

Abstract
IL-12 is a cytokine which showed anti-tumor effects in clinical trials, but also produced serious toxicity. We describe a fusion protein, huBC1-IL12, designed to achieve an improved therapeutic index by specifically targeting IL-12 to tumor and tumor vasculature. huBC-1 is a humanized antibody that targets a cryptic sequence of the human ED-B-containing fibronectin isoform, B-FN, present in the subendothelial extracellular matrix of most aggressive tumors. B-FN is oncofetal and angiogenesis-associated, and is undetectable in most normal adult tissues. The original murine BC-1 antibody has been used successfully for immunoscintigraphy to image brain tumor mass in glioblastoma patients. In huBC1-IL12, each of the IgG heavy chains is genetically fused to the N-terminus of the IL-12 p35 subunit, which in turn is disulfide-bonded to the p40 subunit, resulting in a hexameric molecule of MW of ∼300 kDa. Since human IL-12 has no biological activity in mice, we produced huBC1-muIL12 as a surrogate molecule for animal tumor models. Despite the relatively poor PK profile of this molecule in mice and the apparent drawbacks of xenogeneic models in SCID mice, which lack T and B cells, one cycle of treatment with huBC1-muIL12 was efficacious in the PC3mm2, A431, and HT29 subcutaneous tumor models and PC3mm2 lung metastasis model. This molecule also was found to have surprisingly low toxicity in immunocompetent mice. A fusion protein that contains human IL-12 (huBC1-huIL12), which is a suitable molecule for investigation as a therapeutic, has also been produced. This protein has been shown to have a longer serum half-life than huBC1-muIL12 in mice, and retains both antigen binding and IL-12 activity in in vitro assays.
Keywords: HuBC1-IL12, Immunocytokine, ED-B fibronectin, IL-12
Introduction
Interleukin-12 (IL-12) is a pleiotropic cytokine that mediates both innate and adaptive immunity. It stimulates the cellular cytotoxicity of NK cells and CTLs, and induces the development of Th1 cells [35]. The resultant increase in IFNγ production by these cells in turn activates macrophages and induces the expression of Mig (monokine induced by interferon gamma) and IP-10 (interferon-inducible protein 10), which are chemokines with potent anti-angiogenic and anti-tumor activities [34]. Since human IL-12 is not active in mice [38], murine IL-12 had been tested in its place in metastasis and skin tumor models and was shown to be highly efficacious [3]. Tumor responses have been observed in clinical trials with IL-12 [13], although the use of recombinant human IL-12 in the clinic has been hampered by its severe toxicity [20, 26].
One approach to limit systemic toxicity is to deliver IL-12 to the tumor site, such as by direct intratumoral injection. However, in most clinical settings, especially when multiple tumors are involved, intravenous administration is more practical. The concept of using tumor-specific antibodies to target cytokines to the tumor microenvironment in order to enhance the immune response has been validated in many mouse models (reviewed in [7]). The same approach has been applied successfully to deliver IL-12 to metastases in the prostate and colon by targeting the pan-carcinoma antigen EpCAM, which is expressed at high levels on virtually all epithelial cancers [11]. However, for large solid tumors, because of high interstitial pressure, the ability of an antibody to effectively penetrate the tumor mass is always a challenge, and the task of delivering a more complex antibody-IL12 fusion protein is even more formidable. In light of this, we used an alternative approach that takes advantage of the proximity and accessibility of the extracellular matrix (ECM) of the neovasculature. We utilized a monoclonal antibody, BC-1, which specifically recognizes a fibronectin isoform, B-FN [25]. This is an oncofetal antigen highly expressed in fetal and tumor tissues, but with extremely restricted distribution in normal adult tissues, e.g., endometrium. It is produced by tumor cells and deposited in the subendothelial ECM in solid tumors [27], possibly playing a role to promote angiogenesis to support tumor growth [24]. Bone-marrow-derived circulating endothelial progenitor cells are now known to contribute significantly to the neovasculature, especially in the repair of the tumor endothelium after chemotherapy [1], and it has been suggested that ECM components such as fibronectin play a role in the recruitment and differentiation of such cells [31]. There is also substantial evidence that integrin-mediated attachment of tumor cells and bone-marrow-derived hematopoietic progenitor cells to fibronectin is important for invasion and metastasis [18, 30]. Therefore, B-FN-specific delivery of IL-12, which has potent immunostimulatory and anti-angiogenic activities, to the ECM of the neovasculature, is an attractive approach to inhibit both tumor growth and metastatic spread, as well as angiogenesis.
The B-FN isoform, as a result of alternate splicing in oncofetal tissues, contains an extra domain B (ED-B), a complete type III homology repeat composed of 91 amino acid residues, which are identical in mouse, rat, rabbit, dog, monkey and human [28]. The epitope recognized by the murine monoclonal BC-1 is not in the ED-B domain of the human B-FN, but in the proximal region of the adjacent domain seven, the sequence of which is different in humans and mice. This epitope is cryptic in normal fibronectin, but the presence of ED-B causes a conformational change that unmasks it [4]. The use of B-FN as a neoplastic vasculature marker has been validated in preclinical and clinical studies [28, 32]. Indeed it is the best available indicator to differentiate between high- and low-grade astrocytoma [5]. In this regard, the murine BC-1 antibody has been used successfully for immunoscintigraphy to image brain tumor mass in glioblastoma patients [25]. For potential therapeutic application, the variable regions of the antibody have been humanized by CDR grafting. The resulting huBC-1 antibody, with human gamma-1 and kappa constant regions, has increased binding affinity to B-FN of about seven-fold (unpublished results).
Here we describe the high-level expression and anti-tumor activity of a huBC-1 antibody-IL-12 (huBC1-IL12) fusion protein. IL-12 is a heterodimeric molecule composed of a p35 and a p40 subunit linked by a disulfide bond. In producing the huBC1-IL12 fusion protein, we took advantage of the fact that the p35 subunit cannot be secreted independent of the p40, and genetically fused the end of the Ig H chain to the mature N-terminus of the p35 subunit [11]. Since the H chain, analogous to the p35 subunit, also cannot be secreted in the absence of the L chain, purification of the antibody-IL12 fusion protein by Protein A, which binds to the Fc region of the Ig, ensures that stoichiometric amounts of p40 and the L chain are present in the hexameric product. For preclinical efficacy studies, we used several human tumor cell lines in xenogeneic tumor models, because huBC-1 recognizes only the human B-FN and does not cross-react with the murine B-FN. In addition, since human IL-12 has no biological activity in mice, we produced the fusion protein with both the human and murine IL-12, i.e., huBC1-huIL12 as a clinical candidate, and huBC1-murine IL-12 as a surrogate molecule for evaluation in murine models.
Materials and methods
Cell lines, reagents and animals
The mouse myeloma NS/0 cell line was obtained from the European Collection of Animal Cell Cultures (Salisbury, UK). The NK-92 human lymphoblast, A431 human epidermoid carcinoma, HT29 human colon adenocarcinoma, and human astrocytoma U-87 MG cell lines were obtained from American Type Culture Collection (Manassas, VA). The PC3mm2 human prostate adenocarcinoma cell line was a gift from Dr. Ralph Reisfeld at Scripps Research Institute. All cell culture media were purchased from Invitrogen (Carlsbad, CA). All cytokines and antibodies, including the antibody pair (duoset) used for the human IFNγ ELISA, were purchased from R&D Systems (Minneapolis, MN). Phytoagglutinin PHA-P was purchased from Sigma (St. Louis, MO). The human IL-12 standard was obtained from National Institute for Biological Standards and Control (NIBSC, Hertfordshire, UK). SCID CB17 mice (male) and Balb/c (female) mice (8–9 weeks old) were purchased from Charles River Labs (Wilmington, MA).
Construction of the expression vectors for huBC1-IL12
The DNA construction and expression of antibody-IL12 fusion proteins already have been described [11]. For the construction of the expression plasmid for huBC1-p35, the DNA encoding the variable region of the light chain (VL) of huBC1 in plasmid RKA.pMMR010 was adapted by Polymerase Chain Reaction (PCR) with the forward primer 5′-C TTA AGC GAA ATT GTG TTG ACG CAG TC-3′, where CTTAAG is an AflII restriction site and GAA is the N-terminal amino acid residue of the mature VL, and the reverse primer 5′- GGATCCACTTACG TTT GAT CTC CAG CTT GG-3′, where the underlined sequence hybridized to the 3′ end of the sense strand of the VL and GGATCC adds a BamHI restriction site downstream of the VL splice donor site. The DNA encoding the variable region of the heavy chain (VH) of huBC-1 in plasmid RHA.pGamma1was adapted by PCR using the forward primer 5′-C TTA AGC GAG GTG CAG CTG GTG CAG TC-3′, where CTTAAG is an AflII restriction site and GAG is the N-terminal amino acid residue of the mature VH, and the reverse primer 5′-AAGCTTACT TAC CTG AGG AGA CGG AGA CC-3′, where the underlined sequence hybridized to the 3′ end of the sense strand of VH and AAGCTT adds a HindIII restriction site downstream of the VH splice donor site. The AflII site at the 5′ end of the VL and VH restriction fragments was used to join the variable region to a genomic signal peptide sequence from a mouse immunoglobulin (IgG) light chain gene used for secretion [23]. The BamHI and the HindIII sites at the 3′ end of the VL and VH restriction fragments, respectively, were used to join the variable regions to the constant regions of either the L chain or the H chain-p35 already present in the expression vector [11]. The DNA sequence encoding the huBC-1 H chain-human p35 fusion junction LSLSPGKRNLPV (where LSLSPGK is the C-terminus of the human IgG1 H chain and RNLPV is the mature N-terminus of the human IL-12 p35 subunit) was modified to ATATPGAANLPV, in which the KR to AA substitutions removed a potential proteolytic site and the LSLS to ATAT substitutions removed a potential novel T cell epitope identified in silico, using a process called deImmunization [17].
The huBC1-human IL-12 fusion protein (huBC1-huIL12) was expressed by transfection of the huBC1-human p35 subunit fusion construct into an NS/0 transfectant (selected for neomycin resistance) already expressing high levels of the p40 subunit of human IL-12, followed by selection in methotrexate-containing medium [11]. Similarly, the huBC1-murine IL-12 fusion protein (huBC1-muIL12) was expressed by transfection of the huBC1-murine p35 subunit fusion construct into an NS/0 transfectant already expressing high levels of the p40 subunit of murine IL-12.
IL-12 bioassays
Three bioassays were used to measure IL-12 bioactivity: proliferation of human PBMC, and induction of IFNγ from human PBMC or the human cell line NK-92 [29]. In the first assay, PBMC were cultured with PHA-P for 5 days, washed and then plated in 96-well plates containing dilutions of IL-12 proteins. After incubation for 48 h at 37°C and 5% CO2, proliferation was measured by pulsing with 3H–Thymidine in the last 16 h of the assay [11]. In the second assay, PBMC were cultured with PHA-P for 3 days and then 25 IU/ml of human IL-2 was added for an additional 24 h. The cells were washed and plated in 96-well plates containing 20 IU/ml of human IL-2 and dilutions of IL-12 proteins. Twenty-four hours later, the concentration of IFNγ was measured by ELISA [19]. The third assay was similar to the second assay, except that the culturing step with PHA-P for 3 days and human IL-2 for an additional 24 h was not necessary for NK-92 cells. Recombinant human IL-12 made in Chinese hamster ovary cells from NIBSC, and human or mouse IL-12 made in baculovirus (R&D Systems), were used as internal reference standards in every assay to attain the maximum response used for ED50 calculations. The ED50 concentration was determined using least squares analysis (TREND analysis from Excel). Activities of IL-12 and IL-12 fusion proteins were normalized to IL-12 concentrations in ng/ml.
Pharmacokinetic analysis
Balb/c mice were injected with 25 μg of huBC1-huIL12 or huBC1-muIL12 in a volume of 0.2 ml in the tail vein using a slow push. At various time points, small blood samples were taken by retro-orbital bleeding and collected in tubes coated with heparin to prevent clotting. After centrifugation to remove the cells, the plasma was assayed by capture with anti-human IgG H&L antisera and detection with an anti-human or anti-murine IL12 antibody. Results were normalized and plotted as percentages of the initial concentration in the plasma of each mouse taken within 30 s after injection (t = 0).
Tumor models
Experimental lung metastases were induced by tail vein injection of 2 × 106 viable single PC3mm2 cells in 0.2 ml PBS into SCID CB17 mice on day 0. On Day 11, the mice were treated by i.v. injection of huBC1-muIL12 (5 daily doses of 8 or 16 μg) or control PBS (n = 8). On day 28, when the control mice started to get sick, all mice were sacrificed. Lungs were removed, weighed and fixed in Bouin’s solution. Metastases in the lungs were counted and scored, and organ weights were measured as previously described [11]. For the skin tumor models, SCID mice were injected subcutaneous dorsa with 1 × 106 A431 or HT29 cells, or 2 × 106 PC3mm2 cells. When the tumor size reached about 100 mm3, the mice were sorted into four groups (n = 8), and received seven daily intravenous injections of PBS control, huBC1-muIL12 fusion protein (20 μg), murine IL12 (1.5 μg), or a combination of huBC-1 antibody (10 μg) and murine IL-12 (1.5 μg).
Tumor volumes were determined at different time points using the formula width2 × length × 0.5236. Anti-tumor efficacy was reported as a T/C ratio, where T and C are the average tumor volumes of the fusion protein-treated and PBS control-treated mice, respectively.
Immunohistochemistry with huBC-1
Selected frozen tissues were cut with a cryotome into 2–4 μm thin sections, mounted on silanized glass slides (Sigma), air-dried and fixed in − 20°C cold acetone for 10 min, and washed in distilled water. Sections were incubated in PBS for 5 min at room temperature (RT). Endogenous peroxidases were blocked by incubation in a freshly prepared solution of 0.3% H2O2 in methanol for 15 min at RT followed by washing in distilled water and in PBS for 5 min each.
Prior to application of antibody huBC-1 to the section, the antibody was biotinylated (working dilution 1:450, 24 mg/ml). Biotinylation of huBC-1 was done using biotinylated anti-human Fab fragments (Jackson Immuno Research, West Grove, PA). Equal amounts of antibody [1:450 in antibody dilution solution (DAKO, Santa Barbara, CA)] and Fab fragments were mixed in a test tube and incubated for 15 min at RT. Then normal human serum (Jackson Immuno Research) was added (10% of total volume) and the mixture was incubated for 5 min at RT. The biotinylated antibody was then ready to use.
Calculations were done with help of the ARKulator programme from DAKO. Sections were immediately incubated with this biotinylated huBC-1 antibody for 1 h in a humidified chamber at RT. After three washes in TBS, the sections were incubated with Streptavidin/HRP (DAKO, 1:150 dilution) for 30 min at RT. After washing as before the sections were incubated with DAB solution (DAKO; 1:50 dilution in substrate buffer) for 10 min at RT. Finally the slides were rinsed in water, counterstained with Harris’ hematoxylin, covered with a glass slide and read.
Results
Expression of huBC1-IL12 fusion proteins
In huBC1-IL12, each of the IgG heavy chains is genetically fused to the N-terminus of the IL-12 p35 subunit, which in turn is disulfide-bonded to the p40 subunit. To express the protein, a stable NS/0 clone expressing a high level of the p40 subunit was first obtained using G418 for selection. This was followed by a second transfection with an expression plasmid containing separate transcription units for the IgG light chain and the heavy chain-p35 subunit, using methotrexate for selection [11]. The resulting hexameric molecule, consisting of two of each of the IgG light chains, the IgG heavy chain-p35, and the p40, has a MW of approximately 300 kDa (Fig. 1). The multiple bands between the light and heavy chains have previously been shown to react with anti-p40 antibody by Western blotting analysis and are presumably glycosylation variants [11], with the human p40 (MW of 34.7 kDa for the polypeptide backbone and four potential N-glycosylation sites) significantly smaller than the murine p40 (MW of 35.8 kDa for the polypeptide backbone and five potential N-glycosylation sites). Both fusion proteins were expressed at high levels and secreted efficiently into the culture medium. For the stable clones producing huBC1-huIL12, a titer of 300 mg/L was attained in conditioned medium.
Fig. 1.

Size exclusion chromatography and SDS-PAGE analysis of huBC1-IL12. The chromatogram depicts the analytical SEC behavior of huBC1-huIL12, which runs primarily as a monomeric species. The inset SDS-PAGE gel compares the electrophoretic mobility of 1 huBC1-huIL12 and 2 huBC1-muIL12. The heavy chain-p35 (HC-p35), light chain (LC) and p40 subunits are indicated. The different sets of bands for the human and murine p40 are due to the multiple glycosylation patterns of the subunits
Since huBC1-huIL12 is intended for clinical development, additional genetic modifications were made to enhance its biological properties. The C-terminal residue of the CH3 domain is lysine and the N-terminal residue of the mature p35 is arginine. A direct fusion will bring these two basic residues together, forming a potential proteolytic site. Therefore, both residues were mutagenized to alanine to minimize proteolysis at the fusion junction, which, in the case of IL-2 immunocytokine, has been shown to extend serum half-life [12]. Furthermore, the peptide sequence at the junction, LSLSPGAANLPV, where AA are the two alanine substitutions, is novel and potentially immunogenic. Indeed, in silico analysis showed that this sequence binds to a number of MHC Class II alleles on T helper cells. Therefore we tried to minimize the potential immunogenicity of huBC1-huIL12 by mutating the LSLS to ATAT, which removed the potential T-cell epitope.
Bioactivity of huBC1-IL12
The bioactivity for huBC1-IL12 was determined by three different cell-based assays. The first two measured proliferation and induction of IFNγ using human PBMC, whereas, the third measured induction of IFNγ from the human lymphoblast cell line NK-92 [29]. All three assays can be used to measure the activity of both human and mouse IL-12. In the human PBMC proliferation assay, huBC1-huIL12 and huBC1-muIL12 showed about 10-fold and 5-fold less activity, respectively, than the free IL-12 standards (Fig. 2a, b).
Fig. 2.

Relative bioactivity of huBC1-IL12 and huBC1-muIL12 compared to free IL-12 reference standards. (a, b) Proliferation responses from human PBMC PHA blasts. (c, d) Induction of IFNγ from human PBMC PHA blasts. (e) Induction of IFNγ from the human NK cell line NK-92. In a, c and e, recombinant human IL-12 made in Chinese hamster ovary cells from NIBSC (filled squares) and made in baculovirus (solid circles) were used as reference standards for huBC1-IL12 (open circles). In b and d, recombinant murine IL-12 made in baculovirus (solid circles) was used as a reference standard for huBC1-muIL12 (open triangles). Data presented are typical of at least three experiments and all concentrations were in terms of IL-12. Error bars indicate standard errors
It is well known that IL-12 has a critical role in the development of CD4+ T helper cells into Th1 cells through the induction of IFNγ [37]. Using activated human PBMC in this assay, huBC1-huIL12 and huBC1-muIL12 showed about 3–9 fold and 7–14 fold less induction of IFNγ, respectively, than the free IL-12 standards (Fig. 2c, d).
Since the activity of human PBMC can vary from donor to donor, and the activation of NK cells may be important for anti-tumor activity, we developed a bioassay using the human lymphoblast cell line NK-92, which is an IL-2 dependent NK cell line that secretes newly synthesized IFNγ in response to IL-12 [29, 36]. In this assay, huBC1-huIL12 had 17% activity compared to the human IL-12 standards (Fig. 2e). Since the murine IL-12 standard has 10-fold less activity than human IL-12 (data not shown), the activity of the huBC1-muIL12 was not determined in this assay.
Immunohistochemistry
Fifteen fresh-frozen samples of renal cell carcinoma samples [6 cases were clear cell tumors, 4 cases were clear cell compact tumors, 4 were tumors of chromophil type and one oncocytoma of the kidney (benign non-metastatic tumor)] were stained with the huBC-1 antibody and respective control according to the described protocol for fresh-frozen tissue and evaluated by a pathologist (Klinikum Kassel GmbH, Germany). The staining looked clear and specific. We observed staining of tumor blood vessels and tumor cells (Fig. 3).
Fig. 3.
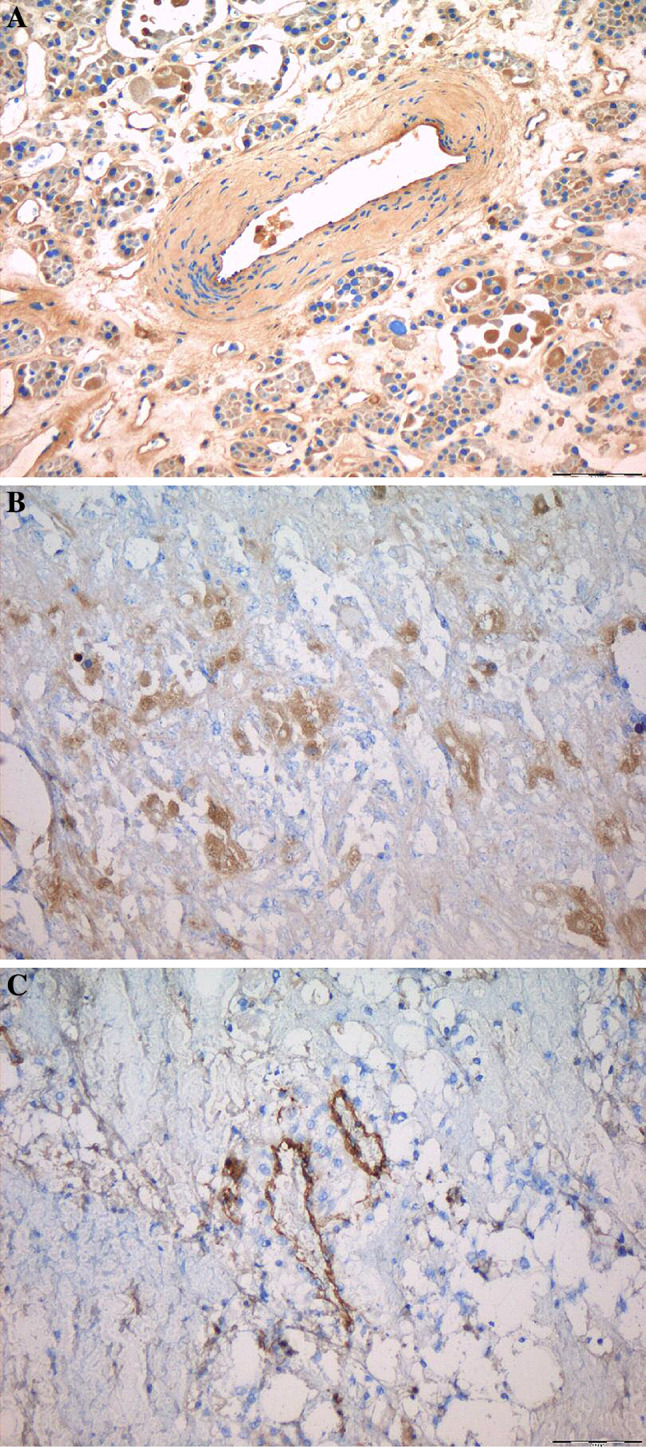
Immunohistochemistry of renal tumors with huBC-1. Figure shows photomicrographs of 2–4 μm thin sections of fresh-frozen renal tumors. All samples showed staining of tumor blood vessels (represented by a 10× huBC-1 oncocytoma, b 10× huBC-1 chromophil type tumor and c 10× huBC-1 clear cell tumor), and there was also evidence of staining of tumor cells (represented by a 10× huBC-1 oncocytoma)
In vivo pharmacokinetic studies
We used a two-compartment model to determine the pharmacokinetic behavior of huBC1-muIL12 and huBC1-huIL12 in mice. The plasma level-time curve shows a biexponential curve of two first-order processes (Fig. 4). At t = 0, the injected protein had distributed instantaneously throughout the blood compartment. In the first 30 min, there was a steep decline in plasma level as the fusion protein distributed rapidly from the blood compartment into the tissue compartment, with a half-life (t ½ α) of 0.12 h for huBC1-muIL12 and 0.18 h for huBC1-huIL12. After the steep α phase, both molecules had a fairly long circulating half-life (t ½ β) of about 19 h. One reason for the long circulating half-life may be because, as is the case with most small-molecule drugs, some of the fusion protein initially distributed into the tissue was being returned into circulation, e.g., via the FcRn [10].
Fig. 4.

Pharmacokinetic analysis of huBC1-IL12 in Balb/c mice after intravenous bolus dose administration. The concentrations of huBC1-muIL12 (open triangles) and huBC1-huIL12 (open circles) present in the plasma at different time points were assayed by an ELISA that detected only the intact fusion protein. The protein concentrations shown in the plasma level-time curve were normalized as percent of the initial concentration in the blood compartment of each mouse taken immediately after injection (t = 0). Error bars indicate standard errors
In vivo efficacy studies
Designing a relevant model for testing the clinical candidate huBC1-huL12 presented a few problems. Since human IL-12 is highly species specific and does not have any biological activity in mice, we first produced huBC1-muIL12 as a surrogate molecule for animal tumor models. In addition, the model has to be xenogeneic since the huBC-1 recognizes only the human B-FN secreted by human tumor cells and does not cross-react with the murine B-FN.
Lung metastasis model in SCID mice
The first model tested was a human prostate carcinoma PC3mm2 lung metastasis model in SCID mice. Treatment began 11 days after intravenous injection of tumor cells, allowing ample time for metastases to establish. Despite the lack of functional T and B cells in the SCID mice, five daily i.v. injections of huBC1-muIL12 at 16 μg almost completely eradicated the established metastases in all the mice and prevented their outgrowth, as measured by the lung surface covered by metastasis and tumor burden (Fig. 5a). Even the 8 μg dosage was very effective, reducing the lung metastases by about 85%, relative to the PBS control.
Fig. 5.

Efficacy in human tumor models in SCID mice. a PC3 metastasis model. Upper panel, percent lung surface covered by metastases; lower panel, percent lung weight normalized to body weight (lung weight/initial body weight × 100%). Normal lungs weigh about 0.18–0.2 g, which is approximately 1% of the body weight of a mouse (line across in lower panel). b PC3 skin tumor model. c HT29 skin tumor model. d A431 skin tumor model. In b, c and d, tumor-bearing mice were treated with seven daily i.v. injections of PBS (filled triangles), 20 μg of huBC1-muIL12 (filled diamonds), a combination of 10 μg of huBC-1 antibody and 1.5 μg of muIL12 (open diamonds), or 1.5 μg of muIL12 (crosses). Error bars indicate standard errors
Subcutaneous human tumor xenograft in SCID mice
We next used three different human tumor cell lines, the PC3mm2, A431 epidermoid carcinoma, and HT29 colon carcinoma to form skin tumors in SCID mice. When the tumor size reached about 100 mm3, the mice received a single-cycle of seven daily consecutive intravenous injections of PBS control, huBC1-muIL12 fusion protein (20 μg), murine IL-12 (1.5 μg), or a combination of huBC-1 antibody (10 μg) and murine IL-12 (1.5 μg). Based on in vitro IFNγ induction assays, the 1.5 μg of free murine IL-12 (20 pmoles of IL-12) and the 20 μg of the huBC1-muIL12 fusion protein (containing 133 pmoles of IL-12) have about equivalent IL-12 activity.
The single-cycle treatment with huBC1-muIL12 was effective against the PC3mm2 prostate tumors, achieving a T/C ratio of 0.33 on day 21 (P < 0.001) (Fig. 5b). The free muIL-12 and the combination free antibody and mu IL-12 were less effective, with T/C ratios of 0.63 and 0.60, respectively (P < 0.001 versus the control group, for both), suggesting that the physical linkage of the antibody to the cytokine is important to confer anti-tumor efficacy (P < 0.001 versus the fusion protein group, for both). Similar results were obtained against the HT29 and A431 skin tumors. In the HT29 tumor model, the T/C ratios of 0.48, 0.72 and 0.71 on day 19 for the groups receiving the fusion protein, free muIL-12, and the combination, respectively, were all statistically significant (P < 0.001) (Fig. 5c). The fusion protein was again significantly better than the free muIL-12 and the combination groups (P = 0.001). In the A431 tumor model, the T/C ratios of 0.44, 0.61 and 0.60 on day 18 for the groups receiving the fusion protein, free muIL-12, and the combination, respectively, were all statistically significant (P < 0.02) (Fig. 5d). However, the fusion protein was not significantly better than the free muIL-12 and the combination groups in this model (P = 0.2).
Discussion
Despite the complexity of the hexameric molecule, we achieved a production level of 300 mg/L in stationary culture for our clinical candidate huBC1-huIL12. Our strategy also ensures that no excess p40, which can be secreted in the form of a disulfide-linked homodimer, is present in the purified product. Both human and murine IL-12 p40 homodimers had been shown to be able to block the function of the heterodimer in vitro and in vivo [8, 22], and in order to minimize the homodimer formation, Ha et al. [14] took a different approach by mutagenizing an N-glycosylation site in p40 to decrease its secretion. Another consideration in our molecular design is to have a free p40 amino terminus, because studies on single-chain human and murine IL-12 showed that in order to maintain biological activity, the p40 had to be at the N-terminus in both cases [21].
Using human PBMC PHA-blast and the NK-92 cell line, the human IL-12 moiety in BC1-huIL12 was found to have several-fold lower activity than the free human IL-12 in both the proliferation and the IFNγ induction assays. The murine counterpart was also found to have several-fold lower activity than the free murine IL-12 in the assays with human PBMC PHA-blasts. It is interesting to note that while both human and murine IL-12 have similar ED50 with human PBMC PHA-blasts, which are mostly activated T cells, the latter had 10-fold less activity than the former with the human NK cell line NK-92. This difference may be explained by differential binding of murine IL-12 to human IL-12 receptor subunits [9]. Overall, the BC1-IL12 fusion proteins have similar bioactivity as other Ab-IL12 fusion proteins, previously reported, and the reduced activity was likely a result of steric hindrance due to the large MW of this cytokine [11]. Given the highly toxic nature of IL-12, this may be an advantage because lower activity would allow for the administration of a higher dosage. This may be clinically relevant because in addition to the potent immunostimulatory activities of IL-12, the IgG1 isotype of the antibody fusion protein also has potent effector functions such as ADCC, which has been shown to play an important role in the mechanism of action for Rituxan and Herceptin [6]. While the activity of IL-12 is dose-limiting, effector functions should increase with a higher dosage.
Since the clinical candidate huBC1-huIL12 cannot be evaluated in murine tumor models due to the species-specificity of human IL-12, we produced the surrogate molecule huBC1-muIL12. Nevertheless, the models are limited to xenogeneic tumors in SCID mice, because BC-1 recognizes only human B-FN. Inspite of the fact that these mice lacked functional T and B cells, huBC1-muIL12 was efficacious in various metastasis and subcutaneous tumor models. A single-cycle treatment of 5–7 daily i.v. injections of huBC1-muIL12 almost completely eradicated established metastases and inhibited tumor growth by 67, 52 and 56%, respectively, in the PC3, HT29, and A431 established skin tumor models. Previously we demonstrated that the anti-EpCAM antibody-huIL12 was superior to an irrelevant antibody-huIL12 control with the same IL-12 activity and pharmacokinetic properties, in a SCID mouse xenograft model reconstituted with human PBMCs [11]. Due to the lack of a non-targeting antibody-muIL12 control in the current study, we cannot rule out the possibility that the improved efficacy of huBC1-muIL12, relative to huBC1 antibody plus free murine IL-12, was a result of prolonging the serum half-life of IL-12. However, the relationship between serum half-life and anti-tumor efficacy is complicated by the fact that IL-12 (both murine and human) binds to heparin and heparan sulphate, which are abundant in the ECM [16]. This binding can partly explain the very steep α phase of huBC1-muIL12 observed in mice. Presumably the fusion protein may be deposited in the ECM of the tissue compartment, and a slow release mechanism that may involve FcRn contributes to the long β-half life. In this respect, the β-half life of 19 h for huBC1-mIL12 is significantly longer than that of another B-FN targeting fusion protein, mIL12-L19, comprising a single-chain murine IL12 fused with scFv(L19) [15], which does not have the benefit of the protective effect of the Fc-FcRn interaction [10]. Although the β-half life of the latter was not reported, a similar molecule, L19 mTNFα, had a β-half life of only 3.9 h, as compared to 106.7 h for the L19 IgG [2].
Another possible explanation for the better efficacy of huBC1-muIL12 is that the systemically administered fusion protein can undergo proteolysis to yield a more active free form of IL-12, especially at the tumor site where proteinase activities are very high. Evidence for such cleavage in vivo was provided in our pharmacokinetic analyses, in which ELISA showed that the amount of intact fusion protein detected in the serum samples was less than that of the human antibody component (representing both intact and cleaved species) [11]. This was supported by a western blot analysis, probed with anti-human Fc, which showed the presence of a degradation product with an apparent molecular weight of the human IgG H chain (data not shown).
It is interesting to note that all the antibody-muIL12 fusion proteins we have tested (such as with antibody targeting human EpCAM, GD2 or histone/DNA complex), distributed into the tissue compartment to a much greater extent than the huIL12 counterparts (data not shown). It is tempting to suggest that this is a result of binding of the muIL-12 moiety to IL-12 receptors on mouse cells, to which the huIL-12 does not bind. However, it is not expected that many cells in a naive mouse express enough IL-12 receptors to account for this large difference in the distribution phase.
Although the observation that human B-FN was present in the vasculature of the human tumor implants in mice led to the conclusion that B-FN is produced by the tumor cells, this finding in itself does not preclude the possibility that murine B-FN is also produced by murine endothelial or accessory cells in the tumor vasculature. If this is indeed the case, targeting by huBC-1 in a xenograft model will not be as efficient and complete as L19, which recognizes the conserved ED-B domain and hence binds both human and murine B-FN.
Preliminary toxicity experiments in mice showed that the huBC1-muIL12 had surprisingly low toxicity in mice. Balb/c and C57 mice survived five daily doses of 1 and 0.3 mg, respectively. The detected serum IFNγ levels of about 1 ng/ml for Balb/c and 4 ng/ml for C57 mice shortly after the last dose (data not shown) were in the same range as the 2 ng/ml observed with Balb/c mice treated with 2.5 μg of IL12-L19 [15]. Given that the efficacious dose for huBC1-muIL12 is about 20 μg, this translates into a very favorable therapeutic index. We expect huBC1-huIL12, with a longer serum half-life, to show even better efficacy in the clinic, where patients during or post-chemotherapy still have a somewhat functional immune system that would benefit from multiple-cycles of treatment. This may be particularly relevant for huBC1-IL12 because anti-tumor activity is critically dependent on CD8, macrophage, and NK cell-mediated cytotoxicity, all of which are potently stimulated by the IL12-induced Th1 response. Indeed tumor masses in mice treated with IL12-L19 have been shown to be infiltrated with these cell types and have elevated levels of the Th1 cytokine IFNγ [15]. Furthermore, CD4+, CD8+ and NK cells are all necessary to mediate the full anti-angiogenic effect of IL-12, which was shown to be dependent on cross-talk between lymphocytes and endothelial cells [33]. Induction of immune responses to human tumors by IL-12 has been shown clinically to be a viable therapeutic approach. By targeting both tumor and tumor vessel ED-B, huBC1-IL12 has the potential to provide enhanced IL-12 effects at the tumor site. Clinical trials of huBC1-IL12 will determine whether this translates into additional therapeutic benefit. These are expected to start during 2007.
References
- 1.Bertolini F, Paul S, Mancuso P, Monestiroli S, Gobbi A, Shaked Y, Kerbel RS. Maximum tolerable dose and low-dose metronomic chemotherapy have opposite effects on the mobilization and viability of circulating endothelial progenitor cells. Cancer Res. 2003;63:4342–6. [PubMed] [Google Scholar]
- 2.Borsi L, Balza E, Carnemolla B, Sassi F, Castellani P, Berndt A, Kosmehl H, Biro A, Siri A, Orecchia P, Grassi J, Neri D, Zardi L. Selective targeted delivery of TNF alpha to tumor blood vessels. Blood. 2003;102:4384–92. doi: 10.1182/blood-2003-04-1039. [DOI] [PubMed] [Google Scholar]
- 3.Brunda MJ, Luistro L, Rumennik L, Wright RB, Dvorozniak M, Aglione A, Wigginton JM, Wiltrout RH, Hendrzak JA, Palleroni AV. Antitumor activity of interleukin 12 in preclinical models. Cancer Chemother Pharmacol. 1996;38(Suppl):S16–21. doi: 10.1007/s002800051031. [DOI] [PubMed] [Google Scholar]
- 4.Carnemolla B, Leprini A, Allemanni G, Saginati M, Zardi L. The inclusion of the type III repeat ED-B in the fibronectin molecule generates conformational modifications that unmask a cryptic sequence. J Biol Chem. 1992;267:24689–92. [PubMed] [Google Scholar]
- 5.Castellani P, Borsi L, Carnemolla B, Biro A, Dorcaratto A, Viale GL, Neri D, Zardi L. Differentiation between high- and low-grade astrocytoma using a human recombinant antibody to the extra domain-B of fibronectin. Am J Pathol. 2002;161:1695–700. doi: 10.1016/S0002-9440(10)64446-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. 2000;6:443–6. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 7.Davis CB, Gillies SD. Immunocytokines: amplification of anti-cancer immunity. Cancer Immunol Immunother. 2003;52:297–308. doi: 10.1007/s00262-002-0349-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gately MK, Carvajal DM, Connaughton SE, Gillessen S, Warrier RR, Kolinsky KD, Wilkinson VL, Dwyer CM, Higgins GF, Jr, Podlaski FJ, Faherty DA, Familletti PC, Stern AS, Presky DH. Interleukin-12 antagonist activity of mouse interleukin-12 p40 homodimer in vitro and in vivo. Ann N Y Acad Sci. 1996;795:1–12. doi: 10.1111/j.1749-6632.1996.tb52650.x. [DOI] [PubMed] [Google Scholar]
- 9.Gately MK, Renzetti LM, Magram J, Stern AS, Adorini L, Gubler U, Presky DH. The interleukin-12/interleukin-12-receptor system: role in normal and pathologic immune responses. Annu Rev Immunol. 1998;16:495–521. doi: 10.1146/annurev.immunol.16.1.495. [DOI] [PubMed] [Google Scholar]
- 10.Ghetie V, Ward ES. Multiple roles for the major histocompatibility complex class I- related receptor FcRn. Annu Rev Immunol. 2000;18:739–66. doi: 10.1146/annurev.immunol.18.1.739. [DOI] [PubMed] [Google Scholar]
- 11.Gillies SD, Lan Y, Wesolowski JS, Qian X, Reisfeld RA, Holden S, Super M, Lo KM. Antibody-IL-12 fusion proteins are effective in scid mouse models of prostate and colon carcinoma metastases. J Immunol. 1998;160:6195–203. [PubMed] [Google Scholar]
- 12.Gillies SD, Lo KM, Burger C, Lan Y, Dahl T, Wong WK. Improved circulating half-life and efficacy of an antibody-interleukin 2 immunocytokine based on reduced intracellular proteolysis. Clin Cancer Res. 2002;8:210–6. [PubMed] [Google Scholar]
- 13.Gollob JA, Mier JW, Veenstra K, McDermott DF, Clancy D, Clancy M, Atkins MB. Phase I trial of twice-weekly intravenous interleukin 12 in patients with metastatic renal cell cancer or malignant melanoma: ability to maintain IFN-gamma induction is associated with clinical response. Clin Cancer Res. 2000;6:1678–92. [PubMed] [Google Scholar]
- 14.Ha SJ, Chang J, Song MK, Suh YS, Jin HT, Lee CH, Nam GH, Choi G, Choi KY, Lee SH, Kim WB, Sung YC. Engineering N-glycosylation mutations in IL-12 enhances sustained cytotoxic T lymphocyte responses for DNA immunization. Nat Biotechnol. 2002;20:381–6. doi: 10.1038/nbt0402-381. [DOI] [PubMed] [Google Scholar]
- 15.Halin C, Rondini S, Nilsson F, Berndt A, Kosmehl H, Zardi L, Neri D. Enhancement of the antitumor activity of interleukin-12 by targeted delivery to neovasculature. Nat Biotechnol. 2002;20:264–9. doi: 10.1038/nbt0302-264. [DOI] [PubMed] [Google Scholar]
- 16.Hasan M, Najjam S, Gordon MY, Gibbs RV, Rider CC. IL-12 is a heparin-binding cytokine. J Immunol. 1999;162:1064–70. [PubMed] [Google Scholar]
- 17.Jones TD, Hanlon M, Smith BJ, Heise CT, Nayee PD, Sanders DA, Hamilton A, Sweet C, Unitt E, Alexander G, Lo KM, Gillies SD, Carr FJ, Baker MP. The development of a modified human IFN-alpha2b linked to the Fc portion of human IgG1 as a novel potential therapeutic for the treatment of hepatitis C virus infection. J Interferon Cytokine Res. 2004;24:560–72. doi: 10.1089/jir.2004.24.560. [DOI] [PubMed] [Google Scholar]
- 18.Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Zhu Z, Hicklin D, Wu Y, Port JL, Altorki N, Port ER, Ruggero D, Shmelkov SV, Jensen KK, Rafii S, Lyden D. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–7. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, Loudon R, Sherman F, Perussia B, Trinchieri G. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med. 1989;170:827–45. doi: 10.1084/jem.170.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, Sosman JA, Dutcher JP, Vogelzang NJ, Ryan JL. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 1997;90:2541–8. [PubMed] [Google Scholar]
- 21.Lieschke GJ, Rao PK, Gately MK, Mulligan RC. Bioactive murine and human interleukin-12 fusion proteins which retain antitumor activity in vivo. Nat Biotechnol. 1997;15:35–40. doi: 10.1038/nbt0197-35. [DOI] [PubMed] [Google Scholar]
- 22.Ling P, Gately MK, Gubler U, Stern AS, Lin P, Hollfelder K, Su C, Pan YC, Hakimi J. Human IL-12 p40 homodimer binds to the IL-12 receptor but does not mediate biologic activity. J Immunol. 1995;154:116–27. [PubMed] [Google Scholar]
- 23.Lo KM, Sudo Y, Chen J, Li Y, Lan Y, Kong SM, Chen L, An Q, Gillies SD. High level expression and secretion of Fc-X fusion proteins in mammalian cells. Protein Eng. 1998;11:495–500. doi: 10.1093/protein/11.6.495. [DOI] [PubMed] [Google Scholar]
- 24.Mariani G, Lasku A, Balza E, Gaggero B, Motta C, Di Luca L, Dorcaratto A, Viale GA, Neri D, Zardi L. Tumor targeting potential of the monoclonal antibody BC-1 against oncofetal fibronectin in nude mice bearing human tumor implants. Cancer. 1997;80:2378–84. doi: 10.1002/(SICI)1097-0142(19971215)80:12+<2378::AID-CNCR7>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 25.Mariani G, Lasku A, Pau A, Villa G, Motta C, Calcagno G, Taddei GZ, Castellani P, Syrigos K, Dorcaratto A, Epenetos AA, Zardi L, Viale GA. A pilot pharmacokinetic and immunoscintigraphic study with the technetium-99 m-labeled monoclonal antibody BC-1 directed against oncofetal fibronectin in patients with brain tumors. Cancer. 1997;80:2484–9. doi: 10.1002/(SICI)1097-0142(19971215)80:12+<2484::AID-CNCR20>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 26.Masiero L, Figg WD, Kohn EC. New anti-angiogenesis agents: review of the clinical experience with carboxyamido-triazole (CAI), thalidomide, TNP-470 and interleukin-12. Angiogenesis. 1997;1:23–35. doi: 10.1023/A:1018301031580. [DOI] [PubMed] [Google Scholar]
- 27.Midulla M, Verma R, Pignatelli M, Ritter MA, Courtenay-Luck NS, George AJ. Source of oncofetal ED-B-containing fibronectin: implications of production by both tumor and endothelial cells. Cancer Res. 2000;60:164–9. [PubMed] [Google Scholar]
- 28.Neri D, Bicknell R. Tumour vascular targeting. Nat Rev Cancer. 2005;5:436–46. doi: 10.1038/nrc1627. [DOI] [PubMed] [Google Scholar]
- 29.Pirhonen J, Matikainen S, Julkunen I. Regulation of virus-induced IL-12 and IL-23 expression in human macrophages. J Immunol. 2002;169:5673–8. doi: 10.4049/jimmunol.169.10.5673. [DOI] [PubMed] [Google Scholar]
- 30.Qian F, Zhang ZC, Wu XF, Li YP, Xu Q. Interaction between integrin alpha(5) and fibronectin is required for metastasis of B16F10 melanoma cells. Biochem Biophys Res Commun. 2005;333:1269–75. doi: 10.1016/j.bbrc.2005.06.039. [DOI] [PubMed] [Google Scholar]
- 31.Rafii S, Lyden D, Benezra R, Hattori K, Heissig B. Vascular and haematopoietic stem cells: novel targets for anti-angiogenesis therapy? Nat Rev Cancer. 2002;2:826–35. doi: 10.1038/nrc925. [DOI] [PubMed] [Google Scholar]
- 32.Santimaria M, Moscatelli G, Viale GL, Giovannoni L, Neri G, Viti F, Leprini A, Borsi L, Castellani P, Zardi L, Neri D, Riva P. Immunoscintigraphic detection of the ED-B domain of fibronectin, a marker of angiogenesis, in patients with cancer. Clin Cancer Res. 2003;9:571–9. [PubMed] [Google Scholar]
- 33.Strasly M, Cavallo F, Geuna M, Mitola S, Colombo MP, Forni G, Bussolino F. IL-12 inhibition of endothelial cell functions and angiogenesis depends on lymphocyte-endothelial cell cross-talk. J Immunol. 2001;166:3890–9. doi: 10.4049/jimmunol.166.6.3890. [DOI] [PubMed] [Google Scholar]
- 34.Teruya-Feldstein J, Jaffe ES, Burd PR, Kanegane H, Kingma DW, Wilson WH, Longo DL, Tosato G. The role of Mig, the monokine induced by interferon-gamma, and IP-10, the interferon-gamma-inducible protein-10, in tissue necrosis and vascular damage associated with Epstein-Barr virus-positive lymphoproliferative disease. Blood. 1997;90:4099–105. [PubMed] [Google Scholar]
- 35.Trinchieri G. Interleukin-12 and its role in the generation of Th1 cells. Immunol Today. 1993;14:335–8. doi: 10.1016/0167-5699(93)90230-I. [DOI] [PubMed] [Google Scholar]
- 36.Uherek C, Tonn T, Uherek B, Becker S, Schnierle B, Klingemann HG, Wels W. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood. 2002;100:1265–73. [PubMed] [Google Scholar]
- 37.Watford WT, Moriguchi M, Morinobu A, O’Shea JJ. The biology of IL-12: Coordinating innate and adaptive immune responses. Cytokine Growth Factor Rev. 2003;14:361–8. doi: 10.1016/S1359-6101(03)00043-1. [DOI] [PubMed] [Google Scholar]
- 38.Zou JJ, Schoenhaut DS, Carvajal DM, Warrier RR, Presky DH, Gately MK, Gubler U. Structure-function analysis of the p35 subunit of mouse interleukin 12. J Biol Chem. 1995;270:5864–71. doi: 10.1074/jbc.270.11.5864. [DOI] [PubMed] [Google Scholar]