OX40 treatment of mice harboring s.c. tumors improved overall tumor-free survival in a number of different tumor types: sarcoma, colon carcinoma, breast tumors, melanoma, and glioma [22, 51]. The therapeutic anti-tumor effects were dose dependent, generated tumor-specific memory, and required both CD4 and CD8 T cells [15, 22, 43, 51]. We hypothesize that anti-OX40-mediated tumor regression involves enhancement of the effector function and persistence of both CD4 and CD8 tumor-specific T cells, while concomitantly abrogating the suppressive effects of Tregs (Fig. 1).
Fig. 1.

Hypothesized model of anti-OX40-mediated tumor destruction. Anti-OX40 administration can enhance the function, accumulation, and survival of differentiated effector T cells (CD4 and CD8). The anti-OX40 enhancement of CD4 T cell function leads to increased IL-2 and IFNγ secretion, “helping” the effector CD8 T cells (CTL). In addition, OX40-engement increases differentiated effector CD8 T cell accumulation and production of IFNγ and granzymes that ultimately mediate the killing of tumor cells. Coinciding with the previous enhancements, anti-OX40 can abrogate the suppressive activity of Tregs on the anti-tumor response
The encouraging preclinical results provided the impetus for clinical studies using agonist OX40 antibodies in cancer patients. Treatment of non-human primates with a murine antibody specific for human OX40 increased the number of lymphocytes in immune organs (lymph nodes and spleen) and augmented antigen-specific T and B cell responses, suggesting that anti-OX40 initiated measurable immunological changes [52]. This study provided the rationale for the current Phase I clinical trial in which advanced stage cancer patients with metastatic or locally advanced tumors have been given anti-OX40 at various doses to measure safety, immunological responses, pharmacokinetics, and to assess tumor regression. The results from this clinical study will provide the basis for follow-up Phase II studies using anti-OX40 as a single agent.
Agonist OX40-enhanced tumor immunotherapy and aging
The potential for agonist OX40 antibodies and other emerging immunotherapies to mediate successful tumor regression in cancer patients will depend on their ability to generate immune responses that not only overcome tolerance and the immunosuppressive tumor environment, but they must also overcome age-related changes in immunity (i.e., immune senescence). It is well established that overall immunity decreases with increasing age, which reduces the effectiveness of vaccines [11, 19, 27]. The effects of age must also be addressed when developing immune augmenting cancer therapies, considering the majority of cancer patients are over 65 years old. Thus, to assess the impact of age on anti-OX40-mediated tumor immunity, we undertook studies in aged mice. Mice, ranging in age from 2 to 20 months, were challenged with sarcoma (MCA205-C57BL/6 background) or colon carcinoma (CT26-BALB/c background) tumors and then treated with two doses of anti-OX40. Systemic anti-OX40 administration in both tumor models had been previously shown to improve tumor-free survival in young mice compared to IgG-treated control mice (Table 1). We found that the same treatment delivered to 12- and 20-month-old tumor-bearing mice was not able to increase tumor rejection (Table 1) [38]. The dramatic age-related loss in efficacy occurred despite the presence of similar levels of OX40+ tumor infiltrating T cells. Our findings demonstrated that 20-month old (elderly) and 12-month old (middle-aged) tumor-bearing mice were less responsive to the anti-OX40 therapy.
Table 1.
Agonist OX40-mediated tumor-free survival in mice of various ages
| Tumor | Age (months) | Tumor-free survival (%)a | References |
|---|---|---|---|
| MCA205 | 2 | 40–80 | Kjaergaard et al. [22], Ruby and Weinberg [38] |
| 6 | 75 | Ruby and Weinberg [38] | |
| 12 | >5 | Ruby and Weinberg [38] | |
| 20 | 0 | Ruby and Weinberg [38] | |
| CT26 | 2–3 | 40–50 | Weinberg et al. [51], Ruby and Weinberg [38] |
| 12 | 0 | Ruby and Weinberg [38] |
aControl IgG-treatment resulted in 0% tumor-free survival in all age groups
Identifying potential age-related dysfunctions
To better understand the nature of the age-related deficiency observed in tumor-bearing aged mice treated with anti-OX40, we examined the status of differentiated effector T cell populations in anti-OX40-treated mice [38]. Enhanced function and accumulation of differentiated effector T cell populations are central to OX40-mediated tumor rejection [15, 22, 43], thus age-related changes in this population of T cells could ultimately affect tumor rejection. The examination of T cell populations in the local tumor environment of 2- and 12-month old anti-OX40-treated mice revealed a notable age-related decrease in the number of cytokine-producing T cells (CD8+IFNγ+, CD4+IL-2+ and CD4+IFNγ+) [38]. The numerical reduction in cytokine-producing T cells in the older anti-OX40-treated mice corresponded with the dramatic decrease in tumor rejection in these mice and initially gave the impression of an age-related deficiency or defect that resided in the responding T cells.
Further analysis of the underlying deficiencies contributing to the diminished accumulation of differentiated effector T cells in the older tumor-bearing mice following anti-OX40 administration employed the CD4 transgenic TCR T cell model (DO11.10). This model allowed for a comparison of young and old T cells in similar aged hosts (via adoptive transfer) and directly addressed the question of whether the age-related dysfunction seen following anti-OX40 administration was T cell intrinsic or extrinsic. The results from the criss-cross adoptive transfer experiments showed a significant decrease in the accumulation of older cytokine-producing T cells compared to younger T cells in age-matched recipients, verification of our previous results [38]. Yet the most surprising finding was that older T cells transferred into young recipients equally acquired a differentiated effector phenotype and accumulated at the same levels as young T cells transferred into young recipients, whereas, young T cells adoptively transferred into older recipients experienced a decrease in the differentiated effector T cell population co-producing IFNγ+ and IL-2+ (Fig. 2). Therefore, the dominant age-related deficiencies underlying the diminished OX40-enhanced T cell responses in older mice appeared to reside outside of the T cell component and within the cells that promote and support T cell differentiation. Other studies in aged mice have also shown that deficiencies in the host environment contributed to impaired immune responses in combination with intrinsic T cell defects [6, 26, 28]; however, our results suggest a much greater contribution of the host environment to age-related immune dysfunction, after anti-OX40 stimulation. In contrast to our findings, results from a similarly designed criss-cross study reported antigen-specific T cell responses (expansion and differentiation) were unaffected by the age of the host environment, but rather were due to intrinsic T cell deficiencies [31]. The difference in the two conclusions may stem from the use of CFA by Mittler and Lee, which can establish a pro-inflammatory condition, potentially compensating for any age-related deficiencies in the host environment, and our use of an agonist OX40 stimuli that mainly enhances T cell function. Curiously, the disparate findings suggest anti-OX40 may alleviate age-related intrinsic T cell defects and pro-inflammatory mediators may overcome age-related T cell extrinsic host defects.
Fig. 2.
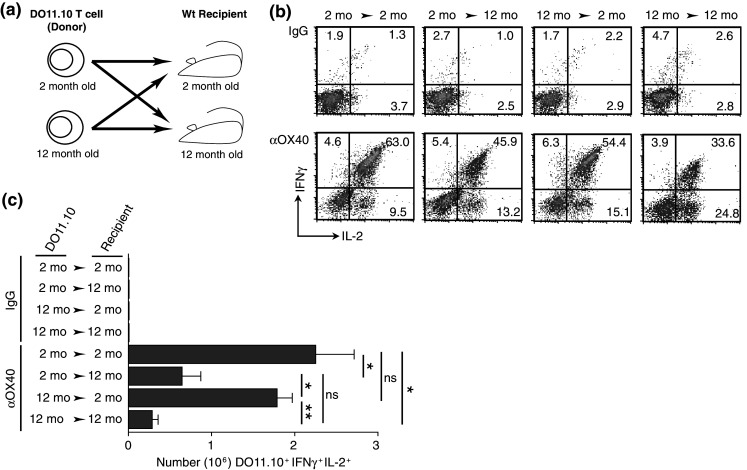
The age of the host environment determines the extent of an OX40-enhanced antigen-specific transgenic TCR T cell immune response. Two- or 12-month-old transgenic TCR CD4 T cells specific for a peptide of ovalbumin (323–339) were isolated from the spleens of DO11.10 mice and adoptively transferred into 2- or 12-month-old BALB/c recipients. Adoptively transferred mice were immunized s.c. with 500 μg whole ovalbumin and 50 μg anti-OX40 or rat IgG. A second dose of anti-OX40 or rat IgG was also given the next day. a Schematic of the adoptive transfer of transgenic TCR T cells. b Representative FACs plots of DO11.10 T cells (gated on CD4+ and the transgenic TCR+) expressing both IFNγ+ and IL-2+ from the antigen-draining LNs 4 days after challenge. c Enumeration of the DO11.10 T cells that co-expressed the cytokines IL-2 and IFNγ. Statistical representation: Student’s t test (two-tailed). For analysis, values of P ≤ 0.05 were considered significant and expressed as follows: *P ≤ 0.05 and **P ≤ 0.001
Based on the hypothesis that the cells of the aging host environment primarily contributed to the OX40-specific age-related defects, we sought to restore immunological function with the pro-inflammatory cytokine, IL-12. IL-12, produced by innate immune cells, not only plays a key role in effector T cell differentiation and survival [45, 49], but it can synergize with OX40 signaling to mediate tumor eradication [24, 36]. The combination of IL-12 and anti-OX40 boosted levels of differentiated effector DO11.10 T cells (IFNγ+) in middle-aged hosts (Fig. 3) and partially restored tumor-free survival in mice treated with anti-OX40 alone [38]. Pro-inflammatory mediators have been proven to produce effective anti-tumor immune responses in aged animals. Lustgarten and colleagues have administered a number of TLR agonists, including Poly I:C and CpG-ODN, to restore T cell responses (CTL) and tumor regression in aged mice [10, 40]. Finally, although IL-12 only partially restored effective OX40-enhanced tumor immunity in older mice, it and other pro-inflammatory agents may help reverse the negative impact of aging on anti-OX40-mediated tumor immune responses.
Fig. 3.
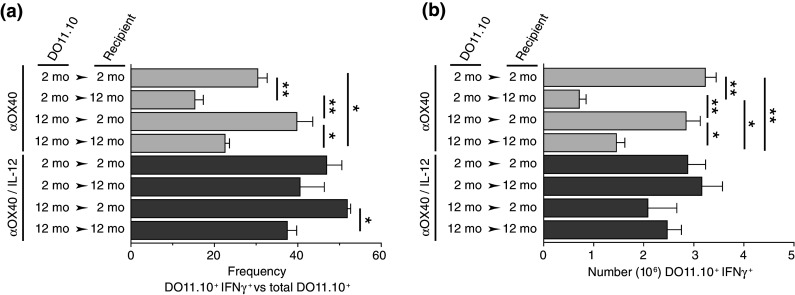
IL-12 increases the acquisition of an effector CD4 T cell phenotype and accumulation of differentiated effector CD4 T cells in aged anti-OX40 treated recipients. Two- or 12-month-old BALB/c mice were adoptively transferred with 2- or 12-month-old DO11.10 T cells and immunized s.c. with 500 μg ovalbumin and 50 μg anti-OX40. A second dose of anti-OX40 was also given the next day. Mice were injected s.c. with IL-12 (100 ng) or control (PBS) daily for 4 days, starting at immunization. Antigen-draining LNs were harvested 4 days after challenge and analyzed. a Frequency of DO11.10 T cells (CD4+ transgenic TCR+) producing IFNγ. b Enumeration of IFNγ-producing DO11.10 T cells. Statistical representation: Student’s t test (two-tailed). For analysis, values of P ≤ 0.05 were considered significant and expressed as follows: *P ≤ 0.05 and **P ≤ 0.001 (adapted from [38])
Conclusion
Work in our laboratory has exploited the activation of OX40 co-stimulatory molecules by agonist antibodies to enhance T cell immune responses and we have identified this pathway as a promising cancer immunotherapy. Recently we measured the activity of an agonist OX40 agent in middle-aged (12-month old) and elderly (20-month old) tumor-bearing mice, to assess the influence of age on this OX40-activating therapy. The study revealed the effects of immune senescence significantly reduced anti-OX40-mediated anti-tumor responses, which correlated with diminished numbers of differentiated effector T cells. The age-related dysfunction appeared to not be associated with the responding T cells, but more likely the cells of the host environment that promote and support T cell responses. Indeed, administration of IL-12, an innate cytokine, restored the critical T cell responses and tumor regression when combined with anti-OX40. Even with this promising finding, is unclear as to the exact biological explanation, whether a particular impaired signaling pathway or deficient cell type, may account for the age-related effects seen in both our tumor and criss-cross experiments. Previous studies have shown significant age-related deficiencies in DC number, maturation, and migration [18, 28, 41], therefore, the focus of our future studies will be on the role of aging on DC function/activation in both the deficient anti-OX40-mediated tumor immune response and in the TCR transgenic “criss-cross” experiments.
Finally, the clinical success of drugs designed to enhance T cell co-stimulation, via CTLA-4, 4-1BB, and OX40, to induce tumor regression, may ultimately hinge on also addressing potential age-related deficiencies in the tumor-bearing host. The initial approach of the various animal models and clinical trials of these drugs was as single agents to regress tumors; yet our recent findings in aged tumor-bearing animals have raised some important questions about this approach. In light of the significant age-related abrogation of OX40-mediated tumor regression seen here, the use of OX40 agonist as a single agent should be further assessed. Although, our data and others [29] have demonstrated that T cells from aged animals respond in a comparable fashion as young T cells to agonist OX40 stimulation, in some cases additional signals/stimuli may be required in older subjects to generate the desired anti-tumor responses. One potential approach is to target the non-T cell environment (e.g., innate immune cells), using at least one of a host of pro-inflammatory TLR-based reagents (monophosphoryl lipid A, CpG, Poly I:C, and resiquimod/imiquimod). These TLR-activators have been shown to augment DC and macrophage activation, including the induction of pro-inflammatory cytokines (e.g., IL-12), and perhaps more importantly, are currently in various levels of clinical application for the enhancement of tumor immunity [3, 13, 23, 34, 46]. Co-administration of TLR-activating agents with OX40 agonists would be hypothesized to provide signals to overcome age-related deficiencies in antigen presentation by APC and to enhance tumor-specific effector T cell persistence and function, respectively, in tumor-bearing subjects. Thus, overcoming immune senescence in the treatment of cancer may require treatments that utilize multiple immunotherapeutic approaches that can restore both adaptive and innate responses lost during the aging process.
Acknowledgments
The authors would like to thank Drs Walter Urba and Michael Gough for their critical reading of the manuscript.
Footnotes
This article is part of the Symposium in Writing on “Impact of Ageing on Cancer Immunity and Immunotherapy”.
References
- 1.Akiba H, et al. Critical contribution of OX40 ligand to T helper cell type 2 differentiation in experimental leishmaniasis. J Exp Med. 2000;191:375–380. doi: 10.1084/jem.191.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arch RH, et al. Translocation of TRAF proteins regulates apoptotic threshold of cells. Biochem Biophys Res Commun. 2000;272:936–945. doi: 10.1006/bbrc.2000.2873. [DOI] [PubMed] [Google Scholar]
- 3.Atanackovic D, et al. Vaccine-induced CD4+ T cell responses to MAGE-3 protein in lung cancer patients. J Immunol. 2004;172:3289–3296. doi: 10.4049/jimmunol.172.5.3289. [DOI] [PubMed] [Google Scholar]
- 4.Brocker T, et al. CD4 T cell traffic control: in vivo evidence that ligation of OX40 on CD4 T cells by OX40-ligand expressed on dendritic cells leads to the accumulation of CD4 T cells in B follicles. Eur J Immunol. 1999;29:1610–1616. doi: 10.1002/(SICI)1521-4141(199905)29:05<1610::AID-IMMU1610>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 5.Cheever MA, Creekmore S (eds) (2007) National Cancer Institute agent workshop proceedings. http://web.ncifcrf.gov/research/brb/workshops.asp
- 6.Clise-Dwyer K, et al. Environmental and intrinsic factors lead to antigen unresponsiveness in CD4(+) recent thymic emigrants from aged mice. J Immunol. 2007;178:1321–1331. doi: 10.4049/jimmunol.178.3.1321. [DOI] [PubMed] [Google Scholar]
- 7.Coley WB. Contribution to the knowledge of sarcoma. Ann Surg. 1891;14:199–220. doi: 10.1097/00000658-189112000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Compaan DM, Hymowitz SG. The crystal structure of the costimulatory OX40-OX40L complex. Structure. 2006;14:1321–1330. doi: 10.1016/j.str.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 9.Croft M. Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat Rev Immunol. 2003;3:609–620. doi: 10.1038/nri1148. [DOI] [PubMed] [Google Scholar]
- 10.Dominguez AL, Lustgarten J. Implications of aging and self-tolerance on the generation of immune and antitumor immune responses. Cancer Res. 2008;68:5423–5431. doi: 10.1158/0008-5472.CAN-07-6436. [DOI] [PubMed] [Google Scholar]
- 11.Effros RB, Walford RL. The immune response of aged mice to influenza: diminished T-cell proliferation, interleukin 2 production and cytotoxicity. Cell Immunol. 1983;81:298–305. doi: 10.1016/0008-8749(83)90237-X. [DOI] [PubMed] [Google Scholar]
- 12.Evans DE, et al. Engagement of OX40 enhances antigen-specific CD4(+) T cell mobilization/memory development and humoral immunity: comparison of alphaOX-40 with alphaCTLA-4. J Immunol. 2001;167:6804–6811. doi: 10.4049/jimmunol.167.12.6804. [DOI] [PubMed] [Google Scholar]
- 13.Ewel CH, et al. Polyinosinic–polycytidylic acid complexed with poly-l-lysine and carboxymethylcellulose in combination with interleukin 2 in patients with cancer: clinical and immunological effects. Cancer Res. 1992;52:3005–3010. [PubMed] [Google Scholar]
- 14.Fujita T, et al. Functional characterization of OX40 expressed on human CD8+ T cells. Immunol Lett. 2006;106:27–33. doi: 10.1016/j.imlet.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 15.Gough MJ, et al. OX40 agonist therapy enhances CD8 infiltration and decreases immune suppression in the tumor. Cancer Res. 2008;68:5206–5215. doi: 10.1158/0008-5472.CAN-07-6484. [DOI] [PubMed] [Google Scholar]
- 16.Gramaglia I, et al. The OX40 costimulatory receptor determines the development of CD4 memory by regulating primary clonal expansion. J Immunol. 2000;165:3043–3050. doi: 10.4049/jimmunol.165.6.3043. [DOI] [PubMed] [Google Scholar]
- 17.Gramaglia I, et al. Ox-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J Immunol. 1998;161:6510–6517. [PubMed] [Google Scholar]
- 18.Grolleau-Julius A, et al. Effect of aging on bone marrow-derived murine CD11c+ CD4-CD8alpha-dendritic cell function. J Gerontol A Biol Sci Med Sci. 2006;61:1039–1047. doi: 10.1093/gerona/61.10.1039. [DOI] [PubMed] [Google Scholar]
- 19.Haynes L, Eaton SM. The effect of age on the cognate function of CD4+ T cells. Immunol Rev. 2005;205:220–228. doi: 10.1111/j.0105-2896.2005.00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaleeba JA, et al. The OX-40 receptor provides a potent co-stimulatory signal capable of inducing encephalitogenicity in myelin-specific CD4+ T cells. Int Immunol. 1998;10:453–461. doi: 10.1093/intimm/10.4.453. [DOI] [PubMed] [Google Scholar]
- 21.Kawamata S, et al. Activation of OX40 signal transduction pathways leads to tumor necrosis factor receptor-associated factor (TRAF) 2- and TRAF5-mediated NF-kappaB activation. J Biol Chem. 1998;273:5808–5814. doi: 10.1074/jbc.273.10.5808. [DOI] [PubMed] [Google Scholar]
- 22.Kjaergaard J, et al. Therapeutic efficacy of OX-40 receptor antibody depends on tumor immunogenicity and anatomic site of tumor growth. Cancer Res. 2000;60:5514–5521. [PubMed] [Google Scholar]
- 23.Krieg AM. Development of TLR9 agonists for cancer therapy. J Clin Invest. 2007;117:1184–1194. doi: 10.1172/JCI31414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuriyama H, et al. Mechanism of third signals provided by IL-12 and OX-40R ligation in eliciting therapeutic immunity following dendritic-tumor fusion vaccination. Cell Immunol. 2006;243:30–40. doi: 10.1016/j.cellimm.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 25.Lee SW, et al. Functional dichotomy between OX40 and 4–1BB in modulating effector CD8 T cell responses. J Immunol. 2006;177:4464–4472. doi: 10.4049/jimmunol.177.7.4464. [DOI] [PubMed] [Google Scholar]
- 26.Li SP, et al. Early antigen-specific response by naive CD8 T cells is not altered with aging. J Immunol. 2002;168:6120–6127. doi: 10.4049/jimmunol.168.12.6120. [DOI] [PubMed] [Google Scholar]
- 27.Linton PJ, et al. From naive to effector—alterations with aging. Immunol Rev. 1997;160:9–18. doi: 10.1111/j.1600-065X.1997.tb01023.x. [DOI] [PubMed] [Google Scholar]
- 28.Linton PJ, et al. Intrinsic versus environmental influences on T-cell responses in aging. Immunol Rev. 2005;205:207–219. doi: 10.1111/j.0105-2896.2005.00266.x. [DOI] [PubMed] [Google Scholar]
- 29.Lustgarten J, et al. Aged mice develop protective antitumor immune responses with appropriate costimulation. J Immunol. 2004;173:4510–4515. doi: 10.4049/jimmunol.173.7.4510. [DOI] [PubMed] [Google Scholar]
- 30.Maxwell JR, et al. Danger and OX40 receptor signaling synergize to enhance memory T cell survival by inhibiting peripheral deletion. J Immunol. 2000;164:107–112. doi: 10.4049/jimmunol.164.1.107. [DOI] [PubMed] [Google Scholar]
- 31.Mittler JN, Lee WT. Antigen-specific CD4 T cell clonal expansion and differentiation in the aged lymphoid microenvironment. I. The primary T cell response is unaffected. Mech Ageing Dev. 2004;125:47–57. doi: 10.1016/j.mad.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 32.Murata K, et al. Constitutive OX40/OX40 ligand interaction induces autoimmune-like diseases. J Immunol. 2002;169:4628–4636. doi: 10.4049/jimmunol.169.8.4628. [DOI] [PubMed] [Google Scholar]
- 33.Ohshima Y, et al. Expression and function of OX40 ligand on human dendritic cells. J Immunol. 1997;159:3838–3848. [PubMed] [Google Scholar]
- 34.Prins RM, et al. The TLR-7 agonist, imiquimod, enhances dendritic cell survival and promotes tumor antigen-specific T cell priming: relation to central nervous system antitumor immunity. J Immunol. 2006;176:157–164. doi: 10.4049/jimmunol.176.1.157. [DOI] [PubMed] [Google Scholar]
- 35.Redmond WL, et al. Defects in the acquisition of CD8 T Cell effector function after priming with tumor or soluble antigen can be overcome by the addition of an OX40 agonist. J Immunol. 2007;179:7244–7253. doi: 10.4049/jimmunol.179.11.7244. [DOI] [PubMed] [Google Scholar]
- 36.Ruby CE, et al. IL-12 is required for anti-OX40-mediated CD4 T cell survival. J Immunol. 2008;180:2140–2148. doi: 10.4049/jimmunol.180.4.2140. [DOI] [PubMed] [Google Scholar]
- 37.Ruby CE, et al. Anti-OX40 stimulation in vivo enhances CD8(+) memory T cell survival and significantly increases recall responses. Eur J Immunol. 2007;37:157–166. doi: 10.1002/eji.200636428. [DOI] [PubMed] [Google Scholar]
- 38.Ruby CE, Weinberg AD. OX40-enhanced tumor rejection and effector T cell differentiation decreases with age. J Immunol. 2009;182:1481–1489. doi: 10.4049/jimmunol.182.3.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salek-Ardakani S, Croft M. Regulation of CD4 T cell memory by OX40 (CD134) Vaccine. 2006;24:872–883. doi: 10.1016/j.vaccine.2005.07.108. [DOI] [PubMed] [Google Scholar]
- 40.Sharma S, et al. CpG-ODN but not other TLR-ligands restore the antitumor responses in old mice: the implications for vaccinations in the aged. Cancer Immunol Immunother. 2008;57:549–561. doi: 10.1007/s00262-007-0393-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shurin GV, et al. Regulation of dendritic cell expansion in aged athymic nude mice by FLT3 ligand. Exp Gerontol. 2004;39:339–348. doi: 10.1016/j.exger.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 42.So T, Croft M. Cutting edge: OX40 inhibits TGF-beta- and antigen-driven conversion of naive CD4 T cells into CD25+ Foxp3+ T cells. J Immunol. 2007;179:1427–1430. doi: 10.4049/jimmunol.179.3.1427. [DOI] [PubMed] [Google Scholar]
- 43.Song A, et al. Cooperation between CD4 and CD8 T cells for anti-tumor activity is enhanced by OX40 signals. Eur J Immunol. 2007;37:1224–1232. doi: 10.1002/eji.200636957. [DOI] [PubMed] [Google Scholar]
- 44.Takeda I, et al. Distinct roles for the OX40—OX40 ligand interaction in regulatory and nonregulatory T cells. J Immunol. 2004;172:3580–3589. doi: 10.4049/jimmunol.172.6.3580. [DOI] [PubMed] [Google Scholar]
- 45.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 46.Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 47.Valzasina B, et al. Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood. 2005;105:2845–2851. doi: 10.1182/blood-2004-07-2959. [DOI] [PubMed] [Google Scholar]
- 48.Vu MD, et al. OX40 costimulation turns off Foxp3+ Tregs. Blood. 2007;110:2501–2510. doi: 10.1182/blood-2007-01-070748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Watford WT, et al. The biology of IL-12: coordinating innate and adaptive immune responses. Cytokine Growth Factor Rev. 2003;14:361–368. doi: 10.1016/S1359-6101(03)00043-1. [DOI] [PubMed] [Google Scholar]
- 50.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 51.Weinberg AD, et al. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. J Immunol. 2000;164:2160–2169. doi: 10.4049/jimmunol.164.4.2160. [DOI] [PubMed] [Google Scholar]
- 52.Weinberg AD, et al. Anti-OX40 (CD134) administration to nonhuman primates: immunostimulatory effects and toxicokinetic study. J Immunother. 2006;29:575–585. doi: 10.1097/01.cji.0000211319.00031.fc. [DOI] [PubMed] [Google Scholar]
- 53.Weinberg AD, et al. OX-40: life beyond the effector T cell stage. Semin Immunol. 1998;10:471–480. doi: 10.1006/smim.1998.0146. [DOI] [PubMed] [Google Scholar]
- 54.Weinberg AD, et al. Blocking OX-40/OX-40 ligand interaction in vitro and in vivo leads to decreased T cell function and amelioration of experimental allergic encephalomyelitis. J Immunol. 1999;162:1818–1826. [PubMed] [Google Scholar]