

Abstract
Neuroblastoma (NB) is often described as an unfavorable target for both HLA-restricted and death receptor-mediated elimination by cytotoxic T lymphocytes (CTLs) due to low or absent HLA class I and caspase-8 expression. We investigated the effects of soluble factors released by CTLs activated by TCR triggering (named as activated supernatant; AS) on the levels and composition of cell surface molecules involved in HLA-restricted and HLA-independent NB cell recognition (surface immune phenotype). Using a panel of long-term propagated NB cell lines and freshly isolated primary human NB cells, we analyzed surface expression of the (1) cognate receptors for TNFα, Fas and TRAIL; (2) HLA class I and II heterodimers; (3) adhesion molecules; (4) the intracellular expression and activation of caspase-8, as well as (5) the susceptibility of NB cells to death receptor-mediated killing prior to and after exposure to AS. The exposure of NB cells to soluble factors released by activated CTLs skewed the surface immune phenotype of both long term cultured and primary NB cells, induced the expression and activation of caspase-8 and increased the susceptibility of tumor cells to lysis by TRAIL and Fas-agonistic antibody. Blocking experiments identified IFNγ and TNFα as main factors responsible for modulating the surface antigens of NB cells by AS. Our data suggest that recruitment of CTLs activated on third party targets into the vicinity of the NB tumor mass, may override the “silent” immune phenotype of NB cells via the action of soluble factors.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-007-0412-2) contains supplementary material, which is available to authorized users.
Keywords: Neuroblastoma, CTL, Cytokines, Death receptor, Caspase-8
Introduction
Neuroblastoma (NB) is the most frequent neoplasia in infants and the third most common tumor in young children. The present treatment modalities, including high-dose chemotherapy with stem cell rescue and the use of differentiating agents such as retinoids, have only limited impact on the prognosis for children with high-risk neuroblastoma, since still the majority of these children die from the disease [4]. Accordingly, new treatment options are warranted and among the new strategies, immunotherapy has attracted attention as a potential treatment modality of high-risk neuroblastoma.
Cytotoxic T lymphocytes (CTLs) play a critical role in tumor elimination in many experimental models (reviewed in [23]). CTLs induce target cell death through two major pathways, the cytotoxic granule exocytosis- and the death receptor-mediated pathway. While an efficient T cell-mediated lysis via the release of perforin/granzyme-containing cytototoxic granules requires expression of the relevant HLA class I complexes and adhesion molecules at the target cell surface, killing via the death receptor pathway involves binding of the ligands to death receptors in the tumor cell membrane, and requires an intact intracellular molecular machinery conveying the signal and executing tumor cell death (reviewed in [38]).
A large body of research data defines NB as a tumor of low immuno- and antigenicity; and, in particular, as a poor target for HLA class I-restricted CTL-recognition and killing. Several studies have shown that human NB cells express very low or undetectable levels of HLA class I [27, 49]. This is consistent with the origin of NB cells, evolving from neuronal tissue, which naturally displays low levels of HLA molecules [28]. In addition, NB cells were shown to be resistant to death-receptor mediated apoptosis due to the absence of caspase-8, the initiator caspase contributing to the transfer of the signal from the surface death receptor to the intracellular death-inducing machinery [11, 12, 18, 19]. The absence of caspase-8 protein expression has mainly been associated with hypermethylation of the caspase-8 gene and, in some cases, with gene deletion [42].
However, strategies to increase the immunogenicity of NB cells could render them into good targets for CTL-mediated immunotherapy. We have recently demonstrated that retinoids increase the cell surface expression of HLA class I and ICAM-1 and enhance the susceptibility of NB cells to HLA class I-restricted CTL-mediated killing [47]. Furthermore, we have demonstrated that CTLs can control NB cells in a HLA-independent by-stander fashion and that soluble factors released by CTLs (referred to as “activated supernatant”, AS) can induce caspase-dependent and -independent cell death in NB cells [10]. The NB cell death induced by CTLs was partially blocked by antibodies neutralizing the cytokines TNFα and interferon-gamma (IFNγ), and was reduced by reagents blocking FasL. However, it remained unclear if and how soluble factors released by activated CTLs contributed to the modulation of the death receptor pathways in NB cells at the molecular level.
In the current study, we investigated the effects of soluble factors, released by T cells following specific T-cell receptor (TCR) triggering, on the expression of a set of molecules relevant for both HLA-restricted and HLA-independent recognition of NB cells by cytotoxic lymphocytes. These include classical HLA class I and HLA class II complexes; the non-classical HLA class I molecule human leukocyte antigen-G (HLA-G), a well-characterized inhibitory ligand for NK receptors (reviewed in [29]), the MHC class I chain-related A (MICA) molecule, known as the ligand for the activating NKG2D NK receptors [6], ICAM-1,TNF- TNF-receptor1 (-R1) and -R2, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-receptors-1, -2, -3, and -4, and the Fas molecule. This set of molecules is hereafter collectively referred to as the surface immune phenotype.
We demonstrate that exposure of NB cells to the supernatant of activated CTLs skews the surface immune phenotype of NB cells. Moreover, exposure to AS induces the expression and activation of caspase-8 in NB cells which otherwise constitutively lack caspase-8 expression. This is paralleled by an increased susceptibility of NB cells to lysis by TRAIL and to a Fas-agonistic antibody. Taken together, our data suggest that soluble factors released into the NB tumor milieu by CTLs activated on third party targets may predispose these tumors to elimination by membrane-bound death ligands expressed by lymphocytes.
Materials and methods
Cell lines
The human NB cell lines MC-IXC, CHP-212, SK-N-SH, SH-SY5Y, SK-N-BE(2), SK-N-FI, and SK-N-AS were purchased from the American Type Culture Collection (ATCC) (Manassas, VA, USA); the human NB cell lines LAN-5 and SHEP-1 were generously provided by Dr. M. Arsenian-Henriksson (MTC, Karolinska Institutet (KI), Stockholm, Sweden); the human NB cell line FL-2 was provided by Dr. Marianne Ifversen (Rigshospitalet, Copenhagen, Denmark). All cell lines were maintained in IMDM medium (Gibco BRL, Life technologies, Grand Island, NY, USA) supplemented with 10% heat-inactivated fetal calf serum (FCS) (GibcoBRL), 100 IU/ml penicillin and 100 μg/ml streptomycin (complete medium).
Patient material
Primary tissue samples from NB tumor lesions were isolated from neuroblastoma patients undergoing surgery at Astrid Lindgren’s Children Hospital, Stockholm, Sweden. Informed consent was obtained from all patients/parents. Freshly isolated single cell suspensions were kept in AIM-V containing 10% heat-inactivated fetal calf serum, 100 IU/ml penicillin, 100 μg/ml streptomycin and 20 μg/ml of cyproxin (complete AIM-V) during the experimental procedures.
Antibodies and reagents
The following antibodies and reagents were used for FACS analysis in this study: the primary antibodies allophycocyanin (APC)-conjugated anti-ICAM-1, Fluorescein isothiocyanate (FITC)-conjugated anti-CD56, APC-conjugated anti-HLA-A,B,C, R-phycoerythrin (RPE)-conjugated anti-HLA-DR, and RPE-conjugated anti-Fas; the isotype control antibodies IgG2b-FITC, IgG1-RPE and IgG1-APC and the secondary APC- and RPE-conjugated goat anti-mouse Igs were all purchased from BD Pharmingen (San Diego, CA, USA). Anti-NB84 and isotype control antibodies IgG1 and IgG2a were purchased from Dakopatts AB (Älvsjö, Sweden). Anti-TrkA, anti-TNF-R1 and anti-TNF-R2 were purchased from R&D Systems Ltd. (Abingdon, UK). Antibodies against TRAIL-R1, -R2, -R3 and -R4 and anti-HLA-G antibody were from Alexis Biochemicals (Lausen, Switzerland). Rabbit polyclonal serum specific to human HLA class I heavy chain (HC) was a kind gift from Dr. H. Ploegh (Whitehead Institute for Biomedical Research, Cambridge, MA, USA). The anti-MICA mAb was from Immatics Biotechnologies GmbH (Tübingen, Germany). Rabbit polyclonal anti-human IFNγ-specific neutralizing antibody from Nordic Biosite (Täby, Sweden) and Enbrel® (Etanercept), a soluble TNFα receptor (TNF-R2)/Fc fusion protein, was obtained from Wyeth Ltd. (Berkshire, UK). killerTRAIL was from Alexis Biochemicals, the Fas-agonistic mouse IgM antibody CH-11 was from MBL (Nagoya, Japan) and mouse IgM isotype control was from Sigma–Aldrich Sweden AB (Stockholm, Sweden). Recombinant human TNFα was obtained from Immunotools (Friesoythe, Germany). The FAM caspase-8 Fluorochrome Inhibitor of Caspases (FLICA) kit was purchased from Serotec Ltd (Oxford, UK) and FITC-conjugated Annexin-V was obtained from BD Pharmingen. The following antibodies and reagents were used for Western blot analysis: the mouse monoclonal antibody specific to β-actin was purchased from Sigma Chemical Co (St Louis, USA) and the mouse anti-human caspase-8 mAb was from BD Pharmingen. The horseradish peroxidise (HRP)-conjugated rabbit anti-mouse secondary antibodies were obtained from Dakopatts AB.
Generation of CTL supernatants
C1R/A11 cells were pre-pulsed with the HLA-A11-restricted Epstein Barr virus (EBV) peptide IVTDFSVIK (IVT) at a final concentration of 10−6 M for 1 h at 37°C and then co-cultured with CAR13 CTLs at a 5:1 effector-to-target ratio. Culture supernatants were collected 24 h after TCR triggering (AS). Supernatants from CTLs co-cultured with non-pulsed antigen presenting cells were prepared in parallel and used as controls (control supernatant, CS).
Treatment of NB cells with CTL supernatants and cytokine blocking
Neuroblastoma cells or primary tumor cells were cultured in their respective complete medium (as detailed above) alone or in the presence of 10% volume-to-volume (v/v) of CS or AS for the indicated periods of time prior to the different assays. Since CS failed to induce changes of neuroblastoma cells when compared to samples kept in complete medium alone, CS samples were omitted from some of the experiments. In cytokine blocking experiments the complete medium containing 10% AS (v/v) was preincubated for 1 h at room temperature with 20 μg/ml of Enbrel®, 2000 units/mLneutralization units/ml of anti-IFNγ-specific neutralizing antibody or a combination of the two.
Flow cytometric analysis
Expression of surface molecules was measured by incubating tumor cells with either a directly fluorochrome-conjugated primary antibody or a relevant isotype control antibody. After washing in PBS containing 0.1% BSA, expression of relevant molecules was analyzed on a FACScan flow cytometer (Becton Dickinson, Mountain View, CA, USA). When non-conjugated primary antibodies were used, samples were incubated with an APC- or RPE-conjugated secondary antibody following the incubation with the relevant primary antibody and then analyzed by flow cytometry. Tumor cells in freshly isolated NB lesions usually constituted the vast majority of the tumor sample preparations and were defined as “CD56-bright” as previously described [48].
Western blot analysis
Western blot was performed as previously described [47]. Briefly, cells were lysed in the electrophoresis sample buffer and total cell lysates corresponding to 1 × 105 cells were separated on precast polyacrylamide gels using the Pharmacia Multiphor II electrophoresis system (Amersham Pharmacia Biotech, Uppsala, Sweden). Proteins were transferred onto a PVDF membrane (Millipore AB, Sundbyberg, Sweden), blocked in PBS containing 5% milk and 0.1% Tween-20, and probed with the relevant primary and secondary antibodies according to the manufacturer’s suggestions. Specific bands were visualized by enhanced chemiluminescence (ECL, GE Healthcare Biosciences AB, Uppsala, Sweden) and digitally captured by a Fujifilm LAS-1000 Image Reader system (Science Imaging Scandinavia AB, Nacka, Sweden).
Analysis of caspase-8 activity
Levels of activated caspase-8 were detected in NB cells using a fluorescein-labelled inhibitor of caspase-8 (Caspase-8 FLICA). NB cells, either untreated or treated with CS or AS for the indicated periods of time, were labeled according to the manufacturer’s instructions in medium containing FLICA solution for 1 h at 37°C, washed and analyzed directly by flow cytometry to detect caspase-8 activity.
Treatment with death receptor ligands
Following propagation of NB cell lines in either complete medium alone or in the presence of 10% v/v AS for indicated periods of time (MC-IXC, 12 h; CHP-212, SK-N-SH and LAN-5, 24 h), cells were washed extensively in complete medium and then incubated for designated periods of time (MC-IXC, 12 h; CHP-212 and LAN-5, 24 h; SK-N-SH, 48 h) at 37°C either in medium alone or in the presence of IgM isotype control antibody (500 ng/ml), killerTRAIL (100 ng/ml for MC-IXC and CHP-212 and 500 ng/ml for SK-N-SH and LAN-5), CH-11 (500 ng/ml) or TNFα (100 ng/ml). Cells were then analyzed by flow cytometry following Annexin-V staining according to the manufacturer’s instructions.
Statistical analysis
All statistical analyses were done using Analyse-It® software for Excel (version 7.2; Analyse-It Software, Ltd. Leeds, UK). P values for differences between independent tumor groups in the correlation analysis of Fig. 1c were calculated using the non-parametric Mann–Whitney U test. Reported P values in Fig. 4 were calculated using the Student’s paired t test. P values * ≤ 0.05, ** ≤ 0.01, *** ≤ 0.001 were considered statistically significant.
Fig. 1.
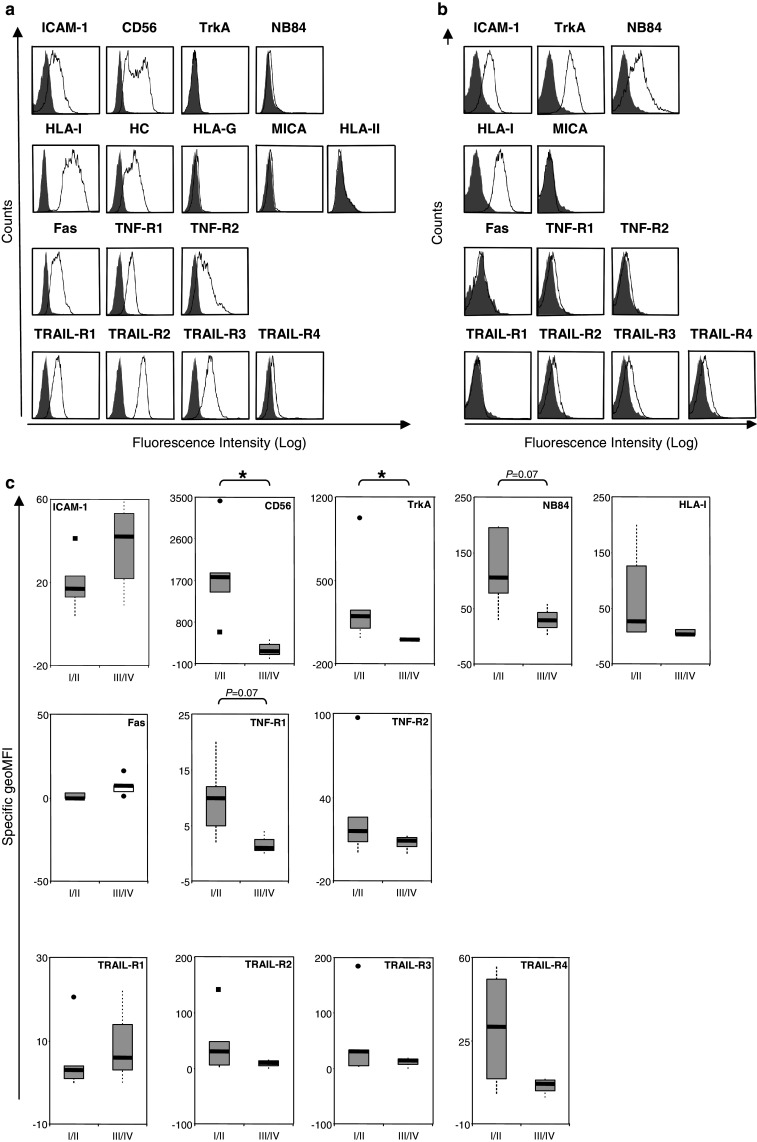
a Cell surface immune phenotype of the neuroblastoma cell line CHP-212. The expression of the molecules indicated in the figure was assessed by flow cytometry, following incubation with specific antibodies (open histograms) or relevant isotype controls (filled histograms) as described in Materials and methods. b, c Cell surface immune phenotype of tumor cells derived from neuroblastoma patients. b Cell surface phenotype of tumor cells derived from neuroblastoma sample NB-KI-3. Tumor cells were analyzed by flow cytometry at the day of tumor excision for the surface expression of the molecules indicated in the figure. Immunostainings were performed using specific antibodies (open histograms) or relevant isotype controls (filled histograms). c Statistical analysis of the tumor cell surface phenotype in low stage (I/II) versus high stage (III/IV) NB patients. The surface expression levels of the molecules indicated in the figure were calculated as specific geometric mean fluorescence intensity (sgMFI = Abgeo mean − Isogeo mean) as determined by flow cytometry. The boxes represent the interquartile range with the lower and upper quartiles surrounding a thicker line representing the median. The vertical lines connect the nearest observations within 1.5 IQRs (inter-quartile ranges) of the lower and upper quartiles. Squares (filled square) and circles (filled circle) indicate possible outliers more than 1.5 IQRs (near outliers) and 3.0 IQRs (far outliers) from the quartiles, respectively. P values for differences between independent groups were calculated using the non-parametric Mann–Whitney U test (*P < 0.05)
Fig. 4.
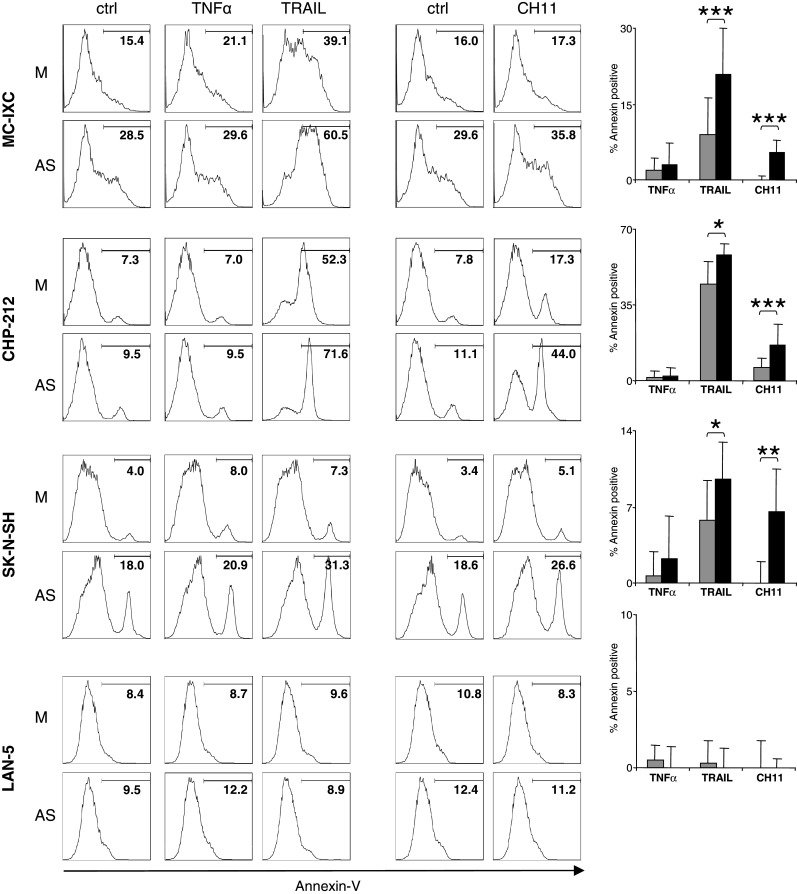
Pre-treatment of NB cells with AS renders them more sensitive to death receptor mediated killing. NB cell lines propagated in complete medium alone or in the presence of 10% v/v of activated supernatant (AS) were exposed to death-inducing signals for the periods of time described in Materials and Methods, and then stained with FITC-conjugated Annexin-V and analyzed by flow cytometry. Data from one representative experiment are shown as histograms for each cell line. The bar graphs show percentages of Annexin-V positive cells induced by the respective death inducer where gray bars represent medium-pretreated cells and black bars represent AS-pretreated cells. Data is presented as the difference between the numbers of Annexin V-positive cells in control cultures (kept in complete medium alone for TNFα and killerTRAIL or in the presence of IgM for CH-11) and cultures exposed to the relevant death inducer. Statistical significance (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001) was determined using the Student’s paired t test
Results
Analysis of the surface immune phenotype of neuroblastoma cell lines and primary neuroblastoma tumor cells
We assessed the surface immune phenotype of both NB cell lines and primary NB tumor cells. The latter were directly analyzed on the day of surgical intervention to limit skewing of the phenotype due to in vitro cell propagation. In addition, we monitored the presence of the NB-associated molecular markers including NB84, the receptor for nerve growth factor TrkA, and CD56 (neural adhesion molecule, NCAM), the two latter being known to be associated with progression and/or metastatic spread of NB [2, 3, 25, 34]. The NB cell lines (Table 1) and freshly isolated NB cells (Table 2) were single- or double-labeled, respectively, with the relevant specific antibodies and subsequently analyzed by flow cytometry.
Table 1.
Cell surface phenotype of neuroblastoma cell lines
| Cell line | ICAM-1 | CD56 | TrkA | NB84 | HLA-I | HC | HLA-G | MICA | HLA-II | Fas | TNF-R1 | TNF-R2 | TRAIL-R1 | TRAIL-R2 | TRAIL-R3 | TRAIL-R4 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MC-IXC | ++ | − | (+) | − | ++ | + | − | + | − | − | + | − | + | + | + | + |
| CHP-212 | + | + | − | − | ++ | + | − | − | − | + | + | + | + | ++ | + | − |
| SK-N-SH | + | ++ | + | − | + | − | − | − | − | (+) | (+) | − | (+) | + | (+) | (+) |
| SH-SY5Y | (+) | ++ | (+) | − | + | − | − | − | − | − | − | − | (+) | + | + | + |
| LAN-5 | (+) | ++ | − | − | ++ | + | − | + | − | + | − | − | − | (+) | + | ++ |
| FL-2 | + | − | − | − | ++ | ND | − | + | − | + | − | + | + | + | (+) | + |
| SHEP-1 | ++ | + | − | − | ++ | ND | ND | ND | − | + | + | + | (+) | + | + | + |
| SK-N-AS | ++ | − | + | − | ++ | ND | ND | ND | − | + | + | + | (+) | + | + | + |
| SK-N-FI | + | + | + | − | ++ | ND | ND | ND | − | + | + | − | + | + | + | − |
Scoring: −: 0–5 sgMFI; (+): ≤ 5 sgMFI but positive on overlay; +: 6–100 sgMFI; ++: > 100 sgMFI (sgMFI: specific geometric mean fluorescence intensity (sgMFI = Abgeo mean − Isogeo mean)). Data from 2 to 5 independent experiments for each cell line are shown in the table
ND not done
Table 2.
Cell surface phenotype of freshly isolated neuroblastoma samples
| Sample ID | ICAM-1 | CD56 | TrkA | NB84 | HLA-I | MICA | HLA-II | Fas | TNF-R1 | TNF-R2 | TRAIL-R1 | TRAIL-R2 | TRAIL-R3 | TRAIL-R4 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NB-KI-1 | + | ++ | ++ | ++ | + | ND | − | − | + | + + | ++ | ++ | + | |
| NB-KI-2 | − | − | − | (+) | − | ND | ND | + | − | − | − | − | − | − |
| NB-KI-3 | + | ++ | ++ | + | ++ | − | ND | − | − | − | − | (+) | (+) | (+) |
| NB-KI-4 | + | + | + | ++ | ++ | − | − | − | + | + | − | + | + | + |
| NB-KI-5 | − | ++ | ++ | ++ | − | − | − | − | − | + | − | + | (+) | + |
| NB-KI-6 | + | ++ | ++ | + | + | − | − | − | + | ++ | − | + | + | + |
| NB-KI-7 | + | + | − | + | − | − | − | + | − | + | + | + | + | + |
| NB-KI-8 | + | + | − | + | + | − | − | − | − | + | + | + | + | − |
Scoring: -: 0–5 sgMFI; (+): ≤ 5 sgMFI but positive on overlay; +: 6–100 sgMFI; ++: > 100 sgMFI
ND not done
Cells were double-stained for CD56 and the indicated molecules. The shown expression levels are from the CD56high population [46]
We observed that the molecules comprising the surface immune phenotype (see Introduction) were heterogeneously expressed by both the long-term in vitro propagated cell lines (Table 1; Fig. 1a) and the primary tumor cells (Table 2; Fig. 1b). All NB cell lines analyzed expressed detectable levels of HLA class I at the cell surface (Table 1; Fig. 1a). In contrast to previously published data, where no HLA class I expression could be observed in NB specimens [27, 49], ex vivo analyzed primary NB cells did express, albeit at variable levels, surface HLA class I as measured by the W6/32 antibody specific for assembled HLA class I complexes [35]. In agreement with previously reported data [32], we also observed a detectable pool of β2-microglobulin((β2m)-free heavy chains recognized by the HC-10 antibody [36] at the surface of NB cell lines. Unfortunately, we were not able to analyze free heavy-chain expression profiles in the primary NB cells due to the limited amount of material available.
In general, the immune phenotypic profiles of the ex vivo analyzed tumor samples largely resembled those observed for the NB cell lines. However, it is difficult to exclude that long-term in vitro manipulation have shifted both the pattern and expression levels of the selected surface molecules in tumor cell lines, which may be exemplified by the higher levels of HLA class I observed on the NB cell lines compared to primary tumor cells. Nevertheless, we did not observe any major differences in the expression of ICAM-1, CD56, MICA, and HLA class II (Tables 1, 2) between the NB cell lines and primary tumor cells. Importantly, similar to the cell lines, each of the primary NB cell samples expressed, although at different levels, at least one of the death domain-containing surface receptors (Table 2).
In order to analyze whether any particular feature of the surface immune phenotype correlates with the progression of the disease, primary tumor samples were compiled into “low-stage” (stage I and II) and “high-stage” (stage III and IV) tumors [5]. As shown in Fig. 1c and summarized in Supplementary Table 2, “high-stage” tumors expressed significantly lower levels of TrkA and CD56 as compared to “low-stage” tumors, which is consistent with results reported in other studies [2, 3, 25, 34]. We also found that “high-stage” tumors exhibit a decreased expression of TNF-R1 (Supplementary Table 2), however due to the limited number of samples analyzed up to date this correlation did not reach statistical significance (P = 0.07).
Soluble factors released from activated CTLs alter the surface immune phenotype of neuroblastoma cells
We have recently shown that NB cell death may be induced by activated CTLs in a by-stander manner [10]. To analyze whether soluble factors, released by CTLs upon activation, can modulate the sensitivity of the tumor cells to death receptor triggering, we first compared the surface expression of death receptors in a panel of NB cell lines and freshly isolated NB tumor samples prior to and after exposure to AS (described in [10]). We found that AS induced TNF-R2 and Fas, but not TNF-R1 and TRAIL-receptors in both NB cell lines (Fig. 2a) and primary NB cells (Fig. 2b, c). We detected an upregulation of the surface pool of HLA class I complexes and ICAM-I expression in both primary cells and long-term NB cell lines, however, no modulation of MICA and HLA class II at the cell surface was detected (Fig. 2a–c). Interestingly, not only total HLA class I but also β2m-free heavy chains were induced in all NB cell lines tested, although the functional consequence of this observation needs to be further investigated. We also observed the AS-mediated induction of the nerve growth factor (NGF)-receptor TrkA in two NB cell lines (Fig. 2a) and, moderately, in one NB primary tumor sample (Fig. 2b, c). Blocking experiments identified IFNγ and TNFα as two major factors modulating the surface immune phenotype in NB cells (Fig. 2d).
Fig. 2.
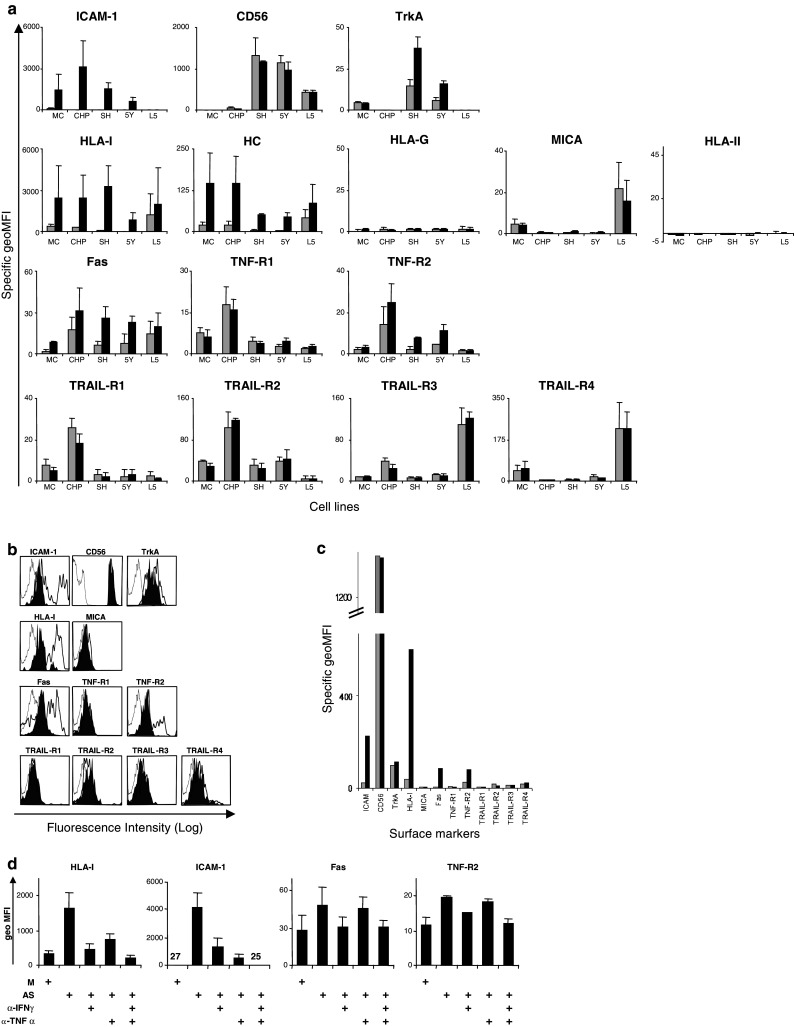
The effect of activated supernatant (AS) on the surface immune phenotype of NB cells. a NB cell lines were cultured overnight in either medium alone (gray bars) or in the presence of 10% v/v of supernatant from activated CTLs (black bars) and subsequently analyzed for the surface expression of the indicated molecules using specific antibodies followed by flow cytometry. Specific geometric MFI was calculated as described in the legend to Fig. 1. Means ± SD from three independent experiments are shown in the figure. (MC, MC-IXC; CHP, CHP-212; SH, SK-N-SH; 5Y, SH-SY5Y; L5, LAN-5). b Tumor cells from patient NB-KI-6 were treated for 84 h with either medium alone (filled histogram) or in the presence of 10% v/v of supernatant from activated CTLs (thick open histogram), and then stained for the indicated surface markers using specific antibodies and analyzed by flow cytometry. The histograms representing fluorescence obtained with the isotype control antibodies on untreated and AS-treated NB cells overlapped and had similar geo mean values, therefore, only one histogram is shown as reference. c Specific geometric MFI values obtained in the experiment shown in b. d CHP-212 cells were cultured overnight in either medium alone (M) or AS. The AS was either non-manipulated or pre-incubated with IFNγ-neutralizing antibodies (α-IFNγ) and/or soluble TNFα receptor (α-TNFα) for 1 h at room temperature. The numbers in the ICAM-1 panel indicates the average geo MFI values
Supernatant from activated CTLs induces the expression and activity of caspase-8 in neuroblastoma cells
Several studies have demonstrated that NB cells often fail to express caspase-8 [42, 44], which is a prerequisite for a functional death signal transduction to be generated upon association of death ligands such as Fas, TRAIL and TNFα with their cognate receptors (reviewed in [46]). Consistent with these findings, defective caspase-8 expression in NB cells correlates with resistance to TRAIL-induced apoptosis [11, 12, 18, 19]. Two of the NB cell lines analyzed in our study were constitutively devoid of caspase-8 expression (SK-N-SH and SH-SY5Y), whereas MC-IXC, CHP-212 and LAN-5 cells expressed detectable levels of caspase-8 protein. We observed that exposure to AS resulted in the induction or further enhancement of the expression (Fig. 3a) as well as activation (Fig. 3a, b) of caspase-8 in all of NB cell lines tested regardless of the steady expression levels, except in LAN-5 cells where very moderate, if any, changes were detected.
Fig. 3.

Supernatant from activated CTLs both induce caspase-8 expression and modulate its activity in NB cells. a NB cell lines were treated for 48 h with either medium alone, or in the presence of 10% v/v of control supernatant (CS) or activated supernatant (AS). The expression of caspase-8 in total cell lystaes was monitored by Western blot using a specific antibody recognizing the 55/50 kD pro-form of caspase-8 as well as the 40/36 and 23 kD cleavage products obtained upon activation. Expression of actin was used as a control of sample loading. b The activity of caspase-8 in NB cells propagated for 24, 48 and 72 h in complete medium alone (thin open histograms), or in the presence of either CS (filled histograms) or AS (bold open histograms) was measured by flow cytometry using fluorescent caspase-8 specific substrates as described in Materials and Methods. Numbers show percentage of AS-treated cells positive for active caspase-8
Soluble factors released from activated CTLs sensitize neuroblastoma cells to death receptor-mediated killing
In order to understand whether the increased levels of surface death receptors as well as the induction of expression and activation of casapase-8 in response to AS have functional consequences, we examined the susceptibility of NB cells to death induced via surface death receptors with and without pre-treatment with AS. We found that NB cells treated with AS were rendered more sensitive to death triggered by TRAIL and by a Fas-agonistic antibody (CH-11), but not by TNFα (Fig. 4). Our data indicate that the expression and activation of caspase-8 (Fig. 3), rather than surface levels of TRAIL receptors-1 and -2 (Fig. 2a) play a role in the enhanced cytotoxic effect exerted by TRAIL on NB cells (reviewed in [1]). The increased sensitivity of NB cells to the Fas-agonistic antibody achieved by pretreatment with AS could potentially result both from the increased surface pool of Fas molecules and the increase in caspase-8 expression by the tumor cells (Figs. 2a, 3a). Interestingly, NB cells remained resistant to TNFα-induced cell death even following AS-treatment (Fig. 4), which correlated with the lack of an effect of AS on the surface TNF-R1 expression (Fig. 2).
Discussion
The interplay between NB tumor cells and the immune system of the patient is still an enigma. Early studies demonstrated that lymphocyte infiltration of the NB tumor mass is associated with better prognosis, suggesting a role for the immune system in limiting the disease [33]. In addition, elevated levels of HLA class I were observed in stage 4S tumors of infants that relatively frequently undergo spontaneous regression, also suggesting a possible involvement of the immune system in this process [39]. Cytotoxic T lymphocytes, key players in the immune response, utilize two major, molecularly different, pathways of target cell killing; namely HLA class I-restricted, perforin/granzyme-mediated lysis and surface death receptor-dependent induction of caspase-mediated apoptosis in target cells (reviewed in [38]). The relative importance of these pathways in the interaction between NB and the host immune system is currently not well understood. In the present study, we assessed the effect of cytokines released by activated T cells on the surface immune phenotype of NB cells as well as on their susceptibility to death receptor-coupled apoptosis-inducing signals.
Neuroblastoma is often referred to as a tumor with a merely detectable, or absent, expression of HLA class I [27, 49]. This contrast with the fact that surface HLA class I expression is easily detectable in long-term in vitro propagated NB cell lines [8, 9, 14, 31, 41, 47]. However, the latter could represent an in vitro artifact resulting from either pre-selection of a limited pool of NB cells with a certain immune phenotype or by propagation of NB cells under conditions that do not match the natural in vivo tumor milieu. In fact, two of the long term NB cell lines, LAN-5 and FL-2, expressed fairly high levels of the stress-induced NKG2D activating ligand MICA, which was not detected in any of the primary NB cells tested in this study. Surprisingly, we detected expression of HLA class I complexes in freshly ex vivo isolated NB cells which had not undergone the procedure of in vitro propagation (Table 2; Fig. 1b). This finding cannot be explained by the contamination of other CD56-positive HLA class I-expressing cell types, such as NK cells or activated CTLs, in primary tumor samples, since the infiltrated lymphocytes constituted a very small proportion of these tumor samples (data not shown). Moreover, the NB population was defined as the CD56high as compared to CD56low NK and T cells, as earlier described [48]. Importantly, in contrast to previous studies based on immunohistochemical analysis, which is unable to discriminate between the total- and the surface pools of the HLA molecules, we showed the presence of the HLA class I complexes on the surface of tumor cells where they can serve as targets for CTL-mediated surveillance. Of note, the W6/32 antibody used for this analysis recognizes both classical and non-classical HLA, such as HLA-E and -G. The possibility that HLA-E may be expressed at the surface of NB cells was supported by the detection of this protein in total cell lysates from primary NB samples analyzed in this study (A. De Geer et al., submitted). However, the detection of surface expression of HLA-E is currently hindered due to the absence of commercially available specific reagents. No HLA-G expression was observed in primary NB samples analyzed in this study (A. De Geer et al., submitted), thus excluding any contribution of HLA-G to the total pool of surface HLA. Unfortunately, the limited amount of available primary tumor material did not allow the assessment of the “stability” (or the off-rate) of HLA class I complexes, which is considered as a key parameter in defining the efficacy of T cell activation. Such data would be of a special importance since the detectable levels of “free” heavy chains were found on NB cell lines in this (Table 1) and in previous studies [32], suggesting that a proportion of HLA complexes reaching the cell surface in NB cells may contain “suboptimal” peptide cargos [7]. Moreover, the surface pool of free heavy chains substantially increased in response to AS in the majority of NB lines tested (Fig. 2). Furthermore, we recently found that soluble factors released from activated CTLs may render some NB cells resistant to perforin-mediated permeabilization and diminish the proportion and/or activity of granzyme B internalized by these cells (A. De Geer, submitted). This indicates that despite the apparent increase in the total amounts of assembled HLA class I complexes on the surface of NB cells, soluble factors released by activated CTLs into the tumor milleu may not always improve HLA class I-restricted recognition of the tumor.
The second type of effector mechanisms executed by CTLs involves TNFα, FasL and TRAIL, which have been evaluated for anticancer activity in preclinical studies [1, 13, 20]. At least one of the receptors specific for either of these death-inducing ligands could be found in NB cell lines (Table 1; Fig. 1a) and, more importantly, in primary NB cells (Table 2; Fig. 1b). While TRAIL showed cytotoxic activity against a majority of the tested NB cell lines, triggering of the Fas pathway in these tumor cells was less efficient in inducing death, whereas TNFα exhibited no or only marginal cytotoxic activity (Fig. 4). The NB cells remained resistant to TNFα-induced cell death even after pre-treatment with AS. The lack of a death-inducing effect of TNFα in AS-pre-treated cells can not be explained by competition for the receptors and/or apoptotic machinery between the recombinant and supernatant-derived TNFα, since pre-treatment MC-IXC cells with supernatant devoid of TNFα did not increase the sensitivity of this cell line to the recombinant cytokine (data not shown). It is generally believed that the cytotoxic effect of TNFα requires engagement of TNF-R1 and caspase-8 recruitment, while TNF-R2 mediates survival signals [51]. While we did not observe any upregulation of TNF-R1 on the NB cells upon treatment with AS, the increase in surface levels of TNF-R2 was detected, suggesting that the skewed ratio between the two receptors may suppress the death-promoting effects of TNF-R1 signaling.
In contrast to cell death mediated by TNFα-receptors, TRAIL-receptor- and, to a lesser extent, Fas-mediated death, was enhanced in NB cells following pretreatment with the pool of soluble factors released by activated CTLs. Interestingly, an increase in the susceptibility to Fas-mediated apoptosis achieved in NB cells by pre-treatment with AS was paralleled by an increase in the surface levels of the Fas receptor, whereas we did not observe a significant correlation between the expression of death domain-containing TRAIL-receptors and the sensitivity of NB cells to killerTRAIL. The latter, however, correlated with the induction of caspase-8 expression and activity induced by AS in the NB cells, suggesting that the rescue of caspase-8-mediated signaling, rather than the actual surface levels of specific receptors, determine the potency of the TRAIL pathway in caspase-8-deficient NB cells. Though the ability of IFNγ (which is present in the AS, data not shown) to serve as an inducer of caspase-8 expression in NB cells is known, we cannot exclude that other components in the AS may play a role in this process [15, 16, 24, 45, 50]. Moreover, the induction of caspase-8 may also contribute to limiting the metastatic spread of NB cells in vivo, since, according to Stupack et al., caspase-8 was defined as a metastasis suppressor gene that together with integrins regulated the survival and the invasive capacity of NB cells in a murine model [40, 43]. Different factors may be responsible for the absence of a sensitizing effect of the AS on death receptor-mediated killing of LAN-5 cells. First, a very moderate induction of caspase-8 activity was triggered in these cells by AS (Fig. 3). Second, a relatively high expression of the TRAIL decoy receptors TRAIL-R3 and -R4 as compared to the death-inducing receptors TRAIL-R1 and -R2 was detected in LAN-5 cells (Table 1). Finally, other proteins such as FLICE-inhibitory protein (FLIP) [22] and protease inhibitor 9 (PI9) [26] can interfere with the caspase-8-mediated signaling acting either at the level of DISC formation or downstream of caspase-8 activation. The involvement of these proteins in the regulation of NB cell death triggered by the death ligands remains to be characterized.
TRAIL has recently gained much interest as a possible anticancer therapeutic agent, due to its ability to selectively induce apoptosis in tumor cells without inducing signals that cause inflammation. The soluble recombinant fragment of TRAIL had no side-effects on normal tissues when used in preclinical studies on primates, and proved efficient against tumor xenografts in mice, which prompted the initiation of phase I trials using recombinant TRAIL (reviewed in [37]). Moreover, agonistic antibodies specific for TRAIL-R1 or TRAIL-R2 have been tested in human phase II clinical trials and exhibited no toxicity [21, 30]. Our data suggest that activated, TRAIL-expressing cytotoxic lymphocytes in the NB tumor vicinity may exert a direct cytolytic activity against the tumor cells. Alternatively, induction of by-stander immune activation in the tumor milleu may be combined with the administration of the TRAIL-receptor agonists mentioned above.
Besides modulating the HLA- and death receptor-mediated pathways, cytokines released by activated CTLs enhanced the expression of TrkA in two of the NB cells lines and primary NB cells (Fig. 2), suggesting a role for bystander immune activation in the NB vicinity in facilitating NGF-induced differentiation of NB cells. This finding is in line with the previously suggested role of the immune system in the induction of spontaneous regression of NB, which could be a result of tumor re-differentiation triggered by bystander immune activation at the tumor site [17]. Thus, a combination of NGF treatment and CTL therapy could have a beneficial effect for NB patients, however, the role of bystander CTL activation in the modulation of NGF-induced signaling needs to be investigated further.
Collectively, our data indicate that enrollment of CTLs, activated on third party targets, in the vicinity of the NB tumor mass, may override the “silent” immune phenotype of NB cells via the action of soluble factors. This in turn, may increase NB cell susceptibility to HLA-independent death-receptor-mediated cytotoxicity by T cells. Although we can not exclude that some of the NB tumors may also become more susceptible to HLA class I-restricted killing as a result of elevated levels of HLA class I and ICAM-1 molecules, current experimental evidence obtained in our laboratory does not support this hypothesis (see above).
We have recently found that activated CD8+ T cells constitute a significant proportion of tumor associated lymphocytes in freshly isolated NB tumors examined at the day of tumor excision (manuscript in preparation). Moreover, a wide panel of pro-inflammatory cytokines, including TNFα and IFNγ was found in the NB tumor milieu, suggesting that immune activation may take place at the NB tumor site. It still needs to be determined whether this immune activation was triggered locally by the specific recognition of tumor cells or reflects a local inflammation developed independently of the tumor-T cell interaction.
The search for drugs that restore the sensitivity of cancer cells to apoptosis represent a challenge in modern tumor biology. Our data suggest that soluble factors released by cytotoxic lymphocytes at the tumor site may be viewed as natural, promising candidates in a new generation of anticancer therapy.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Abbreviations
- AS
Activated supernatant
- APC
Allophycocyanin
- β2m
β2-Microgloblin
- CS
Control supernatant
- CTL
Cytotoxic T lymphocyte
- EBV
Epstein Barr virus
- FITC
Fluorescein isothiocyanate
- FLICA
Fluorochrome inhibitor of caspases
- HC
Heavy chain
- HLA
Human leukocyte antigen
- IFN
Interferon
- MICA
MHC class I chain-related A
- NB
Neuroblastoma
- RPE
R-phycoerythrin
- TCR
T-cell receptor
- TNF-R
TNFα-receptor
- TRAIL
Tumor necrosis factor-related apoptosis-inducing ligand
- v/v
Volume-to-volume
Footnotes
This work was supported by grants from the Swedish Children’s Cancer Foundation, the Swedish Cancer Society, the Cancer Society of Stockholm, the King Gustav the Vth Jubilee Fund, Karolinska Institutet and the Swedish Research Council.
References
- 1.Ashkenazi A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat Rev Cancer. 2002;2:420–430. doi: 10.1038/nrc821. [DOI] [PubMed] [Google Scholar]
- 2.Blaheta RA, Daher FH, Michaelis M, Hasenberg C, Weich EM, Jonas D, Kotchetkov R, Doerr HW, Cinatl J., Jr Chemoresistance induces enhanced adhesion and transendothelial penetration of neuroblastoma cells by down-regulating NCAM surface expression. BMC Cancer. 2006;6:294. doi: 10.1186/1471-2407-6-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blaheta RA, Hundemer M, Mayer G, Vogel JU, Kornhuber B, Cinatl J, Markus BH, Driever PH, Cinatl J., Jr Expression level of neural cell adhesion molecule (NCAM) inversely correlates with the ability of neuroblastoma cells to adhere to endothelium in vitro. Cell Commun Adhes. 2002;9:131–147. doi: 10.1080/15419060214520. [DOI] [PubMed] [Google Scholar]
- 4.Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. . Nat Rev Cancer. 2003;3:203–216. doi: 10.1038/nrc1014. [DOI] [PubMed] [Google Scholar]
- 5.Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, De Bernardi B, Evans AE, Favrot M, Hedborg F, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol. 1993;11:1466–1477. doi: 10.1200/JCO.1993.11.8.1466. [DOI] [PubMed] [Google Scholar]
- 6.Cerwenka A, Lanier LL. Ligands for natural killer cell receptors: redundancy or specificity. Immunol Rev. 2001;181:158–169. doi: 10.1034/j.1600-065X.2001.1810113.x. [DOI] [PubMed] [Google Scholar]
- 7.Cook JR, Myers NB, Hansen TH. The mechanisms of peptide exchange and beta 2-microglobulin exchange on cell surface Ld and Kb molecules are noncooperative. J Immunol. 1996;157:2256–2261. [PubMed] [Google Scholar]
- 8.Cooper MJ, Hutchins GM, Mennie RJ, Israel MA. Beta 2-microglobulin expression in human embryonal neuroblastoma reflects its developmental regulation. Cancer Res. 1990;50:3694–3700. [PubMed] [Google Scholar]
- 9.Corrias MV, Occhino M, Croce M, De Ambrosis A, Pistillo MP, Bocca P, Pistoia V, Ferrini S. Lack of HLA-class I antigens in human neuroblastoma cells: analysis of its relationship to TAP and tapasin expression. Tissue Antigens. 2001;57:110–117. doi: 10.1034/j.1399-0039.2001.057002110.x. [DOI] [PubMed] [Google Scholar]
- 10.De Geer A, Kiessling R, Levitsky V, Levitskaya J. Cytotoxic T lymphocytes induce caspase-dependent and -independent cell death in neuroblastomas in a MHC-nonrestricted fashion. J Immunol. 2006;177:7540–7550. doi: 10.4049/jimmunol.177.11.7540. [DOI] [PubMed] [Google Scholar]
- 11.Eggert A, Grotzer MA, Zuzak TJ, Wiewrodt BR, Ho R, Ikegaki N, Brodeur GM. Resistance to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Cancer Res. 2001;61:1314–1319. [PubMed] [Google Scholar]
- 12.Eggert A, Grotzer MA, Zuzak TJ, Wiewrodt BR, Ikegaki N, Brodeur GM. Resistance to TRAIL-induced apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Med Pediatr Oncol. 2000;35:603–607. doi: 10.1002/1096-911X(20001201)35:6<603::AID-MPO24>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 13.ElOjeimy S, McKillop JC, El-Zawahry AM, Holman DH, Liu X, Schwartz DA, Day TA, Dong JY, Norris JS. FasL gene therapy: a new therapeutic modality for head and neck cancer. Cancer Gene Ther. 2006;13:739–745. doi: 10.1038/sj.cgt.7700951. [DOI] [PubMed] [Google Scholar]
- 14.Feltner DE, Cooper M, Weber J, Israel MA, Thiele CJ. Expression of class I histocompatibility antigens in neuroectodermal tumors is independent of the expression of a transfected neuroblastoma myc gene. J Immunol. 1989;143:4292–4299. [PubMed] [Google Scholar]
- 15.Fulda S, Debatin KM. IFNgamma sensitizes for apoptosis by upregulating caspase-8 expression through the Stat1 pathway. Oncogene. 2002;21:2295–2308. doi: 10.1038/sj.onc.1205255. [DOI] [PubMed] [Google Scholar]
- 16.Fulda S, Debatin KM. 5-Aza-2′-deoxycytidine and IFN-gamma cooperate to sensitize for TRAIL-induced apoptosis by upregulating caspase-8. Oncogene. 2006;25:5125–5133. doi: 10.1038/sj.onc.1209608. [DOI] [PubMed] [Google Scholar]
- 17.Hellstrom KE, Hellstrom I. Immunity to neuroblastomas and melanomas. Annu Rev Med. 1972;23:19–38. doi: 10.1146/annurev.me.23.020172.000315. [DOI] [PubMed] [Google Scholar]
- 18.Hopkins-Donaldson S, Bodmer JL, Bourloud KB, Brognara CB, Tschopp J, Gross N. Loss of caspase-8 expression in highly malignant human neuroblastoma cells correlates with resistance to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 2000;60:4315–4319. [PubMed] [Google Scholar]
- 19.Hopkins-Donaldson S, Bodmer JL, Bourloud KB, Brognara CB, Tschopp J, Gross N. Loss of caspase-8 expression in neuroblastoma is related to malignancy and resistance to TRAIL-induced apoptosis. Med Pediatr Oncol. 2000;35:608–611. doi: 10.1002/1096-911X(20001201)35:6<608::AID-MPO25>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 20.Huang Y, Sheikh MS (2006) TRAIL death receptors and cancer therapeutics. Toxicol Appl Pharmacol (in press) [DOI] [PubMed]
- 21.Ichikawa K, Liu W, Zhao L, Wang Z, Liu D, Ohtsuka T, Zhang H, Mountz JD, Koopman WJ, Kimberly RP, Zhou T. Tumoricidal activity of a novel anti-human DR5 monoclonal antibody without hepatocyte cytotoxicity. Nat Med. 2001;7:954–960. doi: 10.1038/91000. [DOI] [PubMed] [Google Scholar]
- 22.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 23.Kalos M. Tumor antigen-specific T cells and cancer immunotherapy: current issues and future prospects. Vaccine. 2003;21:781–786. doi: 10.1016/S0264-410X(02)00598-4. [DOI] [PubMed] [Google Scholar]
- 24.Kim S, Kang J, Evers BM, Chung DH. Interferon-gamma induces caspase-8 in neuroblastomas without affecting methylation of caspase-8 promoter. J Pediatr Surg. 2004;39:509–515. doi: 10.1016/j.jpedsurg.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 25.Kogner P, Barbany G, Dominici C, Castello MA, Raschella G, Persson H. Coexpression of messenger RNA for TRK protooncogene and low affinity nerve growth factor receptor in neuroblastoma with favorable prognosis. Cancer Res. 1993;53:2044–2050. [PubMed] [Google Scholar]
- 26.Kummer JA, Micheau O, Schneider P, Bovenschen N, Broekhuizen R, Quadir R, Strik MC, Hack CE, Tschopp J. Ectopic expression of the serine protease inhibitor PI9 modulates death receptor-mediated apoptosis. Cell Death Differ. 2007;14:1486–1496. doi: 10.1038/sj.cdd.4402152. [DOI] [PubMed] [Google Scholar]
- 27.Lampson LA, Fisher CA, Whelan JP. Striking paucity of HLA-A, B, C and beta 2-microglobulin on human neuroblastoma cell lines. J Immunol. 1983;130:2471–2478. [PubMed] [Google Scholar]
- 28.Lampson LA, Whelan JP, Fisher CA. HLA-A,B,C and beta 2-microglobulin are expressed weakly by human cells of neuronal origin, but can be induced in neuroblastoma cell lines by interferon. Prog Clin Biol Res. 1985;175:379–388. [PubMed] [Google Scholar]
- 29.Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 30.Le LH, Hirte HW, Hotte SJ, Maclean M, Iacobucci A, Corey A, Fox NL, Oza AM. Phase I study of a fully human monoclonal antibody to the tumor necrosis factor-related apoptosis-inducing ligand death receptor 4 (TRAIL-R1) in subjects with advanced solid malignancies or non-Hodgkin’s lymphoma (NHL) J Clin Oncol (Meeting Abstracts) 2004;22:2533. [Google Scholar]
- 31.Main EK, Monos DS, Lampson LA. IFN-treated neuroblastoma cell lines remain resistant to T cell-mediated allo-killing, and susceptible to non-MHC-restricted cytotoxicity. J Immunol. 1988;141:2943–2950. [PubMed] [Google Scholar]
- 32.Marozzi A, Meneveri R, Bunone G, De Santis C, Lopalco L, Beretta A, Agresti A, Siccardi AG, Della Valle G, Ginelli E. Expression of beta 2m-free HLA class I heavy chains in neuroblastoma cell lines. Scand J Immunol. 1993;37:661–667. doi: 10.1111/j.1365-3083.1993.tb01680.x. [DOI] [PubMed] [Google Scholar]
- 33.Martin RF, Beckwith JB. Lymphoid infiltrates in neuroblastomas: their occurrence and prognostic significance. J Pediatr Surg. 1968;3:161–164. doi: 10.1016/0022-3468(68)91005-1. [DOI] [PubMed] [Google Scholar]
- 34.Nakagawara A, Arima-Nakagawara M, Scavarda NJ, Azar CG, Cantor AB, Brodeur GM. Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N Engl J Med. 1993;328:847–854. doi: 10.1056/NEJM199303253281205. [DOI] [PubMed] [Google Scholar]
- 35.Parham P, Barnstable CJ, Bodmer WF. Use of a monoclonal antibody (W6/32) in structural studies of HLA-A,B,C, antigens. J Immunol. 1979;123:342–349. [PubMed] [Google Scholar]
- 36.Perosa F, Luccarelli G, Prete M, Favoino E, Ferrone S, Dammacco F. Beta 2-microglobulin-free HLA class I heavy chain epitope mimicry by monoclonal antibody HC-10-specific peptide. J Immunol. 2003;171:1918–1926. doi: 10.4049/jimmunol.171.4.1918. [DOI] [PubMed] [Google Scholar]
- 37.Reed JC. Drug insight: cancer therapy strategies based on restoration of endogenous cell death mechanisms. Nat Clin Pract Oncol. 2006;3:388–398. doi: 10.1038/ncponc0538. [DOI] [PubMed] [Google Scholar]
- 38.Russell JH, Ley TJ. Lymphocyte-mediated cytotoxicity. Annu Rev Immunol. 2002;20:323–370. doi: 10.1146/annurev.immunol.20.100201.131730. [DOI] [PubMed] [Google Scholar]
- 39.Squire R, Fowler CL, Brooks SP, Rich GA, Cooney DR. The relationship of class I MHC antigen expression to stage IV-S disease and survival in neuroblastoma. J Pediatr Surg. 1990;25:381–386. doi: 10.1016/0022-3468(90)90375-J. [DOI] [PubMed] [Google Scholar]
- 40.Stupack DG, Teitz T, Potter MD, Mikolon D, Houghton PJ, Kidd VJ, Lahti JM, Cheresh DA. Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature. 2006;439:95–99. doi: 10.1038/nature04323. [DOI] [PubMed] [Google Scholar]
- 41.Sugimoto T, Horii Y, Hino T, Kemshead JT, Kuroda H, Sawada T, Morioka H, Imanishi J, Inoko H. Differential susceptibility of HLA class II antigens induced by gamma-interferon in human neuroblastoma cell lines. Cancer Res. 1989;49:1824–1828. [PubMed] [Google Scholar]
- 42.Teitz T, Lahti JM, Kidd VJ. Aggressive childhood neuroblastomas do not express caspase-8: an important component of programmed cell death. J Mol Med. 2001;79:428–436. doi: 10.1007/s001090100233. [DOI] [PubMed] [Google Scholar]
- 43.Teitz T, Stupack DG, Lahti JM. Halting neuroblastoma metastasis by controlling integrin-mediated death. Cell Cycle. 2006;5:681–685. doi: 10.4161/cc.5.7.2615. [DOI] [PubMed] [Google Scholar]
- 44.Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA, Behm FG, Look AT, Lahti JM, Kidd VJ. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat Med. 2000;6:529–535. doi: 10.1038/75007. [DOI] [PubMed] [Google Scholar]
- 45.Tekautz TM, Zhu K, Grenet J, Kaushal D, Kidd VJ, Lahti JM. Evaluation of IFN-gamma effects on apoptosis and gene expression in neuroblastoma—preclinical studies. Biochim Biophys Acta. 2006;1763:1000–1010. doi: 10.1016/j.bbamcr.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 46.Thorburn A. Death receptor-induced cell killing. Cell Signal. 2004;16:139–144. doi: 10.1016/j.cellsig.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 47.Vertuani S, De Geer A, Levitsky V, Kogner P, Kiessling R, Levitskaya J. Retinoids act as multistep modulators of the major histocompatibility class I presentation pathway and sensitize neuroblastomas to cytotoxic lymphocytes. Cancer Res. 2003;63:8006–8013. [PubMed] [Google Scholar]
- 48.Warzynski MJ, Graham DM, Axtell RA, Higgins JV, Hammers YA. Flow cytometric immunophenotyping test for staging/monitoring neuroblastoma patients. Cytometry. 2002;50:298–304. doi: 10.1002/cyto.10159. [DOI] [PubMed] [Google Scholar]
- 49.Wolfl M, Jungbluth AA, Garrido F, Cabrera T, Meyen-Southard S, Spitz R, Ernestus K, Berthold F. Expression of MHC class I, MHC class II, and cancer germline antigens in neuroblastoma. Cancer Immunol Immunother. 2005;54:400–406. doi: 10.1007/s00262-004-0603-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang X, Merchant MS, Romero ME, Tsokos M, Wexler LH, Kontny U, Mackall CL, Thiele CJ. Induction of caspase 8 by interferon gamma renders some neuroblastoma (NB) cells sensitive to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) but reveals that a lack of membrane TR1/TR2 also contributes to TRAIL resistance in NB. Cancer Res. 2003;63:1122–1129. [PubMed] [Google Scholar]
- 51.Yuan J. Transducing signals of life and death. Curr Opin Cell Biol. 1997;9:247–251. doi: 10.1016/S0955-0674(97)80069-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.