

Abstract
The DNA excision repair protein ERCC1 and the DNA damage sensor protein, XPA are highly overexpressed in patient samples of cisplatin-resistant solid tumors including lung, bladder, ovarian, and testicular cancer. The repair of cisplatin-DNA crosslinks is dependent upon nucleotide excision repair (NER) that is modulated by protein-protein binding interactions of ERCC1, the endonuclease, XPF, and XPA. Thus, inhibition of their function is a potential therapeutic strategy for the selective sensitization of tumors to DNA-damaging platinum-based cancer therapy. Here, we report on new small-molecule antagonists of the ERCC1/XPA protein-protein interaction (PPI) discovered using a high-throughput competitive fluorescence polarization binding assay. We discovered a unique structural class of thiopyridine-3-carbonitrile PPI antagonists that block a truncated XPA polypeptide from binding to ERCC1. Preliminary hit-to-lead studies from compound 1 reveal structure-activity relationships (SAR) and identify lead compound 27o with an EC50 of 4.7 μM. Furthermore, chemical shift perturbation mapping by NMR confirms that 1 binds within the same site as the truncated XPA67-80 peptide. These novel ERCC1 antagonists are useful chemical biology tools for investigating DNA damage repair pathways and provide a good starting point for medicinal chemistry optimization as therapeutics for sensitizing tumors to DNA damaging agents and overcoming resistance to platinum-based chemotherapy.
Graphical Abstract

We have discovered a new chemical series of small molecules which block the protein-protein interaction of ERCC1 and XPA, which are important in DNA damage repair and resistance to chemotherapy. We found hit compound 1 from high-throughput screening. SAR studies from focused library synthesis led to the identification of lead inhibitor 27o with an EC50 of 4.7 μM.
The DNA of living organisms is persistently damaged by endogenous sources such as metabolic byproducts and errors in replication, and also by a diverse array of external causes including exposure to UV radiation, X-rays, and chemical agents. In order to evade the genomic instability that can result from DNA lesions, there are multiple repair pathways in cells that are tailored to eradicate specific types of DNA damage. These repair mechanisms require the coordinated assembly of large protein complexes that help protect the cell from the cytotoxic and mutagenic effects derived from unnaturally modified DNA. Traditional anti-tumor therapy, radiation treatment or chemotherapy, relies on damaging cellular DNA and results in cytotoxicity to both cancer and normal cells. However, cancer cells can develop resistance to DNA-damaging therapy, in part, by upregulating DNA damage repair pathways1 such as nucleotide excision repair (NER). Thus, the identical DNA repair pathways that are in place to help protect normal cells from tumorigenesis can, ironically, lead to tumor cells’ chemoresistance from increased DNA repair function.
Having lost some pathways during tumorigenesis and upregulated other pathways in response to the selective pressure of DNA damage therapy, tumor cells are often hyperdependent on specific repair pathways. As such, DNA repair inhibitors have the potential to target chemoresistant tumors and selectively enhance the chemosensitivity of tumor cells that are hyperdependent on a specific DNA repair pathway, thereby providing therapeutic relevance and specificity. The ability to selectively sensitize tumor cells to DNA damaging therapy is of great clinical interest2, and several combination therapy strategies, including checkpoint kinase (Chk1), poly(ADP-ribose) polymerase (PARP-1), and most recently PDL-1 inhibitors, have been adopted over the past two decades3. The NER pathway involves the assembly of more than 30 proteins into a DNA repair complex that recognizes and removes bulky chemical adducts, such as cisplatin crosslinks, from DNA, NER is highly coordinated through a series of specific protein-protein interactions, including recruitment of the heterodimeric repair endonuclease ERCC1-XPF4 through interactions with the DNA scaffolding protein XPA5. ERCC1-XPF is required for excision of the damaged DNA, and therefore, blocking the recruitment of ERCC1-XPF to the repairosome by deletion or mutagenesis of XPA prevents NER6. ERCC1 also functions in interstrand crosslink repair (ICL) and homologous recombination (HR) DNA repair pathways, for which interaction with XPA is not required6b. Interestingly, ERCC1 and XPF knockout mice are born runted and perish from liver failure while XPA knockout mice6c are not lethal.
Cisplatin is the standard care of treatment for many solid tumors including sarcomas and carcinomas such as lung and ovarian cancer. Cisplatin and other platinating agents act by covalently adding to DNA, generating multiple lesions which impair DNA synthesis and trigger apoptosis, including 1, 2- and 1,3-intrastrand crosslinks and 1,2-interstrand crosslinks between purine bases7. The intrastrand crosslinks generated by cisplatin are repaired primarily through NER, whereas interstrand crosslinks are repaired by ICL8. It is not yet completely understood which type of crosslink damage is primarily responsible for cisplatin cytotoxicity. Expression levels of ERCC1 has been established as a biomarker for cisplatin resistance and can predict for survival of patients with non-small cell lung cancer (NSCLC)9 and other tumor types9e. Mechanisms of cisplatin resistance in tumors10 include changes in drug pharmacokinetics, inhibition of apoptosis, and increased DNA repair function, including NER10b. Cisplatin-resistant tumors, demonstrated in patient samples of multiple tumor types, often exhibit increased DNA repair function from an upregulation of ERCC19 and/or XPA9f by overexpression. Downregulation of ERCC1-XPF expression has also been shown to enhance cisplatin resistance9a. Therefore, inhibitors of ERCC1 function including its interaction with XPF11 and XPA2 are predicted to sensitize tumors and reverse resistance to cisplatin and other platinating agents11.
It was previously reported that a truncated 14-mer peptide corresponding to residues 67-80 of XPA, binds with sub-micromolar affinity to a well-defined binding pocket on ERCC112. Herein, we disclose a new class of small-molecule ERCC1/XPA protein-protein interaction (PPI) antagonists identified from high-throughput screening (HTS) and describe hit to lead identification studies. These new antagonists of ERCC1/XPA are predicted to be reversible blockers of NER activity and represent powerful chemical biology probes useful for investigating the cellular physiology of the DNA damage response modulated by ERCC1.
Results and Discussion
Identification of Small Molecule Antagonists of the ERCC1-XPA Interaction.
Protein-protein interactions (PPIs) such as ERCC1/XPA are a vast yet poorly explored territory for small molecule ligand design13, discovery14 and drug development15. There have been some recent success stories especially in oncology16, but PPI interfaces encompass a large area of relatively solvent-exposed surface which present a challenge for discovering small molecular weight modulators. The ERCC1/XPA protein-protein interface, however, is atypical because it encompasses a small, deep V-shaped groove in ERCC1. This deep V-shaped groove is structurally related to the nuclease active site in XPF12, 17. Mutagenesis of ERCC1 in the XPA binding pocket revealed several hotspots that are responsible for a majority of the binding affinity18. This data was recapitulated using molecular dynamics simulations of XPA peptides19. Therefore, it is plausible that the ERCC1/XPA interaction could be effectively modulated with small molecule antagonists. Others have identified inhibitors through in silico screening20 and in one case have shown sensitization to UV and cisplatin20c. A more recent report also describes inhibitors of the XPA-DNA interaction20d. Prior to these publications, we implemented a high-throughput screening (HTS) assay over ten years ago to identify compounds that directly block the ERCC1/XPA binding interaction.
We constructed a MBP fusion protein of the central XPA binding domain of ERCC1 and developed an inhibition assay. The inhibition assay quantitates the binding of a fluorescein-labeled 14-mer peptide of XPA67-80 (FAM-XPA) to the XPA-binding domain of ERCC1 by fluorescence polarization (FP) or anisotropy (Figure 1a). High throughput screening of five commercially available chemical libraries containing 20,830 diverse compounds was performed at a single point 20 μM concentration of compound. From the screen, we identified 31 primary hits 1-4 (Figure 1b) which displaced greater than 20% FAM-XPA from ERCC1, of which four were confirmed (Figure 2) to inhibit the XPA67-80-ERCC1 interaction in a dose-dependent manner. Interestingly, the two top hits identified from virtual screening20c are structurally similar. The most potent and chemically attractive of these confirmed hits, thiopyridine-3-carbonitrile 1, disrupts the protein-protein interaction with an EC50 of 21 μM and represents a unique structural class of PPI antagonists. After the structural identity and biological activity of compound 1 was established by resynthesis (Scheme 1), we pursued a traditional ligand-based approach in order to elucidate key structure-activity relationships (SAR) and identify analogs with improved potency.
Figure 1.

a) Fluorescence polarization (FP)/anisotropy competitive binding inhibition assay with FAM-XPA67-80; b) Identification of screening hits from compound libraries which inhibit ERCC1/XPA67-80 heterodimerization at 20 μM concentration.
Figure 2.

Confirmed HTS hits of ERCC1/XPA 14-mer.
Scheme 1. Synthesis and initial SAR of nitrile and alcohol.

Reagents and conditions: (a) 3-nitrophenacyl bromide, 10% KOH, DMF; (b) NaBH4, THF/MeOH (1:1)
Synthesis and Initial SAR of HTS hit 1.
Synthesis of compound 1 was achieved by alkylation of 3-cyano-4,6-dimethyl-2-mercaptopyridine with 3-nitrophenacyl bromide using potassium hydroxide in DMF to give ketone 5a (Scheme 1). Subsequent reduction of ketone 5a with sodium borohydride gives 1 in excellent yield. We were first interested in elucidating the importance of the hydroxyl and nitrile functional groups, hypothesizing that these groups form hydrogen bonds with ERCC1. We found that neither the ketone 5 nor the commercially available ester 7 showed any inhibitory effect on XPA binding at concentrations up to 100 μM, indicating the hydroxyl group is required for binding to ERCC1. Des-nitrile 6 also failed to block FAM-XPA binding to ERCC1, suggesting that the nitrile group is also essential for binding. Next, to assess the importance of the sulfide linker, ether analog 10b was synthesized (Scheme 2) in a similar fashion to 1 from 2- hydroxy-4, 6-dimethyl-pyridine-3-carbonitrile, and the amine linker analog 9 was obtained through an aryl substitution reaction of amino alcohol 8 with 2-chloro-3-cynanopyridine. The amine linker analog 9 did not prevent FAM-XPA binding, while the ether analog 10b showed a 2-fold decrease in inhibition (EC50 = 48 μM) relative to sulfide 1. While it is not clear why amine 9 fails to block the ERCC1-XPA interaction, introduction of the NH may affect the ground state conformation or encounter electronic repulsion with ERCC1 that is not conducive to binding. The preference for sulfur over the oxygen linker might indicate that a larger distance from the pyridyl ring to the hydroxyl and/or aryl group results in more optimal contact with ERCC1.
Scheme 2. Evaluation of pyridine ring linker heteroatom.

Reagents and conditions: (a) NaBH4, THF/MeOH (1:1); (b) K2CO3, DMF; (c) i. KOtBu, THF, ii. 3-nitrophenacyl bromide
Lead Identification.
We next focused our attention on synthesizing substituted analogs of the aryl ring (Table 1) in order to further develop SAR and identify potential sites for improving binding affinity to ERCC1. Analogs were synthesized in a similar fashion to 1 by using commercially available alpha-bromo acetophenones. From this set of aryl substituted diverse derivatives, we discovered that removal of the nitro (11) resulted in a 4-fold drop in potency, whereas moving the nitro to the ortho position (12) slightly improved potency (EC50 = 14 μM). In contrast, the para-nitro analog 13 was inactive up to 100 μM prompting further exploration of substituents on the aryl ring. Initially, we designed analogs to assess the steric and electronic effects of the ring system on ERCC1 binding by selecting analogs with a diverse array of electron withdrawing and donating functional groups incorporated on all ortho, meta, and para ring positions (Table 1). We found that a wide variety of functionality is tolerated on either the 2-position (ortho, a) or 3-position (meta, b), but substitution at the 4-position (para, c) completely abrogates inhibition for all six types of analogs tested, indicating that this effect is driven by sterics and not dependent on electronic properties. On the other hand, all three pyridyl analogs 22a-c are inactive up to 100 μM which is similar to the outcome from the des-nitro phenyl analog 11 discussed earlier. Based on these results, we conclude that the substitution of the para position is non-productive for binding to ERCC1, and this position may be sterically blocked by the binding pocket. These results also suggest that substitution of the ortho and/or meta position is required for tight binding to ERCC1, though no significant potency enhancements were realized relative to nitro from the compounds evaluated.
Table 1.
SAR evaluation of aryl ring.

| |||||
|---|---|---|---|---|---|
| Compound | R | EC50 (μM) |
Compound | R | EC50 (μM) |
| 11 | H | 87 | 19b | 3-CF3 | >100 |
| 12 | 2-NO2 | 14 | 19c | 4-CF3 | >100 |
| 1 | 3-NO2 | 21 | 20a | 2-Me | 24 |
| 13 | 4-NO2 | >100 | 20b | 3-Me | 38 |
| 17a | 2-NH2 | 42 | 20c | 4-Me | >100 |
| 17b | 3-NH2 | 28 | 21a | 2-OMe | 24 |
| 17c | 4-NH2 | >100 | 21b | 3-OMe | >100 |
| 18a | 2-Cl | 26 | 21c | 4-OMe | >100 |
| 18b | 3-Cl | 12.5 | 22a | 2-pyridyl | >100 |
| 18c | 4-Cl | >100 | 22b | 3-pyridyl | >100 |
| 19a | 2-CF3 | >100 | 22c | 4-pyridyl | >100 |
In order to increase hydrophobic binding interactions with ERCC1, we pursued a series of analogs bearing larger substituents appended to the aryl ring. We utilized the anilines 17a and 17b as versatile intermediates for a variety of aryl ring substitution. The synthesis of meta-aniline 17b was achieved in good yield through catalytic hydrogenation of meta-nitro ketone 5 followed by borohydride reduction as shown in Scheme 3. However, attempts at the transformation of 2-nitro benzylic alcohol 12 to 17a using this method gave very poor yields of desired product. Therefore, we employed an iron metal-mediated reduction with ammonium chloride which resulted in excellent yields of 17a-c from either the meta, ortho, or para nitro alcohols 1, 12, or 13, respectively. With these key intermediates in hand, we created a small, focused library of mostly aryl amides in the meta position that were synthesized by reaction of aniline 17b with commercially available carboxylic acids using a HATU coupling (Scheme 4). We found that aliphatic derivatives such as 23a and benzamides such as 23b are not able to block the XPA-ERCC1 interaction while in contrast, phenylacetamides such as meta-substituted analog 23c which was equipotent to the starting hit compound 1. The ortho-substituted matched pair phenylacetamide 24, synthesized from aniline 17a, showed enhanced potency with an EC50=11 μM. Presumably, the additional conformational flexibility made possible by the methylene spacer to the aromatic ring in the phenylacetamide series is required to adopt a productive binding conformation to ERCC1.
Scheme 3. Reduction to aniline derivatives of 1, 12, 13.

Reagents and conditions: (a) H2, 10% Pd/C, DCM/MeOH (1:1); (b) NaBH4, THF/MeOH (1:1); (c) Fe, NH4Cl, DCM/MeOH (2:1), water (1% v/v), 40 °C
Scheme 4. Amide substitution of 17a and 17b.
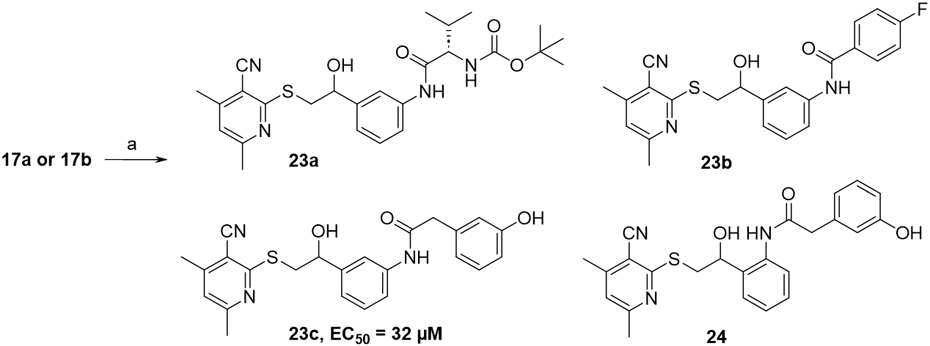
Reagents and conditions: (a) HATU, RCO2H, DIPEA, DMF
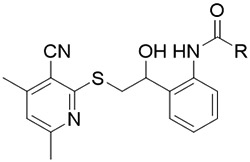
The slightly improved binding affinity of the phenylacetamide compounds prompted is to pursue the synthesis of a larger set of analogs to generate additional SAR and pursue further potency enhancement. Shown in Table 2, a variety of further substitution (25, 26, 27a-r) is tolerated off the ortho position. We found that the phenol of compound 24 is not required for binding to ERCC1 since des-hydroxy analog 26 retains affinity (EC50 = 24 μM). From this study we identified the most potent antagonist, 4-chlorophenylacetamide, 27o with an EC50 of 4.7 μM. Addition of another chlorine 27r results in a 10-fold reduction in activity while nitro (27a) or bromine (27p) replacements for chlorine have no appreciable effect relative to the starting hydroxyl-phenylacetamide 24. It is noteworthy that introducing less conformational flexibility, as seen with the thiophene 28 or indazole 29, leads to a significant reduction in binding affinity to ERCC1. Compound 27o constitutes an attractive new lead ERCC1/XPA protein-protein interaction antagonist suitable for further optimization.
Table 2.
SAR of ortho-substituted phenylacetamides.
|
||
|---|---|---|
| Compound | R | EC50 (μM) |
| 24 | CH2Ph-3-OH | 11 |
| 25 | CH2Ph-4-OH | 9.4 |
| 26 | CH2Ph | 24 |
| 27a | CH2Ph-4-NO2 | 13 |
| 27b | CH2Ph-3-NO2 | 7.0 |
| 27c | CH2Ph-2-F | 9.0 |
| 27d | CH2Ph-3-F | 10 |
| 27e | CH2Ph-4-F | 8.0 |
| 27f | CH2Ph-3-CF3 | >50 |
| 27g | CH2Ph-4-CF3 | >50 |
| 27h | CH2Ph-3-OMe | 30 |
| 27i | CH2Ph-4-OMe | 30 |
| 27j | CH2Ph-2-Me | 7.8 |
| 27k | CH2Ph-3-Me | 7.4 |
| 27l | CH2Ph-4-Me | 8.0 |
| 27m | CH2Ph-2-Cl | 20 |
| 27n | CH2Ph-3-Cl | 20 |
| 27o | CH2Ph-4-Cl | 4.7 |
| 27p | CH2Ph-4-Br | 10 |
| 27q | CH2Ph-3-Br | 20 |
| 27r | CH2Ph-3,4-di-Cl | 42 |
| 28 |

|
>100 |
| 29 |

|
68 |
NMR characterization of Small Molecule Antagonist Binding to ERCC1.
In order to determine if the small molecule PPI antagonists of ERCC1/XPA we have discovered binding in a competitive manner to the same site as XPA and the truncated 14-mer XPA67-80 peptide, we carried out chemical shift perturbation experiments of HTS hit 1 with 15N-labled ERCC1. These studies were intended to enable a rational structure-guided design and optimization of improved analogs with increased binding affinity to ERCC1. Figure 3 summarizes the results of our mapping experiments. Three clusters of residues in ERCC1: 107-110, 140-148, and 172-175 showed pronounced and saturable changes in chemical shifts (Figure 3a). The same residues were observed as the most affected by XPA67-80:ERCC1 binding in NMR mapping experiments by Tripsianes21. To eliminate the possibility of different construct and/or tag effects, we have repeated the XPA67-80:ERCC1 mapping experiments (data not shown) and their results were identical to the published reports. Therefore, we are confident that compound 1 binds to the same site as the truncated XPA 14-mer.
Figure 3.

Backbone chemical shift perturbation mapping of compound 1 binding to ERCC1. a) Plot of combined 1H and 15N amide chemical shift differences for non-proline residues. Ratios of the ERCC1 to compound 1 used in 15N-1H HSQC experiments are shown as the P/S value. Combined 15N and 1H shift differences values were calculated as . Blue, magenta, yellow and green horizontal bars include residues shown on the molecular surface in the same color scheme. b) Proposed model of compound 1 bound to ERCC1 placing in XPA-binding site.
Compounds with measured ERCC1 antagonism were docked to the ERCC1 crystal structure17 using GOLD22 with default parameters. Prioritization of the possible binding modes incorporated our understanding of SAR from the analogs of compound 1 and the NMR binding mode data. Our predicted binding mode (shown in Figure 3b) places the pyridyl ring and the nitrile substituent in proximity to residues 140-148 and the nitro phenyl ring directed at resides 107-110 also placing the nitro group near resides 172-175.
Conclusions
The discovery of low molecular weight protein-protein interaction (PPI) antagonists presents a challenging goal for medicinal chemistry. In this report, we identified thiopyridine-3-carbonitriles as a novel small molecule scaffold which blocks the ERCC1/XPA PPI important in NER DNA damage repair. In the absence of structural biology information, we were able to elucidate initial key SAR for this unique series of PPI antagonists through directed analog synthesis. NMR studies confirmed HTS hit 1 binds to the same central domain of ERCC1 as truncated XPA peptides. These preliminary studies enable further hit to lead identification and optimization using traditional medicinal chemistry coupled with a rational structure-based design using X-ray crystallographic and NMR methods. Current work is directed at elucidation of compound binding to ERCC1 utilizing X-ray, further SAR evaluation, and identifying alternate scaffolds. Future studies will focus on testing for the functional effects of compounds to sensitize chemoresistant tumors to cisplatin and other DNA damaging agents which has been shown for other ERCC1/XPA inhibitors20c.
Methods
Cloning, Expression, and Purification of ERCC1.
For expression, the ERCC1 central domain (residues 96-214) was cloned into an N-terminal 6-His tag. E. coli BL21(DE3) pLyS expressing the ERCC1 central domain were lysed by high-pressure homogenization in Buffer A (50 mM Tris, pH 7.5, 300 mM NaCl, 5 mM β-mercaptoethanol, and 0.1 mM EDTA). The ERCC1 central domain was purified by immobilized metal affinity chromatography on a 5 mL HisTrap nickel column (GE Healthcare Life Sciences), and eluted using Buffer A supplemented with 300 mM imidazole. Fractions containing ERCC1 were combined and loaded onto a Sephadex S200 gel filtration column (GE Healthcare Life Science).
Fluorescence Polarization Measurement of ERCC1-XPA Binding Affinity.
Fluorescence polarization was measured on a BioTek Synergy II multimodal plate reader, using a 485 nm excitation wavelength and a 520 nm emission wavelength. Because the central domain itself did not sufficiently increase polarization for high-throughput screening, ERCC1 was subcloned into pMAL-p4e, creating a fusion protein with a C-terminal 6-His tag and an N-terminal maltose binding protein. This linker between MBP and ERCC1 was mutated to 3 prolines using the QuikChange site-directed mutagenesis kit (Agilent). MBP-ERCC1 was purified using the same protocol as the ERCC1 central domain, as described above. The MBP fusion did not affect the binding affinity of the ERCC1 central domain for a 14mer XPA peptide (data not shown). The binding isotherm of MBP-ERCC1 for XPA was measured by incubating 10 nM fluorescein-labeled XPA peptide (fXPA) with 0-100 μM ERCC1. High throughput screening conditions were chosen to maximize the dynamic range: 10 nM XPA, 2 μM MBP-ERCC1, and 20 μM test compound in 50 mM Tris pH 7.5, 100 mM NaCl, 5 mM β-mercaptoethanol, 0.1 mM EDTA.
High Throughput Screening for Inhibitors of ERCC1-XPA Heterodimerization.
The fluorescence polarization assay described above was used to identify small molecule inhibitors of ERCC1-XPA heterodimerization. Automated high throughput screening of five chemical libraries containing 20,830 compounds was performed in duplicate the Chemical Genetics Screening Core (CGSC) at Washington University School of Medicine. The compound libraries tested were: the ICCB Known Bioactives library (480 compounds, Enzo Life Sciences), the Spectrum collection (2000 compounds, MicroSource Discovery Systems), the Maybridge HitFinder library (14,400 compounds, Maybridge), the Diversity Set (2000 compounds, National Cancer Institute), and the Johns Hopkins Clinical Compound Library (1950 compounds). Assays were conducted in 96-well format, using half-area plates (Corning) containing 100 μL per well. Briefly, 10 nM fluorescein-labeled XPA (fXPA) was mixed with 2 μM ERCC1-MBP in 50 mM Tris pH 7.5, 100 mM NaCl, 5 mM β-mercaptoethanol, 0.1 mM EDTA and incubated at room temperature during dispensing. 2 μL compound (DMSO solutions, 20 μM final concentration) was added to a dry assay plate, then 98 μL ERCC1-fXPA complex was added. Plates were incubated for 10 minutes are room temperature and shaken for 30 seconds before fluorescence intensity and polarization were measured using a BioTek Synergy II plate reader. The median absolute deviation (MAD) were calculated for each plate. Hits were defined as compounds with fluorescence polarization more than 3 MAD below the median, and fluorescence intensity within 5 MAD of the plate median, in both duplicate plates. Hit compounds were re-tested in duplicate using the same conditions as the high-throughput screen. The 50% effective concentration (EC50) of each inhibitor was measured by serially diluting the compound in DMSO prior to performing the fluorescence polarization assay as described above (200 to 20 μM final compound concentration).
Chemical shift perturbation mapping.
The 15N-labeled human ERCC1 fragment containing residues 96-214 was expressed in E.coli BL21(DE3) grown on M9 minimal media supplemented with 15NH4Cl (1 g/L) as the sole nitrogen source. All NMR experiments were done with 0.25 mM ERCC1 in 50 mM Tris, 100 mM NaCl, 3 mM TCEP, 1 mM EDTA, pH 7.5 at 28 °C. Compound 1, 8.5 mM stock in DMSO, was serially diluted to 0.026, 0.053, 0.104, 0.203, and 0.389 mM concentrations. These titration points corresponded to ERCC1 : compound 1 ratios of 10, 5, 2.5, 1.25 and 0.63, respectively. The addition of 5% DMSO alone (approx. the highest amount introduced during titration) to ERCC1 did not have an effect on ERCC1 chemical shifts. 15N-1H HSQC spectra were recorded and processed on a Bruker 600 Avance III spectrometer equipped with a cryoprobe. Each time domain spectrum had 1024 complex t2, and 64 complex t1 points acquired with 16 scans per t1 increment. The frequency spectra were processed to 2048 by 1024 complex size. Assignments of 15N-1H resonances were adapted from BMRB entry 1524021. Peaks in all spectra were picked automatically using routines of Topspin 3.0 software (Bruker), and adjusted manually to center peak positions and remove artefacts. Peak lists were imported, and chemical shift differences were calculated in an Excel spreadsheet. For each assigned HSQC crosspeak, 15N and 1H chemical shift differences in ppm units were calculated according to the formula, .
General Synthetic Methods.
Nuclear magnetic resonance (NMR) spectra were recorded using a 300 MHz Varian Mercury or a 600 MHz Bruker Avance spectrometer equipped with a 5 mm multinuclear probe. 1H chemical shifts are reported in parts per million (ppm) and referenced internally according to residual solvent signals and/or TMS. Coupling constants (J) are reported in Hertz (Hz) and multiplicity is abbreviated as follows: app = apparent, s = singlet, d = doublet, dd = doublet of doublets, dt = doublet of triplets, t = triplet, q = quartet, p = pentet, m = multiplet, br = broad signal. High-pressure liquid chromatography (HPLC) was carried out on a GILSON GX-281 using Waters C18 5μM, 4.6×50 mm and Waters Prep C18 5μM, 19×150 mm reverse phase columns. Mass spectra (MS) were performed on a Waters MicromassZQ (Model MM1) spectrometer. Thin layer chromatography (TLC) was carried out using EMD glass precoated plates with siliga gel (0.25 mm thick, 60F254). Visualization of the plates was accomplished by using UV (254 nm) and/or dyes such as KMnO4, p-anisaldehyde, or CAM (ceric ammonium molybdate). Silica gel flash chromatography was carried out using a Teledyne ISCO purification system using RediSep™ or Thomson Scientific pre-packed silica gel columns (4g-330g sizes). All compounds tested in biological assays are at least of 95% purity based on analytical HPLC results monitored with 220 nm and 254 nm wavelengths, unless otherwise noted. All commercially available materials were used without further purification unless otherwise noted. Anhydrous solvents such as MeOH, DMF, DCM (CH2Cl2), and THF were obtained from Sigma-Aldrich (Sure/Seal™). All reactions requiring anhydrous conditions were carried out under nitrogen atmosphere unless otherwise specified. RT refers to room temperature (~23°C).
Synthesis and Purification of HTS Hit 1 and analogs.
HTS hit 1 and analogs were synthesized as described below using published and standard organic chemistry transformations and procedures from commercially available starting materials. Compounds tested for biological activity were purified by silica gel chromatography or reverse-phase HPLC and encompassed >95% purity as determined by 1H NMR and HPLC/MS.
4,6-dimethyl-2-[2-(3-nitrophenyl)-2-oxo-ethyl]sulfanyl-pyridine-3-carbonitrile (5).
To a pre-mixed suspension (20 min) of 3-cyano-4,6-dimethyl-2-mercaptopyridine (1.6 g, 10 mmol) and 10% aq. KOH (5.6 mL, 11 mmol) in DMF (40 mL) at 0 °C was added 3-nitrophenacyl bromide (4.9 g, 20 mmol) as a solution in DMF (10 mL) via addition funnel slowly under a nitrogen atmosphere. After complete addition, the resulting reaction mixture was warmed to RT and stirred vigorously for 2 h. The reaction mixture was quenched by adding cold water (50 mL), and the crude product precipitated out of solution, and collected by suction filtration on a sintered glass funnel to afford the crude product. The crude solid product was diluted with EtOAc (100 mL), washed with water (3 × 50 mL), and brine solution (2 × 50 mL). The organic layer was dried over Na2SO4, filtered, and evaporated to dryness in vacuo. The product was precipitated by adding cold water and then collected in a sintered glass filter and dried under vacuum overnight to afford the title compound as a yellow solid (90% yield). TLC [Hexanes:EtOAc (7:3), Rf: 0.3]; 1H NMR (300 MHz, DMSO-d6) δ 2.13 (d, J=4.12 Hz, 3 H) 2.39 (d, J=3.85 Hz, 3 H) 4.87 (d, J=4.12 Hz, 2 H) 7.05 (d, J=3.30 Hz, 1 H) 7.88 (td, J=7.97, 4.12 Hz, 1 H) 8.42 - 8.57 (m, 2 H) 8.69 - 8.80 (m, 1 H) ppm; ESI-MS calcd for C16H13N3O3S [M + H]: 328.1; found 328.7.
2-[2-hydroxy-2-(3-nitrophenyl)ethyl]sulfanyl-4,6-dimethyl-pyridine-3-carbonitrile (1).
To 5 (1.2 g, 3.8 mmol) and sodium borohydride (0.14 g, 3.8 mmol) was added anhydrous THF/MeOH (1:1, 24 mL) via syringe under nitrogen atmosphere. Reaction mixture was stirred vigorously at RT overnight. Upon completion, the solvent was evaporated in vacuo, residue was diluted with DCM (150 mL) and extracted with Brine solution (3 × 25 mL). The organic layer was dried over Na2SO4, filtered, and evaporated to dryness in vacuo. The crude product was then purified by flash chromatography (20% EtOAc/Hexanes) to afford the title compound (90%). 1H NMR (300 MHz, DMSO-d6) δ 2.37 (br. s., 3 H) 2.46 (br. s., 3 H) 3.39 - 3.55 (m, 1 H) 3.61 - 3.77 (m, 1 H) 5.01 (br. s., 1 H) 6.09 (br. s., 1 H) 7.06 (br. s., 1 H) 7.61 (t, J=7.83 Hz, 1 H) 7.87 (d, J=6.87 Hz, 1 H) 8.08 (d, J=7.97 Hz, 1 H) 8.28 (br. s., 1 H) ppm; ESI-MS calcd for C16H15N3O3S [M + H]: 330.1; found 330.7.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jim Kiefer for his suggestions on the manuscript.
Funding Sources
The work was funded in part by the NIH grant 5R01GM052504.
Footnotes
Supporting Information. Experimental details on the synthesis, purification and structural characterization of intermediates and all final compounds are provided. Also provided are representative inhibition curves for XPA67-80, compound 1 and compound 11.
REFERENCES
- 1.Martin LP; Hamilton TC; Schilder RJ, Platinum resistance: The role of DNA repair pathways. Clinical Cancer Research 2008, 14 (5), 1291–1295. [DOI] [PubMed] [Google Scholar]
- 2.(a) Groelly FJ; Fawkes M; Dagg RA; Blackford AN; Tarsounas M Targeting DNA damage response pathways in cancer. Nat Rev Cancer 2023, 23 (2), 78–94. DOI: 10.1038/s41568-022-00535-5; [DOI] [PubMed] [Google Scholar]; (b) McPherson KS; Korzhnev DM Targeting protein-protein interactions in the DNA damage response pathways for cancer chemotherapy. RSC Chem Biol 2021, 2 (4), 1167–1195. DOI: 10.1039/d1cb00101a; (c) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Wang Y; Duan M; Peng Z; Fan R; He Y; Zhang H; Xiong W; Jiang W Advances of DNA Damage Repair-Related Drugs and Combination With Immunotherapy in Tumor Treatment. Front Immunol 2022, 13, 854730. DOI: 10.3389/fimmu.2022.854730; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Powell SN; Bindra RS, Targeting the DNA damage response for cancer therapy. DNA Repair (Amst) 2009, 8 (9), 1153–65; [DOI] [PubMed] [Google Scholar]; (c) Jiang M; Jia K; Wang L; Li W; Chen B; Liu Y; Wang H; Zhao S; He Y; Zhou C Alterations of DNA damage response pathway: Biomarker and therapeutic strategy for cancer immunotherapy. Acta Pharm Sin B 2021, 11 (10), 2983–2994. DOI: 10.1016/j.apsb.2021.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Houtsmuller AB; Rademakers S; Nigg AL; Hoogstraten D; Hoeijmakers JH; Vermeulen W, Action of DNA repair endonuclease ERCC1/XPF in living cells. Science 1999, 284 (5416), 958–61; [DOI] [PubMed] [Google Scholar]; (b) Scharer OD ERCC1-XPF endonuclease-positioned to cut. EMBO J 2017, 36 (14), 1993–1995. DOI: 10.15252/embj.201797489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Borszekova Pulzova L; Ward TA; Chovanec M XPA: DNA Repair Protein of Significant Clinical Importance. Int J Mol Sci 2020, 21 (6); [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Krasikova YS; Lavrik OI; Rechkunova NI The XPA Protein-Life under Precise Control. Cells 2022, 11 (23). DOI: 10.3390/cells11233723; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Park CJ; Choi BS, The protein shuffle - Sequential interactions among components of the human nucleotide excision repair pathway. Febs Journal 2006, 273 (8), 1600–1608; [DOI] [PubMed] [Google Scholar]; (d) Li L; Elledge SJ; Peterson CA; Bales ES; Legerski RJ, SPECIFIC ASSOCIATION BETWEEN THE HUMAN DNA-REPAIR PROTEINS XPA AND ERCC1. Proceedings of the National Academy of Sciences of the United States of America 1994, 91 (11), 5012–5016; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sugitani N; Sivley RM; Perry KE; Capra JA; Chazin WJ XPA: A key scaffold for human nucleotide excision repair. DNA Repair (Amst) 2016, 44, 123–135. DOI: 10.1016/j.dnarep.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Li L; Peterson CA; Lu X; Legerski RJ, Mutations in XPA that prevent association with ERCC1 are defective in nucleotide excision repair. Mol Cell Biol 1995, 15 (4), 1993–8; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Orelli B; McClendon TB; Tsodikov OV; Ellenberger T; Niedernhofer LJ; Scharer OD, The XPA-binding domain of ERCC1 Is Required for Nucleotide Excision Repair but Not Other DNA Repair Pathways. Journal of Biological Chemistry 2010, 285 (6), 3705–3712; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) van Steeg H; Mullenders LH; Vijg J, Mutagenesis and carcinogenesis in nucleotide excision repair-deficient XPA knock out mice. Mutat Res 2000, 450 (1-2), 167–80. [DOI] [PubMed] [Google Scholar]
- 7.Jamieson ER; Lippard SJ, Structure, recognition, and processing of cisplatin-DNA adducts. Chemical Reviews 1999, 99 (9), 2467–2498. [DOI] [PubMed] [Google Scholar]
- 8.McCabe KM; Olson SB; Moses RE, DNA Interstrand Crosslink Repair in Mammalian Cells. Journal of Cellular Physiology 2009, 220 (3), 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Arora S; Kothandapani A; Tillison K; Kalman-Maltese V; Patrick SM, Downregulation of XPF-ERCC1 enhances cisplatin efficacy in cancer cells. DNA Repair (Amst) 2010; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cummings M; Higginbottom K; McGurk CJ; Wong OG; Koberle B; Oliver RT; Masters JR, XPA versus ERCC1 as chemosensitising agents to cisplatin and mitomycin C in prostate cancer cells: role of ERCC1 in homologous recombination repair. Biochem Pharmacol 2006, 72 (2), 166–75; [DOI] [PubMed] [Google Scholar]; (c) Chen SF; Zhang JZ; Wang R; Luo XY; Chen HQ, The platinum-based treatments for advanced non-small cell lung cancer, is low/negative ERCC1 expression better than high/positive ERCC1 expression? A meta-analysis. Lung Cancer 2010, 70 (1), 63–70; [DOI] [PubMed] [Google Scholar]; (d) Postel-Vinay S; Soria JC ERCC1 as Predictor of Platinum Benefit in Non-Small-Cell Lung Cancer. J Clin Oncol 2017, 35 (4), 384–386. DOI: 10.1200/JCO.2016.70.5053; [DOI] [PubMed] [Google Scholar]; (e) MA ELB; El Kashef WF ERCC1 Expression in Metastatic Triple Negative Breast Cancer Patients Treated with Platinum-Based Chemotherapy. Asian Pac J Cancer Prev 2017, 18 (2), 507–513. DOI: 10.22034/APJCP.2017.18.2.507; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Cierna Z; Miskovska V; Roska J; Jurkovicova D; Pulzova LB; Sestakova Z; Hurbanova L; Machalekova K; Chovanec M; Rejlekova K; et al. Increased levels of XPA might be the basis of cisplatin resistance in germ cell tumours. Bmc Cancer 2020, 20 (1). DOI: 10.1186/s12885-019-6496-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Rabik CA; Dolan ME, Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treatment Reviews 2007, 33 (1), 9–23; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Duan MR; Ulibarri J; Liu KJ; Mao P Role of Nucleotide Excision Repair in Cisplatin Resistance. International Journal of Molecular Sciences 2020, 21 (23). DOI: 10.3390/ijms21239248; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Suzuki T; Sirimangkalakitti N; Baba A; Toyoshima-Nagasaki R; Enomoto Y; Saito N; Ogasawara Y Characterization of the nucleotide excision repair pathway and evaluation of compounds for overcoming the cisplatin resistance of non-small cell lung cancer cell lines. Oncology Reports 2022, 47 (4). DOI: 10.3892/or.2022.8281; [DOI] [PubMed] [Google Scholar]; (d) Galluzzi L; Senovilla L; Vitale I; Michels J; Martins I; Kepp O; Castedo M; Kroemer G Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31 (15), 1869–1883. DOI: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 11.(a) Manguinhas R; Serra PA; Soares RB; Rosell R; Gil N; Oliveira NG; Guedes RC Unveiling Novel ERCC1-XPF Complex Inhibitors: Bridging the Gap from In Silico Exploration to Experimental Design. Int J Mol Sci 2024, 25 (2). DOI: 10.3390/ijms25021246; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huang MY; Huang YJ; Cheng TL; Jhang WY; Ke CC; Chen YT; Kuo SH; Lin IL; Huang YH; Chuang CH XPF-ERCC1 Blocker Improves the Therapeutic Efficacy of 5-FU- and Oxaliplatin-Based Chemoradiotherapy in Colorectal Cancer. Cells 2023, 12 (11). DOI: 10.3390/cells12111475; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) McNeil EM; Melton DW, DNA repair endonuclease ERCC1-XPF as a novel therapeutic target to overcome chemoresistance in cancer therapy. Nucleic Acids Res 2012, 40 (20), 9990–10004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Elmenoufy AH; Gentile F; Jay D; Karimi-Busheri F; Yang X; Soueidan OM; Weilbeer C; Mani RS; Barakat KH; Tuszynski JA; et al. Targeting DNA Repair in Tumor Cells via Inhibition of ERCC1-XPF. J Med Chem 2019, 62 (17), 7684–7696. [DOI] [PubMed] [Google Scholar]; (e) Weilbeer C; Jay D; Donnelly JC; Gentile F; Karimi-Busheri F; Yang X; Mani RS; Yu Y; Elmenoufy AH; Barakat KH; et al. Modulation of ERCC1-XPF Heterodimerization Inhibition via Structural Modification of Small Molecule Inhibitor Side-Chains. Front Oncol 2022, 12, 819172; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Elmenoufy AH; Gentile F; Jay D; Karimi-Busheri F; Yang X; Soueidan OM; Mani RS; Ciniero G; Tuszynski JA; Weinfeld M; et al. Design, synthesis and in vitro cell-free/cell-based biological evaluations of novel ERCC1-XPF inhibitors targeting DNA repair pathway. Eur J Med Chem 2020, 204, 112658. DOI: 10.1016/j.ejmech.2020.112658. [DOI] [PubMed] [Google Scholar]
- 12.Tsodikov OV; Ivanov D; Orelli B; Staresincic L; Shoshani I; Oberman R; Scharer OD; Wagner G; Ellenberger T, Structural basis for the recruitment of ERCC1-XPF to nucleotide excision repair complexes by XPA. Embo Journal 2007, 26 (22), 4768–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Zhong SJ; Macias AT; MacKerell AD, Computational identification of inhibitors of protein-protein interactions. Curr Top Med Chem 2007, 7 (1), 63–82; [DOI] [PubMed] [Google Scholar]; (b) Walter P; Metzger J; Thiel C; Helms V, Predicting where small molecules bind at protein-protein interfaces. PLoS One 2013, 8 (3), e58583; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen J; Ma X; Yuan Y; Pei J; Lai L, Protein-protein interface analysis and hot spots identification for chemical ligand design. Curr Pharm Des 2014, 20 (8), 1192–200. [DOI] [PubMed] [Google Scholar]
- 14.(a) Wells JA; McClendon CL, Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450 (7172), 1001–1009; [DOI] [PubMed] [Google Scholar]; (b) Berg T, Small-molecule inhibitors of protein-protein interactions. Current Opinion in Drug Discovery & Development 2008, 11 (5), 666–674; [PubMed] [Google Scholar]; (c) Domling A, Small molecular weight protein-protein interaction antagonists - an insurmountable challenge? Current Opinion in Chemical Biology 2008, 12 (3), 281–291; [DOI] [PubMed] [Google Scholar]; (d) Fry DC, Drug-like inhibitors of protein-protein interactions: A structural examination of effective protein mimicry. Current Protein & Peptide Science 2008, 9 (3), 240–247. [DOI] [PubMed] [Google Scholar]
- 15.Ivanov AA; Khuri FR; Fu H, Targeting protein-protein interactions as an anticancer strategy. Trends Pharmacol Sci 2013, 34 (7), 393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nero TL; Morton CJ; Holien JK; Wielens J; Parker MW, Oncogenic protein interfaces: small molecules, big challenges. Nat Rev Cancer 2014, 14 (4), 248–62. [DOI] [PubMed] [Google Scholar]
- 17.Tsodikov OV; Enzlin JH; Scharer OD; Ellenberger T, Crystal structure and DNA binding functions of ERCC1, a subunit of the DNA structure-specific endonuclease XPF-ERCC1. Proceedings of the National Academy of Sciences of the United States of America 2005, 102 (32), 11236–11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Croteau DL; Peng Y; Van Houten B, DNA repair gets physical: mapping an XPA-binding site on ERCC1. DNA Repair (Amst) 2008, 7 (5), 819–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fadda E., Conformational determinants for the recruitment of ERCC1 by XPA in the nucleotide excision repair (NER) Pathway: structure and dynamics of the XPA binding motif. Biophys J 2013, 104 (11), 2503–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Barakat KH; Huzil JT; Luchko T; Jordheim L; Dumontet C; Tuszynski J, Characterization of an inhibitory dynamic pharmacophore for the ERCC1-XPA interaction using a combined molecular dynamics and virtual screening approach. Journal of Molecular Graphics & Modelling 2009, 28 (2), 113–139; [DOI] [PubMed] [Google Scholar]; (b) Neher TM; Shuck SC; Liu JY; Zhang JT; Turchi JJ, Identification of novel small molecule inhibitors of the XPA protein using in silico based screening. ACS Chem Biol 2010, 5 (10), 953–65; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Barakat KH; Jordheim LP; Perez-Pineiro R; Wishart D; Dumontet C; Tuszynski JA Virtual screening and biological evaluation of inhibitors targeting the XPA-ERCC1 interaction. PLoS One 2012, 7 (12), e51329; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gavande NS; VanderVere-Carozza P; Mishra AK; Vernon TL; Pawelczak KS; Turchi JJ Design and Structure-Guided Development of Novel Inhibitors of the Xeroderma Pigmentosum Group A (XPA) Protein-DNA Interaction. J Med Chem 2017, 60 (19), 8055–8070. DOI: 10.1021/acs.jmedchem.7b00780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tripsianes K; Folkers GE; Zheng C; Das D; Grinstead JS; Kaptein R; Boelens R, Analysis of the XPA and ssDNA-binding surfaces on the central domain of human ERCC1 reveals evidence for subfunctionalization. Nucleic Acids Res 2007, 35 (17), 5789–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones G; Willett P; Glen RC; Leach AR; Taylor R, Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol 1997, 267, 727–748. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.