

Summary
The use of Bruton tyrosine kinase (BTK) inhibitors such as ibrutinib achieves a remarkable clinical response in mantle cell lymphoma (MCL). Acquired drug resistance, however, is significant and affects long-term survival of MCL patients. Here, we demonstrate that DNA methyltransferase 3A (DNMT3A) is involved in ibrutinib resistance. We find that DNMT3A expression is upregulated upon ibrutinib treatment in ibrutinib-resistant MCL cells. Genetic and pharmacological analyses reveal that DNMT3A mediates ibrutinib resistance independent of its DNA-methylation function. Mechanistically, DNMT3A induces the expression of MYC target genes through interaction with the transcription factors MEF2B and MYC, thus mediating metabolic reprogramming to oxidative phosphorylation (OXPHOS). Targeting DNMT3A with low-dose decitabine inhibits the growth of ibrutinib-resistant lymphoma cells both in vitro and in a patient-derived xenograft mouse model. These findings suggest that targeting DNMT3A-mediated metabolic reprogramming to OXPHOS with decitabine provides a potential therapeutic strategy to overcome ibrutinib resistance in relapsed/refractory MCL.
Keywords: DNMT3A, gene regulation, OXPHOS, ibrutinib resistance, mantle cell lymphoma
Graphical abstract
Highlights
-
•
DNMT3A expression is required for the growth of MCL cells
-
•
DNMT3A mediates ibrutinib resistance in MCL independently of DNA methylation
-
•
DNMT3A interacts with MEF2B and MYC to mediate OXPHOS independent of DNA methylation
-
•
Targeting DNMT3A with low-dose decitabine overcomes ibrutinib resistance
Hoang et al. shows that DNA methyltransferase 3A (DNMT3A) regulates cellular energy production in mitochondria and contributes to the resistance of mantle cell lymphoma to ibrutinib, an inhibitor of Bruton tyrosine kinase. Targeting DNMT3A with low-dose decitabine is a potential therapeutic strategy to overcome ibrutinib resistance.
Introduction
Mantle cell lymphoma (MCL) is an incurable, rare type of B cell non-Hodgkin lymphoma, accounting for 5%–6% of all lymphoma cases.1,2 A hallmark of MCL is the t(11; 14)(q13; q32) translocation, juxtaposing the cyclin D 1 proto-oncogene (CCND1) to the immunoglobulin heavy-chain gene (IgH), leading to CCND1 constitutive expression at the pre-B cell stage.2 Genomic translocations between CCND2/3 and IgH are also observed but not common.3,4 These genomic alterations, however, are not sufficient to drive tumorigenesis. Additional alterations at the genomic and transcriptional levels during B cell development are required to give rise to MCL.
Constitutively activated B cell receptor (BCR) signaling is required for the survival of MCL cells as demonstrated by the clinical use of selective inhibitors of BCR such as ibrutinib or acalabrutinib.5,6,7 A BCR gene signature can predict the outcomes of MCL patients, although the mechanism for aberrant BCR activation remains unclear.8,9 Ibrutinib covalently binds to the active site at cysteine 481 (C481) of the Bruton tyrosine kinase (BTK), a key component of the early BCR signaling pathway.5 Ibrutinib has been approved as the second line of treatment for relapsed or refractory MCL, with an overall response rate of 68%.5 Unfortunately, almost all patients relapse, with a 1-year survival rate of only 22%.10 The C481-to-S mutation in BTK associated with acquired ibrutinib resistance, which has been frequently identified after treatment of chronic lymphocytic leukemia, is only rarely observed after treatment in MCL.11,12,13 A recent study has identified oxidative phosphorylation (OXPHOS) as a targetable mechanism of ibrutinib resistance in MCL,13 suggesting that nongenetic mechanisms should be explored further to identify therapeutic vulnerabilities to achieve deep and durable responses to BTK inhibitors (BTKis), including ibrutinib.
In addition to the BCR gene signature, methylation signatures cluster MCL patients into two main subtypes with distinct clinical outcomes, with a higher methylation level having worse survival.14 However, the role of DNA methyltransferases (DNMTs) in MCL is less studied. The oncogenic role of inactivating mutations of DNMT3A has been extensively studied in the etiology of acute myeloid leukemia (AML)15,16,17,18,19 and T cell lymphoma,20,21 but DNMT3A is not mutated in MCL.22,23,24 A recent study demonstrated that the expression of DNMT3A is higher in MCL patients who are resistant to ibrutinib compared to patients who are sensitive to ibrutinib,13 but the role of DNMT3A in ibrutinib resistance remains unclear.
Here, we demonstrate a methylation-independent function of DNMT3A as a transcriptional activator. Using MCL as a disease model, we show that DNMT3A is required for MCL cell growth in part by regulating mitochondrial biogenesis. Mechanistically, DNMT3A interacts with myocyte enhancer factor 2B (MEF2B), a transactivator of MYC, to activate the transcription of MYC target genes, including those that govern mitochondrial biogenesis. Targeting DNMT3A with a low dose of decitabine, a pan inhibitor of all DNMTs, significantly reduces OXPHOS and tumor burden as well as prolongs survival in preclinical mouse models. The combination of IM156, a complex I inhibitor, and decitabine induces synergistic lethality in vitro, and this combination is selective for B cell lymphoma cells. In addition, we show that DNMT3A-mediated OXPHOS contributes to ibrutinib resistance in MCL and that decitabine enhances the efficacy of ibrutinib in patient-derived xenografts (PDXs).
Results
DNMT3A expression is required for MCL cell growth in vitro and in vivo without affecting cell cycle or apoptosis
We first tested whether DNMT3A is required for the growth of MCL cell lines in vitro. To knock out DNMT3A, we used an inducible CRISPR-Cas9 system in which Cas9 was constitutively expressed and single-guide RNA (sgRNA) was induced by doxycycline (20 ng/mL). This concentration of doxycycline was not toxic for lymphoma cells, and uninduced cells served as controls as described in our recent studies.25,26
Two distinct sgRNAs were designed to target the second exon and the MTase domain of DNMT3A to knock out both DNMT3A1 and DNMT3A2 isoforms efficiently (Figure 1A). The global methylation was reduced by 20%–50% after knock out, indicating that DNMT3A is important in establishing de novo methylation in MCL cells (Figures 1B and S1A). After the induction of sgDNMT3A, cells from cultures were counted by trypan blue exclusion assay every 3 days, for up to 7 days. The total number of viable cells was calculated relative to day 0. The growth rate of DNMT3A knockout (KO) cells was slower than that of control cells (Figure S1B). After 1 week, all of the DNMT3A KO MCL clonal cell lines exhibited a significant reduction in growth relative to control cells (Figure 1C). Of note, the reduced growth of DNMT3A KO cells was not due to increased cell apoptosis (Figure S1C) or cell-cycle arrest (Figure S1D).
Figure 1.

DNMT3A expression is required for in vitro and in vivo growth of mantle cell lymphoma
(A) The design of 2 sgRNAs targeting DNMT3A (top) and immunoblots (bottom) showing the reduction of DNMT3A expression in 7 distinct clones of MCL cell lines after 6 days of doxycycline treatment (20 ng/mL).
(B) The global methylation was reduced after DNMT3A KO using methylation dot blots. The global methylation was reduced after treating Granta-519 cells with decitabine (1 μM, 48 h), and this served as a positive control for this assay (top). A representative image of reduced global methylation in Rec-1- sgDNMT3A clone no. 6 (Rec-1 no. 6) after doxycycline-induced DNMT3A KO 6 days posttreatment (bottom).
(C) DNMT3A KO inhibited the in vitro growth of MCL cell lines. Error bars represent mean ± SD (Student’s t test).
(D) Z-138 no. 7 (left) and Granta-519 no. 7 (right) were established as subcutaneous tumors in NSG mice. Error bars represent mean ± SEM (2-way ANOVA). All in vitro experiments were performed independently at least 3 times with 3–4 technical replicates.
To test whether DNMT3A is required for MCL cell growth in vivo, two MCL clonal cell lines Z-138 clone 7 (Z-138 no. 7) and Granta-519 clone 7 (Granta-519 no. 7), were subcutaneously injected into immunocompromised NOD scid γc−/− (NSG) mice. Once a tumor reached a palpable size, mice received normal drinking water or water with doxycycline (2 mg/mL) to knock out DNMT3A. The growth rate of tumors deficient in DNMT3A was significantly slower than that in control mice (Figure 1D). The differential growth is likely due to a reduced proliferative capacity of DNMT3A KO cells rather than engraftment potential because doxycycline was given to mice only when tumors had been successfully engrafted. Taken together, these observations suggest that the expression of DNMT3A is required for the growth of MCL cells and that DNMT3A is a potential oncogene that provides a proliferative advantage to MCL cells through a mechanism rather than by promoting the cell cycle or protecting cells from apoptosis.
DNMT3A modulates OXPHOS by controlling mitochondrial biogenesis independently of its methyltransferase activity
To understand the underlying mechanism of DNMT3A-mediated MCL growth, we performed RNA sequencing (RNA-seq) of Z-138 no. 7 cell line. Gene set enrichment analysis (GSEA) revealed that downregulated genes were enriched in OXPHOS, mitochondrial membrane, and target genes of MYC in DNMT3A KO cells as compared to DNMT3A wild-type (WT) cells (Figure 2A). We used functional assays to evaluate mitochondrial activity, mitochondrial membrane potential (MMP), and biogenesis to test whether DNMT3A regulates OXPHOS in MCL by affecting mitochondrial biogenesis as MYC transcriptionally regulates a set of genes required for mitochondrial biogenesis.27,28,29,30,31
Figure 2.

DNMT3A expression is required for OXPHOS in MCL cells by controlling mitochondrial biogenesis independent of its methyltransferase activity
(A) GSEA of DNMT3A KO cells shows downregulation of genes involved in OXPHOS, mitochondrial biogenesis, and MYC transcriptional pathway compared to control cells.
(B) The reduced rate of OCR (pmol/min/cell) and ATP productions in DNMT3A KO cells relative to control cells. All of the OCR values were normalized to total live-cell number. OCR experiments were performed independently at least 2 times, each with 6 technical replicates.
(C) Reduced MMP in DNMT3A KO cells compared to control cells by flow cytometric analysis of JC-1 dye.
(D) Representative images of TEM of Z-138 no. 7 control cells compared to DNMT3A KO cells (top). A significant decrease in mitochondrial mass, approximated by the relative area of mitochondria to cytosol per cell, of DNMT3A KO cells relative to control cells (bottom). Each data point represents an individual cell (n = 60–61 cells). TEM experiments were performed independently twice, each with duplicates.
(E) DNMT3AV716G expression was sufficient to increase MMP in Rec-1 and JeKo-1 cells and the quantification of MMP in Rec-1 cells. Error bars represent mean ± SD (Student’s t test, 2 independent experiments in triplicate).
A mitochondrial stress test showed that the oxygen consumption rate (OCR) and ATP production of DNMT3A KO cells were reduced relative to control cells, indicating that OXPHOS is impaired in DNMT3A-deficient cells (Figure 2B). We next assessed the integrity of the mitochondria by measuring MMP with the JC-1 dye, a cationic dye that aggregates and emits red fluorescence when there is a high concentration of electrons within intact mitochondria (aggregates) or shifts to green fluorescence when the concentration of electrons decreases (monomers). The ratio of aggregates to monomers is an indicator of MMP. The mitochondria of DNMT3A KO cells exhibited reduced MMP compared to that of control cells (Figure 2C). The by-product of mitochondrial activity, mitochondrial reactive oxygen species, approximated by total H2O2, was also decreased in DNMT3A KO cells (Figure S2A). These results indicate that DNMT3A is required for mitochondrial integrity and function.
To test whether DNMT3A affects mitochondrial function by regulating mitochondrial biogenesis, we used transmission electron microscopy (TEM) to capture single-cell images. The mitochondrial mass of each cell was approximated by the ratio of area of mitochondria to that of the cytosol.32,33 The mitochondrial mass was significantly reduced in DNMT3A KO cells compared to control cells (Figure 2D). Flow cytometric analysis using nonyl acridine orange dye, an indicator of mitochondrial mass because it is independent of MMP, was used as an alternative approach to confirm this finding (Figure S2B). The data indicate that DNMT3A is necessary for mitochondrial activity in part by controlling mitochondrial biogenesis.
We next determined whether DNMT3A is sufficient to increase mitochondrial function by overexpressing the cDNA of WT DNMT3A (DNMT3AWT) or a methylation-null mutant DNMT3A (DNMT3AV716G) in MCL cell lines JeKo-1 and Rec-1. Both cell lines express DNMT3AWT. A structural study demonstrated that the DNMT3AV716G mutant is unable to bind the cytosine for base flipping, thus abrogating both CpGs and non-CpG methylation.34 We functionally confirmed this mutant by treating cells with decitabine. Decitabine is an analog of cytosine and forms a covalent bond with DNMTs, leading to their degradation, likely mediated by TRAF6.35 We reasoned that if DNMT3AV716G is null for methylation due to its inability to bind cytosine, DNMT3AV716G should not be degraded by decitabine treatment. Indeed, we observed a significant reduction in DNMT3AWT expression after decitabine treatment but no change in the level of DNMT3AV716G expression relative to DMSO-treated cells (Figure S2C, left). Concordantly, the overexpression of DNMT3AV716G desensitized cells to decitabine to a greater extent than that of DNMT3AWT overexpression (Figure S2C, right). We further confirmed the methylation defect of DNMT3AV716G variant by overexpressing these variants in HEK293T cells. We observed a significant increase in methylation upon the forced expression of DNMT3AWT, but reduced methylation with DNMT3AV716G overexpression by methylation dot blot (Figure S2D). Of note, we were unable to observe any changes in the global methylation of transduced MCL lines, likely due to a higher endogenous level of methylation in the parental cells (Figure S2E). In addition, DNMT3AV716G is unlikely to be a dominant-negative mutant because it was unable to reduce global methylation after transduction in MCL cell lines expressing DNMT3AWT.
Cells that were transduced with DNMT3AWT or DNMT3AV716G showed an increase in OCR compared to cells transduced with an empty vector control (Figure S2E). Because the mammalian expression vector contains GFP that cannot be distinguished with the green fluorescence of monomer in JC-1 assay, we used MitoTracker deep red as an alternative to approximate the MMP of these cells because this dye uses MMP to stain the mitochondria. The MMP of DNMT3AV716G cells was significantly higher than that of empty vector control cells (Figure 2E). Of note, DNMT3AWT expression increased MMP in JeKo-1 cells but not in Rec-1 cells. This may be due to a more saturated expression of endogenous DNMT3A in Rec-1 cells than that in JeKo-1 cells. Together, these data indicate that DNMT3A is sufficient to enhance the mitochondrial activities in a methylation-independent manner.
DNMT3A forms a transcriptional activating complex with MEF2B and MYC to regulate genes involved in mitochondrial biogenesis in MCL
Our data challenge the existing paradigm that DNMT3A acts as a transcriptional repressor. We tested the hypothesis that DNMT3A interacts with other transcription factors to promote the transcription of specific genes. We performed high-throughput sequencing of chromatin immunoprecipitation (ChIP-seq) to search for the footprints of potential factors binding with or in the proximity of DNMT3A. To determine the chromatin-binding profile of DNMT3A in MCL, we used the Z-138 cell line that is WT for DNMT3A and Z-138 DNMT3A KO cells as a negative control. A total of 477 significant binding peaks were identified (Figure 3A). DNMT3A binding sites were primarily enriched at promoter (37.3%) and intergenic (31.35%) regions, followed by intronic regions (22.29%) (Figure 3B).
Figure 3.

DNMT3A, MEF2B, and MYC form a transcriptional activating complex to regulate genes involved in mitochondrial biogenesis in MCL
(A) ChIP-seq of DNMT3A in Z-138 cells. Heatmaps show 477 DNMT3A peaks using Z-138 DNMT3A KO no. 7 cells as a ChIP control after 6 days of sgRNA induction.
(B) Genomic features of DNMT3A binding sites.
(C) Genomic occupancy analysis demonstrated that DNMT3A binds in proximity of MEF2B binding sites.
(D) Endogenous DNMT3A coprecipitated with MEF2B in the Z-138 cell line and in 2 primary MCL samples, MCL-4 and MCL-7.
(E) Biochemical characterization of the interaction between DNMT3A and MEF2B. Immunoprecipitation of FLAG-tagged MEF2B cotransfected with myc-tagged DNMT3AWT or DNMT3AV716G that lacks cytosine base-flipping, or DNMT3AD331A, defective in H3K36me2 recognition.
(F) Endogenous MYC coprecipitated with DNMT3A in the Z-138 cell line.
(G) MEF2B is a transcriptional activator of tfam based on the luciferase reporter assay in HEK293T cells (left). MYC alone decreased the transcription of tfam (center), but MEF2B enhanced MYC transcription of tfam (right). ΔMADSMEF2B failed to increase the transcription of tfam. All of the conditions were cotransfected with β-galactosidase as an internal control.
(H) DNMT3AWT and DNMT3AV716G enhanced MYC and MEF2B-mediated transcriptional activation of tfam despite lacking the MADS domain of MEF2B (first 2). ΔMADSMEF2B variant failed to enhance transcriptional activation of tfam by MYC in the presence of DNMT1 or DNMT3B (last 2). Error bars represent mean ± SD (Student’s t test).
DNMT3A is known to bind other cofactors and transcriptional factors at distal intergenic regions for transcriptional activation.36,37 Given the enrichment in downregulated genes after knocking out DNMT3A, we hypothesized that DNMT3A binds with other factors at enhancer regions to facilitate transcriptional activation. After performing de novo motif analyses, we found significant enrichments of many transcription factors. Among them, MEF2B stood out to us because MEF2B is critical to the development of the germinal center and has been found to be mutated in both indolent and aggressive lymphomas (Figure 3C).38,39,40 The footprint of MEF2B around DNMT3A binding sites indicated two working models: DNMT3A and MEF2B bind in proximity or they physically interact irrespective of DNA context. To delineate these possibilities, DNase was added during all of the coimmunoprecipitation (coIP) experiments to fragment DNA (Figure S3A). We showed that DNMT3A and MEF2B interact despite the fragmentation of DNA in both MCL cell lines and primary cells (Figure 3D). This result favors the latter model that the interaction between DNMT3A and MEF2B is likely to be direct.
We further examined whether methylation and histone-binding activities of DNMT3A are required for this interaction. We took advantage of the DNMT3AV716G variant that is unable to bind cytosine and generated DNMT3AD333A variant that is unable to recognize histone H3 lysine 36 dimethylation (H3K36me2).41 We performed coIP in HEK293T cells in which FLAG-tagged MEF2B and myc-tagged DNMT3AWT/V716G/D333A were cotransfected. The interaction of MEF2B and DNMT3A was preserved in all of the conditions (Figure 3E), indicating that the interaction does not depend on DNA methylation or H3K36me2 binding of DNMT3A.
We tested whether MEF2B selectively interacts with DNMT3A or with other members of the DNMTs as well. We used anti-FLAG antibody to pull down FLAG-tagged MEF2B in HEK293T cells that was cotransfected with FLAG-tagged MEF2B and myc-tagged DNMT1, DNMT3B, or DNMT3A.The interaction between DNMT3A and MEF2B was stronger than those between MEF2B and DNMT1 or MEF2B and DNMT3B (Figure S3B). FLAG pulldown did not immunoprecipitate the endogenous MYC in HEK293T cells (Figure S3B). It is therefore likely that the interaction of DNMTs and MEF2B was direct rather than mediated by MYC.
MEF2B is known to enhance MYC transcriptional activities.42 Given that DNMT3A KO downregulates target genes of MYC and genes involving in mitochondrial membrane (Figure 2A), and that MYC regulates genes required for mitochondrial biogenesis, we postulated that MYC, MEF2B, and DNMT3A form a transcriptional activator complex to regulate genes involved in mitochondrial biogenesis in MCL cells. The immunoprecipitation of endogenous MYC revealed that MYC and DNMT3A interacted in all three MCL cell lines tested (Figures 3F and S3C), consistent with previous reports in other cancers.43,44 Importantly, MYC only interacts with MEF2B, not with other MEF2 family members in lymphoma cell lines (Figure S3D).
To test whether MYC-regulated mitochondrial genes are co-regulated by DNMT3A and MEF2B, we confirmed that the mRNA levels of some of MYC target genes regulating mitochondrial biogenesis were reduced after DNMT3A KO compared to control cells (Figure S3E). To identify putative regulatory regions to which DNMT3A, MEF2B, and MYC cobind, we overlapped multiple ChIP-seq datasets of DNMT3A (Z-138 cells), H3K27ac (cell line: Z-138; GEO: GSE97541), and MEF2B (cell line: GM12878; GEO: GSE127481). We did not overlap these datasets on a genome-wide scale but focused on two mitochondrial MYC target genes that showed overlapping peaks across these ChIP datasets to functionally validate this transcriptional program in the context of MCL (Figures S3F and S3G).
We chose the mitochondrial transcription factor A (tfam) because, as a direct target of MYC,27 tfam is essential for mitochondrial biogenesis and mitochondrial DNA maintenance.45 We cloned the 3.5-kb upstream region of tfam into a luciferase backbone (pGL3-tfam) because this region is cobound by DNMT3A and MEF2B and is marked with H3K27ac. Of note, this region is extensively marked with cis-regulatory elements (Figure S3H; ENCODE). We truncated the MADS (MCM1-agamous-deficiens-serum response factor) domain of MEF2B (residues 8–91; ΔMADSMEF2B). The MADS domain is required for dimerization and DNA binding of the MEF proteins.46 MEF2B harbors 3 hotspot mutations in lymphomas (K4, Y69, and D83: 8%–18% in diffuse large B cell lymphoma,13% in follicular lymphoma, and 3% in MCL), all of which are within the truncated MADS domain.22,47,48,49 The ΔMADSMEF2B variant therefore controls for the transactivation function of MEF2B that is dependent on DNA-binding ability. It is important to note that the C terminus, which is enzymatically responsible for the transactivation activity of MEF2Bs, is intact. As expected, MEF2BWT alone but not ΔMADSMEF2B activated the transcription of pGL3-tfam (Figure 3G, left). Although MYC alone decreased pGL3-tfam transcription, the addition of MEF2BWT but not ΔMADSMEF2B enhanced MYC transcription of pGL3-tfam (Figure 3G, center, right). These results confirmed that MEF2B transactivates MYC to increase the transcription of pGL3-tfam DNA, whereas MYC alone represses this gene.
Both DNMT3AWT and DNMT3AV716G enhanced the expression of pGL3-tfam alone or in the presence of MEF2B and MYC (Figures 3H and S3I). More intriguingly, both DNMT3AWT and DNMT3AV716G supported the ability of ΔMADSMEF2B to transactivate MYC transcription of pGL3-tfam, whereas DNMT3B and DNMT1 could not (Figure 3H). We propose that the biological significance of the interaction between DNMT3A and MEF2B is that it selectively retains the transactivation of MYC by MEF2B even when MEF2B lacks DNA-binding ability. These results collectively favor a model in which DNMT3A selectively forms an activating transcriptional complex with MEF2B and MYC, relative to DNMT1 and DNMT3B, to regulate the expression of genes, including those involved in mitochondrial biogenesis in MCL cells.
Targeting DNMT3A by decitabine reduces OXPHOS and tumor burden in both cell line- and PDXs
We assessed whether inhibiting DNMT3A by a pan-DNMT inhibitor, decitabine, could reduce OXPHOS and tumor growth in vivo. We opted to use decitabine because it is clinically safe and effective in treating several hematological malignancies, including AML.50,51 Decitabine has been shown to induce the degradation of DNMT1, DNMT3A, and DNMT3B by TRAF6 through a lysosome-dependent protein-degradation pathway.35 We validated decitabine-mediated degradation of DNMT1, DNMT3A, and DNMT3B in MCL cells (Figure S4A). Decitabine reduced the growth of MCL cell lines in vitro (Figure S4B). The expression levels of MYC target genes involved in mitochondrial biogenesis were reduced after decitabine treatment (Figure S4C). Importantly, decitabine treatment increased the fraction of cells with low mitochondrial activity, whereas overexpressing DNMT3AWT or DNMT3AV716G reduced the fraction of cells with low mitochondrial activity after decitabine treatment (Figure S4D). Furthermore, decitabine-induced DNMT3A degradation resulted in reduced interaction between MYC and MEF2B (Figure S4E). Taken together, these data indicate that decitabine reduces tumor growth partly by inhibiting mitochondrial activity through the DNMT3A-MEF2B-MYC complex in a DNA-methylation-independent manner.
Given that DNMT3A regulates mitochondrial biogenesis and OXPHOS, we tested whether decitabine reduces OXPHOS in vivo by optical metabolic imaging (OMI) of Z-138 tumors in NSG mice52 (Figures 4A and 4B). Two-photon fluorescence lifetime imaging microscopy (FLIM) of the autofluorescent metabolic coenzymes nicotinamide adenine dinucleotide (phosphate) (NAD(P)H) and flavin adenine dinucleotide (FAD) provides images of cell metabolism at a single-cell level with spatial context conserved. The fluorescence lifetime, the time a molecule remains in its excited state before relaxing to ground state, informs on the protein-binding activity of NAD(P)H and FAD. The fluorescence lifetimes of free versus protein-bound NAD(P)H and FAD are distinct and can be quantified using FLIM. After 6 days of treatment with decitabine, we imaged Z-138 tumor ex vivo and observed a significant reduction in the mean fluorescence lifetime (τm) of FAD in decitabine-treated compared to PBS-treated mice, indicating that FAD is more predominantly in its protein-bound form (<0.1 ns) rather than in its free form (1–2 ns)53,54,55 (Figures 4C and 4D, left). The percentage of bound FAD α1 was higher and free FAD (α2) was correspondingly lower in decitabine-treated mice than in PBS-treated mice (Figure 4D, center, right). In addition, we observed that the optical redox ratio was significantly more oxidized in control compared to decitabine-treated mice, although there were large variations among tested mice (Figure 4E). Overall, decitabine-induced metabolic changes measured by OMI were consistent with the inhibition of OXPHOS because treatment with rotenone and antimycin a, known inhibitors of OXPHOS, elicited similar changes in these parameters in quiescent T cells.56 There was no clear change in NAD(P)H fluorescence lifetimes, suggesting that decitabine had less effect on NAD(P)H enzyme-binding activities.
Figure 4.

Decitabine inhibits OXPHOS and the growth of ibrutinib-resistant MCL cells
(A) A schematic of ex vivo OMI experiments in Z-138 xenografts.
(B) Representative images of the fluorescence intensity of NAD(P)H and FAD in PBS (control, n = 3) and decitabine-treated mice (n = 3). Scale bar, 25 μm.
(C) Representative images of τm of FAD in control and decitabine-treated mice (left) and the quantification of FAD τm (right) at the single-cell level (control = 2,544 cells; decitabine = 1,743 cells). Bars represent mean ± SD. Different colors denote separate imaging days. Scale bar, 25μm.
(D) Single-cell quantifications of the lifetime of bound FAD (τ1) (ps, picosecond), the percentage of bound FAD (α1), the lifetime of free FAD (τ2), and the percentage of free FAD (α2).
(E) Representative of images of the optical redox ratio and the single-cell quantifications of optical redox ratio. Different colors denote separate imaging days. Scale bar, 25 μm. Mann-Whitney U test was used in (C)–(E).
(F) Z138 xenografts, the same as in (A). Red arrows indicate treatment start and end dates. Tumor dimensions were measured with calipers 3 times per week until endpoint, defined as any dimension exceeding 20 mm or mice becoming morbid.
(G) The effect of decitabine in MCL-7 PDXs.
(H) Bioluminescence images of MCL-4 PDX mice (left), growth curves presented as log10 of the average radiance of tumors (ps−1cm−2sr−1), with dashed line indicating that all of the mice are euthanized (center), and the probability of survival (right) are shown. Error bars represent mean ± SEM (2-way ANOVA). sr: steradian or square radian.
We further tested the efficacy of decitabine in vivo by using the Z-138 cell line-derived and two MCL PDXs. The two samples (MCL-4 and MCL-7), together with another two samples (MCL-5 and MCL-9), came from relapsed MCL patients who were heavily treated with chemotherapy drugs and ibrutinib.57 The Z-138 xenograft and the MCL-7 PDX showed a significant reduction in tumor burden (Figures 4F and 4G) but no improvement in survival, which is likely due to the toxicity associated with the dosage of 1 mg/kg decitabine. We reduced the dosage to 0.25 mg/kg and observed both tumor regression and prolonged survival in the MCL-4 PDX model up to 120 days posttumoral injection (Figure 4H). This dose of decitabine in mice can be converted to the human equivalent dose <1 mg/m2/day54, which is much lower than the 20 mg/m2/day that has usually been used in treating hematologic malignancies.58,59,60,61,62,63,64,65
Of note, we performed whole-exome sequencing (WES) on all 4 MCL patient samples (MCL4, MCL-5, MCL-7, and MCL-9) used in this study and found no ibrutinib-resistant related mutations in BTK or PLCG2 or any hotspot mutations of DNMT3A and MEF2B (Table S1). The hotspot mutation in the NSD2 gene (E1099K) was detected in MCL-5 (Table S1). Collectively, the data show that decitabine reduces tumor growth and OXPHOS in vivo, in part due to the degradation of DNMT3A.
Decitabine synergizes with a complex I inhibitor to inhibit the growth of MCL cell lines and primary MCL samples
We next investigated whether the inhibition of both DNMT3A and OXPHOS exerts synergistic lethality in MCL. We used IM156, a metformin derivative.66 IM156 exhibits higher bioavailability than metformin, which therefore requires a lower dose to reach therapeutic efficacy. IM156 has been shown to be safe and effective in patients with refractory advanced solid tumors and preclinical models.67,68 IM156 has moderate cytotoxic effect as a single agent in MCL cells because the half-maximal inhibitory concentration (IC50) of several MCL cell lines are in the micromolar range (Figure S5A). We tested whether DNMT3A is a determinant of sensitivity to IM156 because DNMT3A regulates mitochondrial biogenesis in MCL cells. We first created a reference for sensitivity of IM156 by depleting cellular mitochondria using gamitrinib, a selective mitochondrial HSP90 inhibitor that reduces mitochondrial biogenesis and functions partly in a PINK1/Parkin-dependent mitophagy manner (Figure S5B).69,70,71,72,73 Depleting mitochondria resulted in a 15-fold decrease in sensitivity to IM156 (Figure S5C). Similarly, depleting DNMT3A that resulted in reduced mitochondrial mass decreased the sensitivity to IM156 of MCL cell lines (Figure S5D). Conversely, the forced expression of DNMT3AWT or DNMT3AV716G sensitized cells to IM156, presumably due to the increased dependency of cells on the mitochondria (Figure S5E). Taken together, these data attributes DNA-methylation-independent functions of DNMT3A to the sensitivity of MCL cells to IM156.
We next examined whether the combination of IM156 and decitabine resulted in synergistic toxicity in vitro. Synergistic lethality was achieved across six MCL cell lines tested (Figure 5A). Synergism was quantified by synergistic scores (s-score) calculated using four different models with Synergy Finder+.74 Most of the mean s-scores were above 8, indicating a strong in vitro synergism between decitabine and IM156 in MCL cell lines (Figure S5F). Two primary MCL samples, MCL-4 and MCL-7, demonstrated a dose-dependent sensitivity to decitabine or IM156 (Figure S5G). Low dosages of decitabine and IM156 also induced synergistic lethality in both samples (Figure 5B). Equally important, this combination did not induce synergistic toxicity in normal B cells (Figure 5C). These results indicate that decitabine and IM156 selectively synergize to inhibit MCL cell growth in vitro.
Figure 5.

Decitabine synergizes with a novel complex I inhibitor to inhibit the growth of cell lines and primary MCL cells
(A) Highest single agent synergy scores of decitabine and IM156, an inhibitor of mitochondrial complex I, in MCL cell lines. A 4 × 4 matrix of 2 drugs and all of the possible combinations were designed on a 96-well plate. Synergy scores were calculated using SynergyFinder+.
(B) Decitabine and IM156 synergized to inhibit primary MCL cells.
(C) Decitabine and IM156 did not synergistically kill normal B cells.ns, not significant.
(D) MCL cells were pretreated with gamitrinib (1.25 μM) or solvent control for 4 days; fresh drug was added every 2 days. Drug/solvent control was removed on day 4. Cells were incubated in normal growth media for 24 h before treatment with decitabine, IM156, or the combination of both drugs at indicated concentrations in a 96-well plate. On day 3 of treatment, cell viability was measured using CellTiter Glo. Error bars represent mean ± SD (Student’s t test). All of the experiments were performed independently twice, except for primary normal B cells, each with 3 technical replicates.
To address whether this synergism is dependent on mitochondrial biogenesis, we tested whether decitabine or IM156 alone or the combination of both induces lower toxicity in cells with low mitochondrial mass induced by gamitrinib compared to cells with higher mitochondrial mass. There was a significant reduction in sensitivity to the single drug alone and to the combination treatment in cells with lower mitochondrial mass, indicating that mitochondrial mass is attributed to the synergism between IM156 and decitabine (Figure 5D). In summary, these data suggest that the antitumor response of decitabine and IM156 combination is driven by DNMT3A-regulated mitochondrial biogenesis.
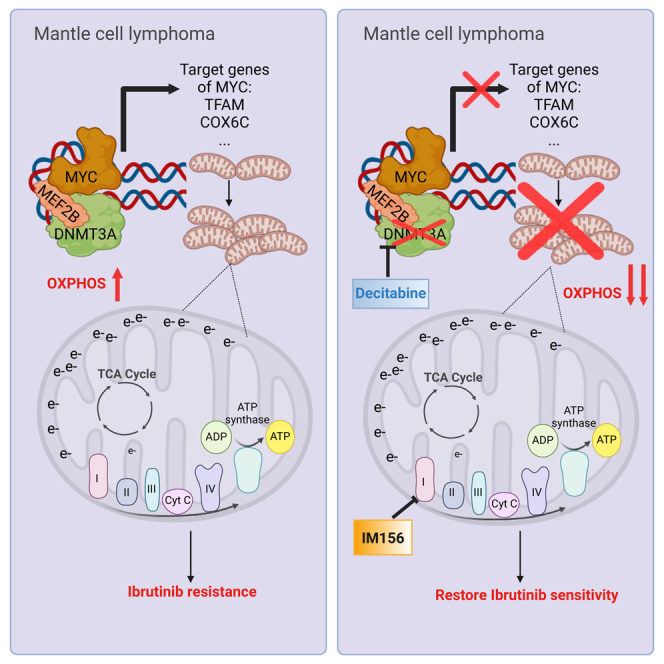
DNMT3A is upregulated after ibrutinib treatment and therefore a therapeutic vulnerability of ibrutinib resistance in MCL
We have established the role of DNMT3A in regulating mitochondrial biogenesis and OXPHOS in MCL. We next tested whether DNMT3A contributes to resistance to ibrutinib in MCL because increased OXPHOS has been identified as a nongenetic driver of ibrutinib-resistance in MCL.13 To test how DNMT3A protein levels change after ibrutinib treatment, we used three primary ibrutinib-resistant MCL samples (MCL-4, MCL-7, and MCL-9) and one control sample (MCL-5) with less ibrutinib resistance, as described in our recent study.75
Primary MCL cells underwent in vivo expansion as described previously.57 For each mouse, spleen and liver were collected at days 30–35 postinjection. Cell identity was confirmed by surface markers (human CD20 and CD19), with tumor purity of both organs being >90% (Figure S6A), although there were variations in the expression of surface markers between organs and among mice inoculated with the same sample. We also observed differential sensitivities to ibrutinib (Figures 6A and S6B). Because of the heterogeneity in the immunophenotypes and sensitivities to ibrutinib, likely due to the clonality of MCL,22,76 we considered cells from each organ as an independent sample.
Figure 6.

DNMT3A is a potential biomarker for and therapeutic vulnerability of ibrutinib resistance
(A) Representative intracellular flow cytometric of DNMT3A. The staining was validated in DNMT3A KO cells (top left). MCL-5 was less ibrutinib resistant (top right). MCL-4, MCL-7, and MCL-9, defined as ibrutinib-resistant samples, showed an increased DNMT3A expression after ibrutinib treatment (2.5 μM, 3 days).
(B) Increased DNMT3A expression was correlated with resistance to ibrutinib.
(C) DNMT3A KO desensitized MCL cell lines to ibrutinib.
(D) The IC50 of ibrutinib in control and DNMT3A KO MCL clones (Student’s t test, ∗∗p < 0.01).
(E) The overexpression of DNMT3AWT or DNMT3AV716G desensitized the ibrutinib-sensitive JeKo-1 cells to ibrutinib. The IC50 of ibrutinib is shown.
(F) Decitabine enhanced the efficacy of ibrutinib in MCL cell lines. Relative viability for each condition, control (uninduced) or DNMT3A KO, were obtained by normalizing the number of live cells in treated conditions to that in the DMSO-treated control. The statistical significance of individual drug concentrations when combined one another is shown. Error bars represent mean ± SD (Student’s t test).
(G) The effect of decitabine/ibrutinib alone or in combination in MCL-7 PDX model. Bioluminescence images (left), the radiance of primary tumors (top right), and mouse body weight of mice before and during treatment as indicated between 2 arrows (bottom right) are shown. Error bars represent mean ± SEM (2-way ANOVA).
Intracellular flow cytometry was used to evaluate the protein level of DNMT3A in live cells (Figure 6A). After 3 days of treatment with ibrutinib at 2.5 μM, there was a significant increase in the level of DNMT3A protein in all 12 individually cultured cells from the spleens and livers of three primary ibrutinib-resistant MCL samples whose viabilities were >90% after treatment (MCL-4, MCL-7, and MCL-9) (Figure 6B). Conversely, DNMT3A expression did not change in the less ibrutinib-resistant MCL-5 sample whose viability was <85% after ibrutinib treatment (Figures 6A and S6B). Importantly, ibrutinib treatment upregulated the protein level of DNMT3A in a dose-dependent manner in two primary MCL samples, two ibrutinib-resistant cell lines, JVM-13 and Z138, but not in two ibrutinib-sensitive cell lines (Figures S6C and S6D). In addition, DNMT3A expression increased in acquired ibrutinib-resistant JeKo-1 and Rec-1 cell lines but not in their respective parental cells (Figure S6E). This result suggests that DNMT3A expression is associated with ibrutinib resistance in MCL.
To test the role of DNMT3A in ibrutinib resistance, we tested the sensitivity to ibrutinib of MCL cells when DNMT3A was knocked out or overexpressed. DNMT3A KO cells were more sensitive to ibrutinib than control cells (Figure 6C) because multiple DNMT3A KO clonal cell lines showed a significantly lower IC50 of ibrutinib than control cell lines (Figure 6D). Overexpressing DNMT3AWT or DNMT3AV617G conferred resistance to ibrutinib with a 4-fold increase in IC50 (Figure 6E). This observation is consistent with our finding that DNA methylation is not necessary for DNMT3A-mediated mitochondrial biogenesis and OXPHOS. Overall, these results establish that DNMT3A contributes to ibrutinib resistance by regulating mitochondrial biogenesis and OXPHOS in a methylation-independent manner.
To test whether decitabine enhanced ibrutinib efficacy, we first demonstrated that the combination of decitabine significantly reduced cell viability to a greater extent than either drug alone in multiple MCL cell lines (Figure 6F). For in vivo study, we used MCL-7 primary cells labeled with luciferase. Nine days after injection, tumor engraftment was documented by bioluminescent imaging. Mice were randomized into four groups: PBS control, ibrutinib, decitabine and a combination of both drugs. The results demonstrated that tumor volumes were significantly reduced in the combination group compared with the single drug or PBS groups (Figure 6G), suggesting that decitabine restored the sensitivity of MCL-7 cells to ibrutinib treatment. No apparent toxicities were observed in the decitabine- and ibrutinib-treated MCL PDX mice, and body weights were not significantly different from each treatment group (Figure 6G). In summary, these data suggest that ibrutinib treatment upregulates DNMT3A expression and that this increase is associated with the lack of response to ibrutinib in primary MCL cells. Furthermore, targeting DNMT3A by a low dose of decitabine is a potential therapeutic strategy to overcome ibrutinib resistance.
Discussion
DNA methyltransferase proteins DNMT3A and DNMT3B have been regarded as tumor suppressors in some hematological malignancies.77,78 The driver mutation R882H of DNMT3A, for example, is associated with a global hypomethylation by inhibiting DNMT3AWT forming active tetramers in AML.15 However, decitabine, which acts as a hypomethylating agent that degrades the endogenous level of all DNMTs, is an effective treatment for AML and other hematological malignancies. We have resolved this paradox. A clue to our resolution came from a study demonstrating that AML patients with DNMT3A mutations are dependent on the activities of electron transport chain complex I and exhibit a high level of OXPHOS.79 We have now revealed that DNMT3A is required for the optimal growth of MCL both in vitro and in vivo by regulating mitochondrial biogenesis in a methylation-independent manner. We suggest that decitabine is effective not by hypomethylating the genomes of cancer cells but by inducing the degradation of DNMTs, which in turns prevents the methylation-independent inhibition of oncogenic pathways such as MYC. Therefore, we provide an example of this effect in which a methylation null DNMT3A variant is sufficient to increase OXPHOS in MCL and provides a survival advantage in the presence of ibrutinib.
Furthermore, our OMI data provide evidence at the single-cell level that decitabine inhibits OXPHOS in vivo, likely due to the degradation of DNMT3A. These data provide an additional mechanistic insight into the impressive efficacy of the combinatory treatment of decitabine/azacitidine and venetoclax, a selective inhibitor of BCL-2, for AML.50,51 It is important to emphasize that additional mechanisms also contribute to the potent antitumor effect of decitabine because decitabine inhibits all DNMTs. For instance, decitabine induced DNA damage, cell-cycle arrest, and apoptosis at nanomolar concentrations in MCL cell lines, likely due to the increase in the activation of interferon-γ through the reactivation of LINE-1, an endogenous retrotransposable element (Figures S6F–S6J).80 These mechanisms of action for decitabine have been reported in other cancers.81,82,83,84,85,86
Mechanistically, we have identified MEF2B as a binding partner of DNMT3A. The interaction of these two proteins transactivate MYC, leading to the expressions of MYC target genes, including those required for the mitochondrial biogenesis. Notably, DNMT3A but not DNMT1 or DNMT3B can maintain the transactivation of a MEF2B variant that is defective in DNA-binding ability.
The function of DNMT3A identified in this study explains the discrepancy of two studies focusing on the effects of the hotspot mutations K4E, D83V, and Y69H in MEF2B.38,40 It is reasonable to expect that such mutations in the DNA-binding domain of MEF2B decrease its ability to bind DNA and therefore reduce transcriptional activity. Indeed, K43E and D83V mutations decrease DNA binding of MEF2B as shown by gel shift assay.38 Our study demonstrates that the interaction between DNMT3A and MEF2B can overcome the defects of these hotspot mutations in binding DNA because DNMT3A is a DNA-binding enzyme itself. In addition, among the MEF2 family members, MEF2B is selectively essential for lymphoid cells, as opposed to the nonessentiality of MEF2A as well as the essentiality of MEF2C and MEF2D found in both lymphoid and myeloid cells (Dependency Map). Furthermore, MYC interacts with MEF2B but not with other MEF2 family members in lymphoma cell lines. Taken together, we propose that DNMT3A-MEF2B interaction is selectively important in the context of MYC-related metabolism in non-Hodgkin lymphomas.
We believe that DNMT3A-MEF2B interaction is of broad interest for several reasons: the hotspot mutations in MEF2B are present in several types of lymphoma, including diffuse large B cell lymphoma and follicular lymphoma in addition to MCL22,47,48,49; the selective dependency of lymphoma cells on MEF2B; and the selective interaction between MYC and MEF2B.
From a translational perspective, the study suggests that decitabine is a potential therapeutic drug for the treatment of MCL patients. We provide evidence that DNMT3A contributes to ibrutinib resistance in MCL by increasing mitochondrial biogenesis and OXPHOS. Recent clinical studies demonstrated the potential of BTKis as a first-line treatment option for MCL.87,88 Given the high relapse rate observed with ibrutinib and other BTKis, there is an urgent need to identify mechanisms of resistance to enhance the efficacy of these BTKis. Here, we have demonstrated that targeting DNMT3A by genetic knockout overcomes ibrutinib resistance in various MCL clonal cell lines. More important, targeting DNMT3A by a low dose of decitabine enhances the efficacy of ibrutinib in an ibrutinib-resistant PDX model.
In summary, our study uncovers a DNA-methylation independent role of DNMT3A. DNMT3A physically interacts with MEF2B as an anchor to DNA; this interaction bypasses the DNA-binding requirement for the transactivation of MYC by MEF2B to increase the transcription of genes involved in mitochondrial biogenesis. We also provide evidence that decitabine is effective in inhibiting tumor growth in PDX models. Because decitabine has already been used in several hematological malignancies and solid tumors, it can be rapidly repurposed to treat relapsed/refractory MCL patients in combination with a BTKi.
Limitations of the study
Our study has several limitations. First, with the development of the next generation of BTKis, it is unclear whether OXPHOS and the upregulation of DNMT3A are associated with resistance to these BTKis. Second, because we used NSG mice to study the effect of decitabine in MCL, the effect of decitabine on normal B cell subtypes remains unclear. Finally, the rarity of MCL precludes us from testing whether DNMT3A upregulation is associated with primary or acquired resistance to ibrutinib because we could not obtain paired, untreated primary MCL samples. Although further studies are needed to address these limitations, our current studies mechanistically demonstrate a methylation-independent function of DNMT3A in mediating OXPHOS and ibrutinib resistance.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-DNMT3A (64B1446) | Novus Biologicals™ | NB120-13888; RRID: AB_1149786 |
| Rabbit polyclonal anti-MEF2B | ThermoFisher Scientific | PA5101143; RRID: AB_2850587 |
| Rabbit monoclonal anti-beta-Actin | Cell signaling | 4967S; RRID: AB_330288 |
| GAPDH-HRP | CST | 3683; RRID: AB_1642205 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell signaling | 7074; RRID: AB_2099233 |
| Anti-mouse IgG, HRP-linked Antibody | Cell signaling | 7076; RRID: AB_330924 |
| Mouse monoclonal anti-FLAG® M2 antibody | Sigma-Aldrich | F1804; RRID: AB_262044 |
| APC mouse anti-human CD38 | BD biosciences | 555462; RRID: AB_398599 |
| PE mouse anti-human CD19 Clone 4G7 | R&D biotechne | MAB4867R |
| anti human CD20 FITC-LT20 clone, mouse IgG1k | Miltenyi Biotec | 130-113-373; RRID: AB_2726142 |
| 5mC antibody | epigentek | A-1014-050 |
| Mouse monoclonal [9E10] to Myc tag | abcam | ab32; RRID: AB_303599 |
| Rabbit monoclonal- MYC antibody (Y69) | abcam | ab32072; RRID: AB_731658 |
| Bacteria and virus strains | ||
| DH5α | Invitrogen™ | 18265017 |
| Biological samples | ||
| B cells from cord blood | Stem Cell Technologies | 70013 |
| Primary MCL cells | Mohanty et al.57 | https://doi.org/10.1182/blood-2018-05-851667 |
| Chemicals, peptides, and recombinant proteins | ||
| Ibrutinib | Selkchem | S2680 |
| IM156 | Targetmol | S9604 |
| Decitabine | Targetmol | T1508-SB100 |
| JC-1 Dye | Invitrogen™ | T3168 |
| MitoSOX™ Red Mitochondrial Superoxide Indicator | Thermo Fisher Scientific | M36008 |
| MitoTracker™ Deep Red | Thermo Fisher Scientific | M22426 |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | 34096 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | 23225 |
| Trizol for RNA extraction | Thermo Fisher Scientific | 15596018 |
| APC Annexin V | BD pharmingen | 550474 |
| PI flow dye (BD) | BD pharmingen | 556463 |
| Doxycycline hyclate | Sigma-Aldrich | D9891 |
| RPMI-1640 medium (1x) -L-glutamine | hyclone | SH30096.LS |
| DMEM/High glucose with L-glutamine, sodium pyruvate | hyclone | SH30243.FS |
| FBS | R&D | S10350 |
| XF RPMI Medium ph7.4 | Agilent | 103576 |
| XF Calibrant, ph 7.4 | Agilent | 100840 |
| Corning® Cell-Tak™ Cell and Tissue Adhesive | Corning | 354240 |
| pen/strep solution | lonza | 17-602E |
| Corning® glutagro™ | Corning | 25-015-CI |
| MEM Non-Essential Amino Acid Solution (100X) | Lonza | 13-114E |
| Sodium Pyruvate Solution | GE Healthcare Life Sciences | SH30239.01 |
| Trypan Blue Solution (w/v) in PBS | corning | 25-900-CI |
| Protease inhibitor cocktail | sigma-aldrich | P8340 |
| PMSF Protease Inhibitor | Thermo Scientific™ | 36978 |
| D-Luciferin, Sodium Salt | Gold Biotechnology | LUCNA |
| Triton X-100 | EMD MILLIPORE | 94101L |
| pierce 16% formaldehyde (w/v), methanol-free | pierce | 28908 |
| PureLink™ RNase A | Invitrogen | 12091021 |
| Critical commercial assays | ||
| Galacto-Star™ β-Galactosidase Reporter Gene Assay System | Thermo Fisher Scientific | T1012 |
| SuperScript™ IV First-Strand Synthesis System | Thermo Fisher Scientific | 18091050 |
| GeneArt® Site-Directed Mutagenesis System | invitrogen | A13282 |
| Maxwell RSC Culture Cells DNA kit | Promega | AS1620 |
| Luciferase Assay System | Promega | E1500 |
| CellTiter-Glo(R) 2.0 Assay | Promega | G9242 |
| APC BrDu flow kit | BD pharmingen | 552598 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent | 103015 |
| QIAquick PCR Purification kit | QIAGEN | 28106 |
| SensiFast SYBR Hi-Rox Kit | Bioline | BIO-92005 |
| TruSeq Stranded mRNA Library Preparation Kit Set B | illumina | RS-122-2102 |
| TruSeq DNA LT Sample Prep Kit | Illumina | FC-121-2001 |
| Ovation Ultralow System V2 | NuGEN | 0344NB |
| Qbit dsDNA Broad range assay | Invitrogen | Q32850 |
| Deposited data | ||
| MCL-4 | This paper | PRJNA1077921 |
| MCL-5 | This paper | PRJNA1077921 |
| MCL-7 | This paper | PRJNA1077921 |
| MCL-9 | This paper | PRJNA1077921 |
| DNMT3A ChIP-seq in Z138 WT | This paper | GSE227976 |
| DNMT3A ChIP-seq in Z138#7 KO | This paper | GSE227976 |
| DNMT3A-Z138sgDNMT3A#7-uninduced-1 | This paper | GSE227976 |
| DNMT3A-Z138sgDNMT3A#7-uninduced-2 | This paper | GSE227976 |
| DNMT3A-Z138sgDNMT3A#7-uninduced-3 | This paper | GSE227976 |
| DNMT3A-Z138sgDNMT3A#7-dox-5 days-1 | This paper | GSE227976 |
| DNMT3A-Z138sgDNMT3A#7-dox-5 days-2 | This paper | GSE227976 |
| DNMT3A-Z138sgDNMT3A#7-dox-5 days-3 | This paper | GSE227976 |
| DNMT3A-Z138sgDNMT3A#7-dox-7 days-1 | This paper | GSE227976 |
| DNMT3A-Z138sgDNMT3A#7-dox-7 days-2 | This paper | GSE227976 |
| DNMT3A-Z138sgDNMT3A#7-dox-7 days-3 | This paper | GSE227977 |
| MEF2B ChIP-seq in GM12878 | Luo et al.89 | GSE127481 |
| H3K27ac in Z138 | Ryan et al.90 | GSE97541 |
| Experimental models: Cell lines | ||
| JeKo-1∗ | Li et al.6 | https://doi.org/10.1038/onc.2016.155 |
| Rec-1∗ | Li et al.6 | https://doi.org/10.1038/onc.2016.155 |
| Z138∗ | Li et al.6 | https://doi.org/10.1038/onc.2016.155 |
| Rec-1∗ | Li et al.6 | https://doi.org/10.1038/onc.2016.155 |
| Mino∗ | Li et al.6 | https://doi.org/10.1038/onc.2016.155 |
| JVM-13 | Zhang et al.13 | https://doi.org/10.1126/scitranslmed.aau1167 |
| Granta-519 | Zhang et al.13 | https://doi.org/10.1126/scitranslmed.aau1167 |
| Maver-1 | Zhang et al.13 | https://doi.org/10.1126/scitranslmed.aau1167 |
| JeKo-1 Ibrutinib resistant | Liu et al.75 | https://doi.org/10.1182/blood.2022018674 |
| Rec-1 Ibrutinib resistant | Liu et al.75 | https://doi.org/10.1182/blood.2022018674 |
| Experimental models: Mouse | ||
| NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | Jackson Laboratory | 5557 |
| Oligonucleotides | ||
| See Table S2 for sequences of oligonucleotides | N/A | |
| Recombinant DNA | ||
| pRSGT16-U6Tet-sg-HTS6C-CMV-TetRep-2A-TagRFP-2A-Puro | Cellecta | #SVCRU6T16-L |
| pR-CMV-Cas9–2A-Hygro | Cellecta | #SVC9-PS |
| phCMV-GalV-MTR | Caeser et al.91 | Addgene #163612 |
| pCDH-DNMT3A WT | This paper | N/A |
| pCDH-DNMT3A V716G | This paper | N/A |
| pCDNA.3-Myc-DNMT3A WT | Chen et al.92 | Addgene plasmid #35521 |
| pCDNA.3-Myc-DNMT3A D333A | This paper | N/A |
| pCDNA.3-Myc-DNMT3A V716G | This paper | N/A |
| pCDNA.3-Myc-DNMT1 | Li et al.93 | Addgene plasmid #36939 |
| pCDNA.3-Myc-DNMT3B1 | Chen et al.92 | Addgene plasmid #35522 |
| pGL3-tfam | This paper | N/A |
| Software and algorithms | ||
| ImageJ 142 software | NIH | https://imagej.nih.gov/ij/index.html |
| GraphPad Prism 9.0 | Graphpad | www.graphpad.com |
| FlowJo Software vX 10.0.7v2 | FLOWJO, LLC | https://www.flowjo.com/ |
| StringTie | Pertea et al.94 | https://ccb.jhu.edu/software/stringtie/ |
| DEseq2 | Love et al.95 | http://bioconductor.org/packages/DESeq2/ |
| Genrich | Gaspar96 | https://github.com/jsh58/Genrich |
| HISAT2 | Kim et al.97 | http://daehwankimlab.github.io/hisat2/ |
| ChIPSeeker | Wang et al.98 Yu et al.99 |
http://bioconductor.org/packages/release/bioc/html/ChIPseeker.html |
| deepTools-Correct GC bias | Benjamini et al.100 | https://deeptools.readthedocs.io/en/latest/ |
| GSEA v3.0 | Broad Institute | https://software.broadinstitute.org/cancer/software/gsea/wiki/index.php/Main_Page |
| BioRender BioRender | BioRender | https://biorender.com/ |
| Igtegrative Gomomic Viewer v2.13.2 | Broad Institute | https://software.broadinstitute.org/software/igv/ |
| Custom Python script for manual correction of tumor segmentation | Pachitariu et al.,101 Stringer et al.102 | https://www.github.com/mouseland/cellpose |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lixin Rui (lrui@medicine.wisc.edu)
Material availability
Request for cell line generated in this study should be directed to and will be fulfilled by the lead contact, Lixin Rui (lrui@medicine.wisc.edu)
Data and code availability
-
•
RNA-seq, ChIP-seq from MCL cell lines, and WES data from MCL patients generated have been deposited at GEO and SRA, respectively, and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Cell lines and culture
Doxycycline-inducible human MCL cell lines (Mino, Rec-1, JeKo-1, Z138), originally obtained from the American Type Culture Collection (ATCC), were engineered and authenticated by gene expression profiling to express the bacterial tetracycline repressor as described previously.6 Wild-type Granta-519, Maver-1, JVM-13 were provided by Dr. Michael Wang at MD Anderson Cancer Center. Doxycycline (20 ng/mL) was used for inducing the expression of sgDNMT3A of interest. The MCL cell lines were grown in RPMI 1640 media (Hyclone) supplemented with 10% FBS (fetal bovine serum, Atlanta Biologicals), 100 U/ml penicillin, 100 μg/mL streptomycin (Corning Cellgro), 2 mM GlutaGRO (Corning Cellgro), 1×MEM-NEAA (Quanlity Biological, Inc.), and 1 mM Sodium Pyruvate Solution (Hyclone). Human embryonic kidney cell line 293T was cultured in DMEM (Dulbecco’s modified Eagle’s medium, Hyclone) with 10% FBS, 100 U/ml penicillinand 100 μg/mL streptomycin (Corning Cellgro). Cell lines were authenticated using short tandem repeat analysis (Idexx BioAnalytics, Westbrook, ME) and per ATCC guidelines using morphology, growth curves, and Mycoplasma testing within 6 months of use with the e-Myco mycoplasma PCR detection kit (iNtRON Biotechnology Inc, Boca Raton, FL). All cell lines were cultured at 37°C in a 5% CO2.
Xenografts
Male and female (8–12 weeks of age) NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) breeder pairs were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and bred under specific pathogen-free conditions in sterile ventilated racks in the animal care facility at the University of Wisconsin-Madison. The study was approved by the Animal Care and Use Committee of the University of Wisconsin-Madison. Tumor cells (7-8 x 106) were injected subcutaneously into the right flank of each mouse.
Human samples
Four viable cryo-preserved of previously treated primary MCL samples were provided by Dr. Vu N. Ngo at the Beckman Research Institute of City of Hope. Samples were originally obtained from the tumor bank of the Pathology Department of City of Hope as de-identified samples. De-identified patient samples were obtained with informed consent and xenografted under Dana-Farber/Harvard Cancer Center Institutional Review Board (IRB)-approved protocol #13–351. MCL-4 (DFBL-98848, male), MCL-5 (DFBL-44685, male), MCL-7 (DFBL-91438, male) and MCL-9 (DFBL-96069, female) patients were relapsed from immunochemotherapy drugs (all four patients) and ibrutinib (except for MCL-4).
Method details
sgDNMT3A experiment
When tumors became palpable (Z138 #7 median: 73.4 mm3; range: 37.4–613.4 mm3; Granta-519 #7 median: 127 mm3; range 44.1–239.2 mm3), mice received normal drinking water or water with doxycycline (2 mg/mL). Tumor measurements were recorded three times per week until endpoint, defined as when tumor exceeded 20 mm in any direction or became morbid. The relative tumor growth was calculated by normalizing tumor volumes to the start day of tumor measurement for each mouse.
Decitabine treatment
Treatment started when palpable tumors were observed (∼300–450 mm3), approximately 10–12 days post tumoral injection. When mice became moribund or when tumor size exceeded 20 mm in any direction, mice were euthanized as required by institutional protocols.
Expansion of primary MCL samples
In vivo expansion was described in our recent study.75 Briefly, 2 x 106 cells were injected into the tail vein of NSG mouse after sublethal irradiation (2 Gy). 35–45 days post tumoral injection, mice were sacrificed. Livers and spleens were collected and processed by mechanical disruption, Ficoll density gradient centrifugation and AKC lysis to remove red blood cells. Flow cytometric analysis using markers for human CD19 and human CD20 confirmed that 85–90% of cells were cancer cells.
In vitro maintenance of primary MCL samples
To maintain primary MCL cells in vitro, we co-cultured them with YK6-CD40lg-IL21 cells (YK6).91 2-4 x 104 of irradiated YK6 cells (30 Gy) were seeded in a 6 well plate 24 h prior to co-culture of MCL cells. Primary MCL cells grew and expanded more than 7 days without observable changes of surface markers.
MCL patient-derived xenografts
All four primary cell samples were used for a recent study.57
MCL-7 PDX
Freshly collected MCL-7 cells, which had been previously tagged with luciferase and expanded in vivo for 30–35 days, were subcutaneously injected into immunocompromised NSG mice. Tumor signals were monitored using the Xenogen IVIS Imaging System (Caliper Life). When they reached approximately 12 x 108 of total flux (ps−1), mice were treated with either PBS or with 1 mg/kg of decitabine (i.p.) every day, 5 days a week for a total of 21 injections. Bioluminescence images of mice before and every week during the course treatment (left) and growth curves are shown (right). Fresh aliquots of decitabine were used for every injection. The red arrow indicates treatment start. All mice died after 10 injections, potentially due to the potency of decitabine when fresh aliquots were used.
MCL-4 PDX
A procedure similar to MCL-4 PDX was followed to establish tumors. Starting bioluminescence signal was approximately 2.5 x 107 total flux (ps−1), bioluminescence images were recorded every 5 days, and decitabine concentration was 0.25 mg/kg.
The endpoint for MCL-4 PDX was reached when tumor signal exceeded 1 x 1010, or there were greater than 2 metastases, or paralysis, or morbidity. The endpoint for MCL-7 PDX was similar with the exception that the total flux exceeds 1 x 1011 because MCL-7 tumors tended to localize at the injection site, whereas MCL-4 tumors had a higher tendency to metastasize.
MCL-7 PDX in decitabine-ibrutinib combination study
A procedure like the above PDX was followed to establish tumors. When tumor signal reached approximately 11.8 x 107 of total flux (ps−1), mice were intraperitoneally (i.p.) treated with decitabine (0.25 mg/kg) or ibrutinib (30 mg/kg), at 5 days a week for both, or with a combination of the two drugs.
DNMT3A knockout with an inducible 2-vector CRISPR-Cas9 knockout system
The Lentiviral plasmid pRSGT16-U6Tet-sg-HTS6C-CMV-TetRep-2A-TagRFP-2A-Puro (Cellecta) was used to express sgRNA targeting a gene of interest and pR-CMV-Cas9-2A-Hygro (Cellecta) was used to generate Cas9-expressing cells. We first transduced Cas9 expressing plasmid into different MCL cell lines and select cells with hygromycin for at least 10 days. Two distinct sgRNAs targeting exon 2 and the MTase domain of DNMT3A were packaged into lentiviral particles in equal molar ration (1:1) using PAX2 and MD2G plasmids. Cells that expressed Cas9 confirmed by immunoblot were then transduced with sgDNMT3A. After puromycin selection, Cas9-sgDNMT3A expressing cells were diluted at a density of 1 cell per 3 well and seeded onto a 96-well plate. Single-cell clones were confirmed by light microscope (ZEISS PrimoVert). sgDNMT3A targeting exon 2 was designed using online software (https://chopchop.rc.fas.harvard.edu/). The sgDNMT3A sequence that targets the MTase domain was from a publication.103 These sequences were inserted into sgRNA construct following the manufacturer’s instructions. Doxycycline (2 ng/mL) was used to induce the expression of sgRNA and the subsequent knockout of the target gene.
DNMT3A plasmid construction
The pCDNA.3-myc-DNMT3A cDNA was subcloned into HindIII and Xho1 restriction sites of the lentiviral vector pCDH.
Mutagenesis of DNMT3A
pCDNA.3-myc-DNMT3A was used as template to mutate D333 to A and V716 to G using ThermoFisher Mutagenesis kits with the primer sequences listed in the key resources table. Mutations were confirmed by Sanger sequencing. pCDNA.3-myc-DNMT3A V716G was further subcloned into HindIII and Xho1 restriction sites of the lentiviral vector pCDH-GFP for further experiments.
Lentivirus transduction
All the lentiviral constructs were packaged and enveloped using PAX2 and MD2G or phCMV-GalV-MTR in HEK293T cells using LentiIT (Mirus) per manufacturer protocol. Supernatant were collected 48 and 72 h post transfection, spun down to remove cell debris, filtered through a 0.45 μm PVDG membrane and aliquoted and stored at −80°C for each use.
IWhen necessary, viruses were concentrated using PEG800 follow protocol from Dr. Sudgen’s laboratory that was modified from MD Anderson Cancer Center. MCL cell lines were transduced in the presence of 8 μg/mL of polybrene and selected with appropriate antibiotics for 10–14 days. Purity is then confirmed using fluorescent markers (GFP or RFP).
RNA-sequencing
Z138 sgDNMT3A #7 (Z183 #7) cells were induced with 20 ng/mL doxycycline starting at staggered timepoints (5 days and 7 days) for concurrent RNA collection. Each pair of RNA-seq sample (control and DNMT3A KO) was collected independently for a total of 3 biological replicates. RNA was kept in TriZol until all samples are collected. The total RNA was extracted using TriZol according to the manufacturer’s protocol. RNA-seq libraries were prepared with the Illumina TruSeq stranded mRNA LT sample preparation kit (Illumina). Paired-end sequencing was performed on Illumina NovaSeq 600 at the length of 150 bp.
Raw reads were mapped to the human reference genome (hg38) using HISAT2 (v2.1). Genes and transcripts were assembled and quantified using StringTie (v1.3.4). Differential expression (DE) was analyzed with DESeq2. DE analysis compared the changes in mRNA abundance in cells deficient in DNMT3A to that in control cells. Gene set enrichment analysis (GSEA) was performed by GSEA software (V4.0.1). For GSEA analysis, a pre-ranked gene list was generated using the formula:
The molecular signatures databases including h.all.v5.2 symbols, along with mitochondria-related gene sets were used.
Chromatin immunoprecipitation sequencing
Chromatin immunoprecipitation (ChIP) experiments were performed as previously described.104 Briefly 20 million cells of either Z138 WT or DNMT3A KO cells (Z138#7; 7 days after doxycycline induction) were cross-linked with formaldehyde (1%) for 10 min at room temperature. The reaction was stopped by adding glycine to the final concentration of 125 mM for 5 min. The nuclear pellets were collected and sonicated with Covaris s220. Then anti-DNMT3A antibody was added and incubated at 4°C for 16 h on a rotator. Dynabeads protein G were prewashed and added for 2 h of incubation at 4°C on a rotator. The immunoprecipitated complex was eluted in elution buffer (1% SDS, 0.1 M NaHCO3) by a 65°C vortex for 15 min. DNA was purified using a Qiagen PCR purification kit according to the manufacturer’s protocol. Ten nanograms of ChIP’ed DNA were used to generate the ChIP-seq library using Ovation Ultralow Library System V2 (NuGen Technologies), according to the manufacturer’s protocol. Paired-end sequencing was performed on an Illumina NovaSeq 600 (150-bp length) at the University of Wisconsin Biotechnology Center DNA sequencing facility.
For ChIP-seq data analysis, raw reads were mapped to the human reference genome (hg38) by HISAT2 (V2.1) using default parameters. Identical reads were collapsed into one. Because sequencing results had high GC bias, including input controls, all datasets were corrected for GC bias using deepTools with computeGCbias.105 ChIP-seq data were visualized in an Integrative Genomic Viewer (V2.3 40). Peaks were called by Genrich (V1.0) using default parameters. The MEME suite (with ±1 kb around a peak summit) was used for motif discovery (meme-suite.org). ChIPseeker was used to annotate and visualize the distribution of peaks. EnrichedHeatmap (Bioconductor V1.14.0) was used to visualize differential peaks.
RNA-seq and ChIP-seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus106 and are accessible through GEO Series accession number GSE227976.
Immunoblotting assay
Cells were lysed using RIPA lysis buffer with protease inhibitor cocktail (Sigma) and phenylmethylsulfonyl flouride. Whole cell lysates were resolved by SDS-PAGE (8–10%), transferred onto PVDF membranes upon blocking in TBS 0.05% Tween 20 (TBS-T) and 5% non-fat dry milk for 1 h, and then incubated with a primary antibody at 4°C overnight. After washing, the membranes were probed with a horseradish peroxidase-conjugated secondary antibody (HPR) for 1 h at room temperature. Membranes were washed with TBS-T for at least 1 h before imaging using the G:Box SynGene imager.
Whole exome sequencing (WES)
Genomic DNA was extracted from frozen MCL samples using the Promega Maxwell RSC Culture Cells DNA kit on the automated Maxwell RSC according to manufacturer’s protocol. Exome enrichment and paired-end sequencing was performed on an Illumina NovaSeq 600 (150-bp length) at the University of Wisconsin Biotechnology Center DNA sequencing facility. Exome was enriched using the human comprehensive exome (twist bioscience). WES FASTQ files were processed by Illumina’s DRAGEN software (version 3.10) using human genome hg38, GENCODE gene definitions (version 42), and annotations from dbSNP (version 155). The limit of somatic variation detection is 5% at 100x and only those with filter status as PASS were kept. Variation’s impact was inferred by VEP (version 108) and its human annotations (version 109) via the vcf2maf.pl script from https://github.com/mskcc/vcf2maf.
Flow cytometry analyses
Cell cycle was analyzed with the APC BrdU Kit (BD Pharmingen) according to manufacturer’s instructions. Cells were pulsed with 10mM with bromodeoxyuridine (BrdU) for 4 h at 37°C. Cells were washed in the staining buffer, fixed/permeabilized with the Cytofix/Cytoperm buffer or Cytofix/Cytoperm buffer plus and washed with the Perm/Wash buffer. After permeabilization, cells were treated with DNase for 1 h at 37°C, and then stained with allophycocyanin (APC)- conjugated anti-BrdU antibody and 7-amino-actinomycin D (7-AAD; 25 mg/mL).
Cell apoptosis was measured using APC-Annexin V and propidium iodide (PI) staining kit following the manufacture’s protocols. Briefly, cells with different treatments were collected and washed twice with cold PBS, and then re-suspended in 1×binding buffer from the kit. APC-Annexin V and PI were added and incubated for 15 min at room temperature in the dark.
Mitochondrial membrane potential (ΔΨ), mitochondrial ROS, or mitochondrial mass
One million cells were washed with warm PBS once and incubated with JC-1 (5 μg/mL) for 10 min or mitoRox (5 μM) for 20 min or MitoTracker Deep Red (20nM) in phenol red free media at 37°C or NAO (10 nM) for 10 min at room temperature in the dark, followed by three washes with warmed PBS and immediately analyzed. DAPI, a viability dye, was immediately added prior to the acquisition on the flow cytometer. All stained samples were measured and analyzed using ThermoFisher Attune and FlowJo (V10.0) software. All measurements were performed in 10,000 live cells and performed independently at least 3 times, each with 3 technical replicates.
Intracellular staining of DNMT3A
Cells were counted and resuspended in PBS at the concentration of 1 x 106 cells/100μL and processed as previously described.107 Individual samples that showed greater than 90% viable cells after 3 days of ibrutinib treatment (2.5μM) were stained for DNMT3A expression. Live cells were gated before measuring DNMT3A level. MCL-4: 3 spleens and 3 livers; MCL-7: 2 spleens and 2 livers; MCL-9: one spleen and one liver.
Immunoprecipitation
Whole cell protein extracts were prepared from MCL cell lines or HEK293T cell lines 48 h post transfection using the immunoprecipitation (IP) buffer. For endogenous IP in MCL cell lines, the buffer included 50mM TrisHCL pH 8.0, 150 mM NaCl, 1mM EDTA, 0.5% NP40, 0.5 mM PMSF, 50 mM sodium fluoride, and protease inhibitor cocktail (Sigma). For IP of co-transfected HEK293T, the buffer included 50 mM TrisHCl pH 7.5, 150 mM NaCl, 2 mM EDTA pH 7.0, 0.1% NP40, 1% Triton X-100, 1% Tween, 0.5 mM PMSF, 50 mM sodium fluoride, 1 mM sodium orthovanadate and protease inhibitor cocktail (Sigma). Lysates were pre-cleared with IgG mouse for 2 h; Dynabeads protein G were added for an additional 2 h. DNAse (6 μg/mL) was freshly added every 2 h. Eluates were incubated with anti-FLAG or anti-DNMT3A or anti-MYC (Y69) for 3–4 h before adding dynabeads protein G for overnight incubation. Fresh DNAse was added prior to overnight incubation. Beads were washed 8 times for a total of 2 h in the same buffer for endogenous IP, and in wash buffer (300mM NaCl) for co-transfected IP. Fifty microliters of the eluate from overnight incubation were collected, digested with proteinase K at 65°C for 2 h and then analyzed on an agarose gel to ensure proper DNA fragmentation. The immunocomplexes were eluted in 1X SSB buffer. Eluates were resolved on SDS-PAGE, similar to immunoblotting protocol. After washing, the membrane was probed with HPR-conjugated, rabbit conformation specific- or mouse light chain specific-secondary antibodies for 1 h at room temperature. The membranes were washed with TBS-T for at least 2 h before imaging using the Odissey LI-COR imager. All immunoprecipitation experiments were performed independently twice except for primary MCL samples.
XF cell mito stress analysis
The mitochondrial respiratory capacity was determined using XF Cell Mito Stress Test Kit (Agilent Technologies). 3-6 x105 cells per well were seeded in parallel into the Cell-Tak-Coated 96-well XF Cell Culture Microplate and a round bottom 96 well plate. To normalize OCR per viable cell, cells in the round-bottom 96 well plate were counted using DAPI on a flow cytometer. Cells in the XF Cell Culture Microplate were incubated in XF RPMI Medium, pH7.4 basal media (Agilent # 103576-100) with the addition of 10 mM glucose, 2 mM sodium pyruvate and 2.5 mM L-glutamine in a CO2-free incubator at 37°C for 1 h prior to assay. The oxygen consumption rate (OCR) was measured by XFe 96 extracellular flux analyzer (Agilent Technologies) with sequential injection of oligomycin A (1 μM), FCCP (1 μM), and rotenone/antimycin A (0.5 μM). ATP production was calculated per manufacturer’s guideline as:
Transmission electron microscopy
Samples were processed and sectioned at ultrathin at the electron microscope facility at University of Wisconsin-Madison. Samples were immersion fixed in a solution of 2.5% glutaraldehyde and 2.0% paraformaldehyde in 0.1M sodium cacodylate buffer, pH 7.4 1 h at room temperature. The tissue was then post fixed in 1% osmium tetroxide (OsO4) in the same buffer for 30 min at room temperature. Following OsO4 post-fixation, the samples were dehydrated in a graded ethanol series, and embedded in Durcupan epoxy resin in small aluminum weighing dishes. After polymerization of the resin, excess resin was scrapped from the bottom of cover slips to expose the glass surface. The samples were then etched with 52% hydrofluoric acid to completely remove the cover slips, leaving the cells exposed at the surface of the preparation. Samples were sectioned for TEM using a Reichert-Jung Ultracut-E Ultramicrotome and contrasted with Reynolds lead citrate and 8% uranyl acetate in 50% EtOH. Ultrathin sections were observed with a Philips CM120 electron microscope and images were captured with a MegaView III side mounted digital camera.
Multiphoton imaging
Z138 cells (7 million cells/mouse) were established as subcutaneous tumors in NSG mice. When tumors became palpable, approximately 10 days post tumoral injection, mice received either PBS or decitabine (1 mg/kg) 5 days a week (n = 8 per group) for a total of 20 injections. The same aliquot of decitabine was used for 5 days (4 cycles of freeze-thaw).
Ex vivo imaging of Z-138 tumors was performed in PBS treated mice (n = 3) and decitabine treated mice (n = 3) on day 6 post treatment. Tumors were well established (150 mm3), 10–12 days after inoculation. Imaging was performed on two separate days. Ex vivo imaging of whole Z-138 tumors was performed immediately following euthanasia via CO2. Excised tumors were secured to an imaging dish with PBS coupling and tape. Ex vivo imaging was complete within one to 2 h post mouse euthanasia, accurately capturing cellular metabolism based on our previous work that showed that ex vivo metabolism is statistically identical to in vivo metabolism for up to 12 h post euthanasia.108
Autofluorescence images were captured with a custom-built multi-photon microscope (Bruker) using an ultrafast femtosecond laser (InSight DSC, Spectra Physics). The laser was tuned to 750 nm for NAD(P)H excitation and tuned to 890 nm for FAD excitation. The average power incident on the sample was ∼2–5 mW for NAD(P)H excitation and ∼2–6 mW for FAD excitation. A pixel dwell time of 4.8 μs was used for all images. NAD(P)H and FAD images were acquired sequentially. A 440/80 nm bandpass filter isolated NAD(P)H emission onto the photomultiplier tube (PMT) detector. A dichroic mirror directed wavelengths greater than 500 nm onto a 550/100 nm bandpass filter, isolating FAD emission onto a second PMT. Fluorescence lifetime measurements were acquired with time-correlated single photon counting electronics (Becker and Hickl) and a GaAsP PMT (Hamamatsu). All images were acquired with a 40×/1.13 NA water-immersion objective (Nikon) at 512 × 512 pixel resolution and an optical zoom of 1.0–3.0 (majority of images taken at zoom of 2.0). NAD(P)H and FAD intensity and lifetime images were acquired to sample metabolic behavior of Z-138 tumor cells across 5–7 fields of view and multiple depths within each tumor.
Multiphoton image analysis
Fluorescence lifetime fitting
The fluorescence lifetimes of free and protein-bound NAD(P)H and FAD are distinct, and these lifetimes along with their weights can be recovered with a two-exponential fit function. Therefore, fluorescence lifetime decays for both NAD(P)H and FAD were fit to the following bi-exponential function in SPCImage:
For NAD(P)H, τ1 corresponds to the free lifetime, τ2 corresponds to the protein-bound lifetime, and the weights (α1, α2; α1 + α2 = 1) correspond to the proportion of free and protein-bound NAD(P)H, respectively.52,109,110 Conversely for FAD, τ1 corresponds to the protein-bound lifetime and τ2 corresponds to the free lifetime.52,54,111 An instrument response function was measured using SHG (900 nm excitation) from urea crystals for input into the decay fit procedure. The following fluorescence lifetime endpoints were calculated from the fitted model: τ1, τ2, α1, and α2 for both NAD(P)H and FAD; along with the optical redox ratio,111,112 defined as the fluorescence intensity of NAD(P)H divided by the sum of fluorescence intensities from NAD(P)H and FAD.
Tumor cell image segmentation
Automated tumor cell image segmentation was performed through Cellpose, with manual corrections made as needed using the Napari viewer through a custom Python script.101,102 Single tumor cells were identified, circled, and segmented as whole cells (nuclei and cytoplasm captured) using NAD(P)H intensity images. The resulting segmented images were saved as masks. Using these masks and the raw imaging data, fluorescence lifetime variables and optical redox ratio values were calculated for each individual cell. Calculations were performed using RStudio.
Statistical analysis
Mann–Whitney statistical tests for non-parametric, unpaired comparisons were performed to assess differences in OMI parameters between treatment groups. This test was chosen because these data distributions were not assumed to be parametric. Results are represented as dot plots showing mean ± standard deviation where each dot represents a single tumor cell (GraphPad Prism 9).
Luciferase reporter assay
The luciferase reporter construct (pGL3-tfam) was generated by cloning the upstream region of tfam that is co-bound by DNMT3A and marked with H3K27ac and MEF2B into the pGL3 basic vector. All final products were verified by enzymatic digestion and Sanger sequencing analysis.
2 x 105 HEK293T cells were seeded 24 h prior to transfection. Cells were co-transfected using PEI (PEI: total plasmid DNA = 4:1; Polysciences) in triplicates with pCMX-β-galactosidase (β-gal) plasmid as internal control for transfection efficiency, along with pGL3-tfam and in the presence or absence of the expression vectors, pCDNA.3-DNMT3AWT or pCDNA.3-DNMT3AV716G or pCDNA.3 DNMT1 or pCDH-DNMT3B. pCDH-GFP was added so that the total quantity of plasmid DNA was equal. Cells were harvested and lysed using lysis buffer 48 h post transfection. Luciferase activity was analyzed using Promega Luciferase Assay System; β-gal activity was detected using Galacto-Star β-Galactosidase Reporter Gene Assay System for Mammalian Cells according to manufacturer’s protocols. The luminescence of firefly luciferase from the pGL3 basic reporter was normalized to that of β-gal.
Luciferase experiments were performed independently, twice each with 3 technical replicates.
Design of drug synergism experiment
A 4 x 4 matrix of two drugs and all possible combinations were designed on a 96-well plate. 10,000–20,000 cells were seeded in each well. 3-day treatment was performed in triplicates. Cells were stained with DAPI and immediately analyzed on the Attune flow cytometer. Live cells were gated as a negative signal for DAPI.
IC50 calculation
All IC50 values in this manuscript were calculated using variable slopes algorithm in PRISM. This algorithm fits the Hill Slope, which measures the steepness of the slope, from the data rather than assuming the standard slope of 1.0. Therefore, the IC50 is the concentration of the antagonist that reduces cell viability halfway between the top and the bottom point. Importantly, this is not the same as the response at Y = 0.5.
Drug formulation
For in vivo studies, ibrutinib was formulated in 5% DMSO, 30% PEG400, 5% Tween 80, and 60% water. Decitabine was diluted in PBS and frozen in aliquots. Fresh aliquots were used for every injection unless noted otherwise.
Generation and maintenance of acquired ibrutinib-resistant MCL cell lines
To generate ibrutinib-resistant MCL cells, JeKo-1 and Rec-1 cells were originally cultured in 1μM ibrutinib. The drug concentrations were increased in a stepwise manner up to 5 μM and 10μM, respectively, for a period of 40 weeks. The stable ibrutinib-resistant phenotype was confirmed by measuring and comparing the IC50 of ibrutinib in parental and resistant cells at multiple time points during and after 40 weeks of culture. Ibrutinib-resistant JeKo-1 and Rec-1 cells were maintained in culture media with 5μM or 10μM of ibrutinib, respectively.
Quantification and statistical analysis
The two-tailed Student’s t test was used to compare two normally distributed groups. The Mann-Whitney U test was used to compare two groups that were not normally distributed. One-way or two-way analysis of variance was used to analyze variance for comparisons between 3 or more groups. Results were presented as mean ± standard deviation or standard error of the mean. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 were used to show statistical significance. GraphPad Prism 9 and R packages were used for data analysis and visualization.
Acknowledgments
We thank Dr. Daniel Hodson for providing YK6-CD40lg-IL21 cells and Dr. Shannon Kenney for providing normal B cells from cord bloods as well as plasmids. We thank the University of Wisconsin (UW)-Madison Biotechnology Center Bioinformatics Core Facility for processing the WES data, the UW-Madison Center for High Throughput Computing for its computing facilities, and the University of Wisconsin Carbone Cancer Center Flow Cytometry Laboratory, supported by P30 CA014520, for use of their facilities and services. This work was supported by the National Institutes of Health/National Cancer Institute (NIH/NCI) grant R01 CA266354 (to L.R.), the ASH Bridge Grant (to L.R.), the UW Center for Human Genomics and Precision Medicine Seed Grant (to L.R.), the Midwest Athletes Against Childhood Cancer (MACC) Fund (to P.D.B. and C.M.C.), the Morgridge Interdisciplinary Postdoctoral Fellowship (to A.R.H.), the Morgridge Institute for Research (to M.C.S.), the Carol Skornicka Chair of Biomedical Imaging (to M.C.S.), the Retina Research Foundation Daniel M. Albert Chair (to M.C.S.), and the UW Carbone Cancer Center pilot grant NIH/NCI P30 CA014520 (to C.M.C., L.R., and P.L.). The contents of this article do not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the US government.
Author contributions
L.R. conceived and designed the study and supervised the research. C.M.C. supervised the xenograft experiments, and M.C.S. supervised the OMI experiments. N.-M.H., Y.L., P.D.B., A.R.H., A.F.L., F.Z., A.K., R.C., X.Z., and P.E.S. performed research. N.-M.H., Y.L., P.L., A.R.H., A.F.L., and L.R. analyzed the data. V.N.N. provided the PDX samples and intellectual input. N.-M.H. and L.R. drafted and revised the manuscript. All of the authors revised and approved the submitted manuscript.
Declaration of interests
C.M.C. receives honorarium for advisory board membership from Bayer, Elephas, Nektar Therapeutics, Novartisn, and WiCell Research Institute, who had no input in the study design, analysis, manuscript preparation, or decision to submit for publication.
Published: March 29, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101484.
Supplemental information
(A) Mutations in DNMT, BTK, MEF2B, and NSD2. (B) Mutations in frequently mutated genes in MCL. (C) Mutations in metabolic-related genes, related to Figure 4
References
- 1.Jain P., Wang M. Mantle cell lymphoma: 2019 update on the diagnosis, pathogenesis, prognostication, and management. Am. J. Hematol. 2019;94:710–725. doi: 10.1002/ajh.25487. [DOI] [PubMed] [Google Scholar]
- 2.Navarro A., Beà S., Jares P., Campo E. Molecular Pathogenesis of Mantle Cell Lymphoma. Hematol. Oncol. Clin. N. Am. 2020;34:795–807. doi: 10.1016/j.hoc.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jares P., Colomer D., Campo E. Molecular pathogenesis of mantle cell lymphoma. J. Clin. Invest. 2012;122:3416–3423. doi: 10.1172/JCI61272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campo E., Jaffe E.S., Cook J.R., Quintanilla-Martinez L., Swerdlow S.H., Anderson K.C., Brousset P., Cerroni L., de Leval L., Dirnhofer S., et al. The International Consensus Classification of Mature Lymphoid Neoplasms: a report from the Clinical Advisory Committee. Blood. 2022;140:1229–1253. doi: 10.1182/blood.2022015851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang M.L., Rule S., Martin P., Goy A., Auer R., Kahl B.S., Jurczak W., Advani R.H., Romaguera J.E., Williams M.E., et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2013;369:507–516. doi: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Y., Bouchlaka M.N., Wolff J., Grindle K.M., Lu L., Qian S., Zhong X., Pflum N., Jobin P., Kahl B.S., et al. FBXO10 deficiency and BTK activation upregulate BCL2 expression in mantle cell lymphoma. Oncogene. 2016;35:6223–6234. doi: 10.1038/onc.2016.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang M., Rule S., Zinzani P.L., Goy A., Casasnovas O., Smith S.D., Damaj G., Doorduijn J., Lamy T., Morschhauser F., et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet. 2018;391:659–667. doi: 10.1016/S0140-6736(17)33108-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bomben R., Ferrero S., D'Agaro T., Dal Bo M., Re A., Evangelista A., Carella A.M., Zamò A., Vitolo U., Omedè P., et al. A B-cell receptor-related gene signature predicts survival in mantle cell lymphoma: results from the Fondazione Italiana Linfomi MCL-0208 trial. Haematologica. 2018;103:849–856. doi: 10.3324/haematol.2017.184325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young R.M., Staudt L.M. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat. Rev. Drug Discov. 2013;12:229–243. doi: 10.1038/nrd3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheah C.Y., Chihara D., Romaguera J.E., Fowler N.H., Seymour J.F., Hagemeister F.B., Champlin R.E., Wang M.L. Patients with mantle cell lymphoma failing ibrutinib are unlikely to respond to salvage chemotherapy and have poor outcomes. Ann. Oncol. 2015;26:1175–1179. doi: 10.1093/annonc/mdv111. [DOI] [PubMed] [Google Scholar]
- 11.Cheng S., Guo A., Lu P., Ma J., Coleman M., Wang Y.L. Functional characterization of BTK(C481S) mutation that confers ibrutinib resistance: exploration of alternative kinase inhibitors. Leukemia. 2015;29:895–900. doi: 10.1038/leu.2014.263. [DOI] [PubMed] [Google Scholar]
- 12.Woyach J.A., Furman R.R., Liu T.M., Ozer H.G., Zapatka M., Ruppert A.S., Xue L., Li D.H.H., Steggerda S.M., Versele M., et al. Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 2014;370:2286–2294. doi: 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L., Yao Y., Zhang S., Liu Y., Guo H., Ahmed M., Bell T., Zhang H., Han G., Lorence E., et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aau1167. [DOI] [PubMed] [Google Scholar]
- 14.Queirós A.C., Beekman R., Vilarrasa-Blasi R., Duran-Ferrer M., Clot G., Merkel A., Raineri E., Russiñol N., Castellano G., Beà S., et al. Decoding the DNA Methylome of Mantle Cell Lymphoma in the Light of the Entire B Cell Lineage. Cancer Cell. 2016;30:806–821. doi: 10.1016/j.ccell.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Russler-Germain D.A., Spencer D.H., Young M.A., Lamprecht T.L., Miller C.A., Fulton R., Meyer M.R., Erdmann-Gilmore P., Townsend R.R., Wilson R.K., Ley T.J. The R882H DNMT3A Mutation Associated with AML Dominantly Inhibits Wild-Type DNMT3A by Blocking Its Ability to Form Active Tetramers. Cancer Cell. 2014;25:442–454. doi: 10.1016/j.ccr.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dai Y.-J., Wang Y.-Y., Huang J.-Y., Xia L., Shi X.-D., Xu J., Lu J., Su X.B., Yang Y., Zhang W.N., et al. Conditional knockin of Dnmt3a R878H initiates acute myeloid leukemia with mTOR pathway involvement. Proc. Natl. Acad. Sci. USA. 2017;114:5237–5242. doi: 10.1073/pnas.1703476114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ley T.J., Ding L., Walter M.J., McLellan M.D., Lamprecht T., Larson D.E., Kandoth C., Payton J.E., Baty J., Welch J., et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoang N.M., Rui L. DNA methyltransferases in hematological malignancies. J Genet Genomics. 2020;47:361–372. doi: 10.1016/j.jgg.2020.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brunetti L., Gundry M.C., Goodell M.A. DNMT3A in Leukemia. Cold Spring Harb. Perspect. Med. 2017;7 doi: 10.1101/cshperspect.a030320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Couronné L., Bastard C., Bernard O.A. TET2 and DNMT3A mutations in human T-cell lymphoma. N. Engl. J. Med. 2012;366:95–96. doi: 10.1056/NEJMc1111708. [DOI] [PubMed] [Google Scholar]
- 21.Odejide O., Weigert O., Lane A.A., Toscano D., Lunning M.A., Kopp N., Kim S., van Bodegom D., Bolla S., Schatz J.H., et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123:1293–1296. doi: 10.1182/blood-2013-10-531509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beà S., Valdés-Mas R., Navarro A., Salaverria I., Martín-Garcia D., Jares P., Giné E., Pinyol M., Royo C., Nadeu F., et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc. Natl. Acad. Sci. USA. 2013;110:18250–18255. doi: 10.1073/pnas.1314608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J., Jima D., Moffitt A.B., Liu Q., Czader M., Hsi E.D., Fedoriw Y., Dunphy C.H., Richards K.L., Gill J.I., et al. The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood. 2014;123:2988–2996. doi: 10.1182/blood-2013-07-517177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hill H.A., Qi X., Jain P., Nomie K., Wang Y., Zhou S., Wang M.L. Genetic mutations and features of mantle cell lymphoma: a systematic review and meta-analysis. Blood Adv. 2020;4:2927–2938. doi: 10.1182/bloodadvances.2019001350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu F., Guo H., Bates P.D., Zhang S., Zhang H., Nomie K.J., Li Y., Lu L., Seibold K.R., Wang F., et al. PRMT5 is upregulated by B-cell receptor signaling and forms a positive-feedback loop with PI3K/AKT in lymphoma cells. Leukemia. 2019;33:2898–2911. doi: 10.1038/s41375-019-0489-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kimpara S., Lu L., Hoang N.M., Zhu F., Bates P.D., Daenthanasanmak A., Zhang S., Yang D.T., Kelm A., Liu Y., et al. EGR1 Addiction in Diffuse Large B-cell Lymphoma. Mol. Cancer Res. 2021;19:1258–1269. doi: 10.1158/1541-7786.MCR-21-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li F., Wang Y., Zeller K.I., Potter J.J., Wonsey D.R., O'Donnell K.A., Kim J.W., Yustein J.T., Lee L.A., Dang C.V. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell Biol. 2005;25:6225–6234. doi: 10.1128/MCB.25.14.6225-6234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim J., Lee J.H., Iyer V.R. Global identification of Myc target genes reveals its direct role in mitochondrial biogenesis and its E-box usage in vivo. PLoS One. 2008;3 doi: 10.1371/journal.pone.0001798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morrish F., Hockenbery D. MYC and mitochondrial biogenesis. Cold Spring Harb. Perspect. Med. 2014;4 doi: 10.1101/cshperspect.a014225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dang C.V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 2013;3:a014217. doi: 10.1101/cshperspect.a014217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morrish F., Noonan J., Perez-Olsen C., Gafken P.R., Fitzgibbon M., Kelleher J., VanGilst M., Hockenbery D. Myc-dependent mitochondrial generation of acetyl-CoA contributes to fatty acid biosynthesis and histone acetylation during cell cycle entry. J. Biol. Chem. 2010;285:36267–36274. doi: 10.1074/jbc.M110.141606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bueno M., Lai Y.C., Romero Y., Brands J., St Croix C.M., Kamga C., Corey C., Herazo-Maya J.D., Sembrat J., Lee J.S., et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Invest. 2015;125:521–538. doi: 10.1172/JCI74942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Callender L.A., Carroll E.C., Bober E.A., Akbar A.N., Solito E., Henson S.M. Mitochondrial mass governs the extent of human T cell senescence. Aging Cell. 2020;19 doi: 10.1111/acel.13067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Z.-M., Lu R., Wang P., Yu Y., Chen D., Gao L., Liu S., Ji D., Rothbart S.B., Wang Y., et al. Structural basis for DNMT3A-mediated de novo DNA methylation. Nature. 2018;554:387–391. doi: 10.1038/nature25477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu J., Qin B., Moyer A.M., Nowsheen S., Liu T., Qin S., Zhuang Y., Liu D., Lu S.W., Kalari K.R., et al. DNA methyltransferase expression in triple-negative breast cancer predicts sensitivity to decitabine. J. Clin. Invest. 2018;128:2376–2388. doi: 10.1172/JCI97924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rinaldi L., Datta D., Serrat J., Morey L., Solanas G., Avgustinova A., Blanco E., Pons J.I., Matallanas D., Von Kriegsheim A., et al. Dnmt3a and Dnmt3b Associate with Enhancers to Regulate Human Epidermal Stem Cell Homeostasis. Cell Stem Cell. 2016;19:491–501. doi: 10.1016/j.stem.2016.06.020. [DOI] [PubMed] [Google Scholar]
- 37.Weinberg D.N., Papillon-Cavanagh S., Chen H., Yue Y., Chen X., Rajagopalan K.N., Horth C., McGuire J.T., Xu X., Nikbakht H., et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature. 2019;573:281–286. doi: 10.1038/s41586-019-1534-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pon J.R., Wong J., Saberi S., Alder O., Moksa M., Grace Cheng S.W., Morin G.B., Hoodless P.A., Hirst M., Marra M.A. MEF2B mutations in non-Hodgkin lymphoma dysregulate cell migration by decreasing MEF2B target gene activation. Nat. Commun. 2015;6:7953. doi: 10.1038/ncomms8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brescia P., Schneider C., Holmes A.B., Shen Q., Hussein S., Pasqualucci L., Basso K., Dalla-Favera R. MEF2B Instructs Germinal Center Development and Acts as an Oncogene in B Cell Lymphomagenesis. Cancer Cell. 2018;34:453–465.e9. doi: 10.1016/j.ccell.2018.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ying C.Y., Dominguez-Sola D., Fabi M., Lorenz I.C., Hussein S., Bansal M., Califano A., Pasqualucci L., Basso K., Dalla-Favera R. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat. Immunol. 2013;14:1084–1092. doi: 10.1038/ni.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weinberg D.N., Papillon-Cavanagh S., Chen H., Yue Y., Chen X., Rajagopalan K.N., Horth C., McGuire J.T., Xu X., Nikbakht H., et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature. 2019;573:281–286. doi: 10.1038/s41586-019-1534-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang K., Saito M., Bisikirska B.C., Alvarez M.J., Lim W.K., Rajbhandari P., Shen Q., Nemenman I., Basso K., Margolin A.A., et al. Genome-wide identification of post-translational modulators of transcription factor activity in human B cells. Nat. Biotechnol. 2009;27:829–839. doi: 10.1038/nbt.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pang Y., Liu J., Li X., Xiao G., Wang H., Yang G., Li Y., Tang S.C., Qin S., Du N., et al. MYC and DNMT3A-mediated DNA methylation represses microRNA-200b in triple negative breast cancer. J. Cell Mol. Med. 2018;22:6262–6274. doi: 10.1111/jcmm.13916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brenner C., Deplus R., Didelot C., Loriot A., Viré E., De Smet C., Gutierrez A., Danovi D., Bernard D., Boon T., et al. Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J. 2005;24:336–346. doi: 10.1038/sj.emboj.7600509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Larsson N.G., Wang J., Wilhelmsson H., Oldfors A., Rustin P., Lewandoski M., Barsh G.S., Clayton D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998;18:231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 46.Potthoff M.J., Olson E.N. MEF2: a central regulator of diverse developmental programs. Development. 2007;134:4131–4140. doi: 10.1242/dev.008367. [DOI] [PubMed] [Google Scholar]
- 47.Morin R.D., Mendez-Lago M., Mungall A.J., Goya R., Mungall K.L., Corbett R.D., Johnson N.A., Severson T.M., Chiu R., Field M., et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lohr J.G., Stojanov P., Lawrence M.S., Auclair D., Chapuy B., Sougnez C., Cruz-Gordillo P., Knoechel B., Asmann Y.W., Slager S.L., et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. USA. 2012;109:3879–3884. doi: 10.1073/pnas.1121343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pasqualucci L., Trifonov V., Fabbri G., Ma J., Rossi D., Chiarenza A., Wells V.A., Grunn A., Messina M., Elliot O., et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat. Genet. 2011;43:830–837. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pollyea D.A., Stevens B.M., Jones C.L., Winters A., Pei S., Minhajuddin M., D'Alessandro A., Culp-Hill R., Riemondy K.A., Gillen A.E., et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018;24:1859–1866. doi: 10.1038/s41591-018-0233-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DiNardo C.D., Jonas B.A., Pullarkat V., Thirman M.J., Garcia J.S., Wei A.H., Konopleva M., Döhner H., Letai A., Fenaux P., et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020;383:617–629. doi: 10.1056/NEJMoa2012971. [DOI] [PubMed] [Google Scholar]
- 52.Datta R., Heaster T.M., Sharick J.T., Gillette A.A., Skala M.C. Fluorescence lifetime imaging microscopy: fundamentals and advances in instrumentation, analysis, and applications. J. Biomed. Opt. 2020;25:1–43. doi: 10.1117/1.JBO.25.7.071203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koziol B., Markowicz M., Kruk J., Plytycz B. Riboflavin as a source of autofluorescence in Eisenia fetida coelomocytes. Photochem. Photobiol. 2006;82:570–573. doi: 10.1562/2005-11-23-RA-738. [DOI] [PubMed] [Google Scholar]
- 54.Ramanujam N. Fluorescence spectroscopy of neoplastic and non-neoplastic tissues. Neoplasia. 2000;2:89–117. doi: 10.1038/sj.neo.7900077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakashima N., Yoshihara K., Tanaka F., Yagi K. Picosecond fluorescence lifetime of the coenzyme of D-amino acid oxidase. J. Biol. Chem. 1980;255:5261–5263. [PubMed] [Google Scholar]
- 56.Walsh A.J., Mueller K.P., Tweed K., Jones I., Walsh C.M., Piscopo N.J., Niemi N.M., Pagliarini D.J., Saha K., Skala M.C. Classification of T-cell activation via autofluorescence lifetime imaging. Nat. Biomed. Eng. 2021;5:77–88. doi: 10.1038/s41551-020-0592-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mohanty A., Sandoval N., Phan A., Nguyen T.V., Chen R.W., Budde E., Mei M., Popplewell L., Pham L.V., Kwak L.W., et al. Regulation of SOX11 expression through CCND1 and STAT3 in mantle cell lymphoma. Blood. 2019;133:306–318. doi: 10.1182/blood-2018-05-851667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kantarjian H.M., O'Brien S., Cortes J., Giles F.J., Faderl S., Issa J.P., Garcia-Manero G., Rios M.B., Shan J., Andreeff M., et al. Results of decitabine (5-aza-2'deoxycytidine) therapy in 130 patients with chronic myelogenous leukemia. Cancer. 2003;98:522–528. doi: 10.1002/cncr.11543. [DOI] [PubMed] [Google Scholar]
- 59.Issa J.P.J., Gharibyan V., Cortes J., Jelinek J., Morris G., Verstovsek S., Talpaz M., Garcia-Manero G., Kantarjian H.M. Phase II study of low-dose decitabine in patients with chronic myelogenous leukemia resistant to imatinib mesylate. J. Clin. Oncol. 2005;23:3948–3956. doi: 10.1200/JCO.2005.11.981. [DOI] [PubMed] [Google Scholar]
- 60.Abaza Y., Kantarjian H., Alwash Y., Borthakur G., Champlin R., Kadia T., Garcia-Manero G., Daver N., Ravandi F., Verstovsek S., et al. Phase I/II study of dasatinib in combination with decitabine in patients with accelerated or blast phase chronic myeloid leukemia. Am. J. Hematol. 2020;95:1288–1295. doi: 10.1002/ajh.25939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kantarjian H., Issa J.P.J., Rosenfeld C.S., Bennett J.M., Albitar M., DiPersio J., Klimek V., Slack J., de Castro C., Ravandi F., et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106:1794–1803. doi: 10.1002/cncr.21792. [DOI] [PubMed] [Google Scholar]
- 62.Malik P., Cashen A.F. Decitabine in the treatment of acute myeloid leukemia in elderly patients. Cancer Manag. Res. 2014;6:53–61. doi: 10.2147/CMAR.S40600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Daver N., Kantarjian H., Ravandi F., Estey E., Wang X., Garcia-Manero G., Jabbour E., Konopleva M., O'Brien S., Verstovsek S., et al. A phase II study of decitabine and gemtuzumab ozogamicin in newly diagnosed and relapsed acute myeloid leukemia and high-risk myelodysplastic syndrome. Leukemia. 2016;30:268–273. doi: 10.1038/leu.2015.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bocchia M., Candoni A., Borlenghi E., Defina M., Filì C., Cattaneo C., Sammartano V., Fanin R., Sciumè M., Sicuranza A., et al. Real-world experience with decitabine as a first-line treatment in 306 elderly acute myeloid leukaemia patients unfit for intensive chemotherapy. Hematol. Oncol. 2019;37:447–455. doi: 10.1002/hon.2663. [DOI] [PubMed] [Google Scholar]
- 65.Filì C., Candoni A., Zannier M.E., Olivieri J., Imbergamo S., Caizzi M., Nadali G., Di Bona E., Ermacora A., Gottardi M., et al. Efficacy and toxicity of Decitabine in patients with acute myeloid leukemia (AML): A multicenter real-world experience. Leuk. Res. 2019;76:33–38. doi: 10.1016/j.leukres.2018.11.015. [DOI] [PubMed] [Google Scholar]
- 66.Lam T.G., Jeong Y.S., Kim S.A., Ahn S.G. New metformin derivative HL156A prevents oral cancer progression by inhibiting the insulin-like growth factor/AKT/mammalian target of rapamycin pathways. Cancer Sci. 2018;109:699–709. doi: 10.1111/cas.13482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Izreig S., Gariepy A., Kaymak I., Bridges H.R., Donayo A.O., Bridon G., DeCamp L.M., Kitchen-Goosen S.M., Avizonis D., Sheldon R.D., et al. Repression of LKB1 by miR-17 approximately 92 Sensitizes MYC-Dependent Lymphoma to Biguanide Treatment. Cell Rep. Med. 2020;1 doi: 10.1016/j.xcrm.2020.100014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Janku F., Beom S.H., Moon Y.W., Kim T.W., Shin Y.G., Yim D.S., Kim G.M., Kim H.S., Kim S.Y., Cheong J.H., et al. First-in-human study of IM156, a novel potent biguanide oxidative phosphorylation (OXPHOS) inhibitor, in patients with advanced solid tumors. Invest. N. Drugs. 2022;40:1001–1010. doi: 10.1007/s10637-022-01277-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kang B.H., Siegelin M.D., Plescia J., Raskett C.M., Garlick D.S., Dohi T., Lian J.B., Stein G.S., Languino L.R., Altieri D.C. Preclinical characterization of mitochondria-targeted small molecule hsp90 inhibitors, gamitrinibs, in advanced prostate cancer. Clin. Cancer Res. 2010;16:4779–4788. doi: 10.1158/1078-0432.CCR-10-1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fiesel F.C., James E.D., Hudec R., Springer W. Mitochondrial targeted HSP90 inhibitor Gamitrinib-TPP (G-TPP) induces PINK1/Parkin-dependent mitophagy. Oncotarget. 2017;8:106233–106248. doi: 10.18632/oncotarget.22287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Siegelin M.D., Dohi T., Raskett C.M., Orlowski G.M., Powers C.M., Gilbert C.A., Ross A.H., Plescia J., Altieri D.C. Exploiting the mitochondrial unfolded protein response for cancer therapy in mice and human cells. J. Clin. Invest. 2011;121:1349–1360. doi: 10.1172/JCI44855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kang B.H., Plescia J., Dohi T., Rosa J., Doxsey S.J., Altieri D.C. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell. 2007;131:257–270. doi: 10.1016/j.cell.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 73.Kang B.H., Plescia J., Song H.Y., Meli M., Colombo G., Beebe K., Scroggins B., Neckers L., Altieri D.C. Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J. Clin. Invest. 2009;119:454–464. doi: 10.1172/JCI37613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ianevski A., Giri A.K., Aittokallio T. SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020;48:W488–W493. doi: 10.1093/nar/gkaa216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu Y., Kimpara S., Hoang N.M., Daenthanasanmak A., Li Y., Lu L., Ngo V.N., Bates P.D., Song L., Gao X., et al. EGR1-mediated metabolic reprogramming to oxidative phosphorylation contributes to ibrutinib resistance in B-cell lymphoma. Blood. 2023;142:1879–1894. doi: 10.1182/blood.2023020142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu F., Yoshida N., Suguro M., Kato H., Karube K., Arita K., Yamamoto K., Tsuzuki S., Oshima K., Seto M. Clonal heterogeneity of mantle cell lymphoma revealed by array comparative genomic hybridization. Eur. J. Haematol. 2013;90:51–58. doi: 10.1111/ejh.12030. [DOI] [PubMed] [Google Scholar]
- 77.Vasanthakumar A., Lepore J.B., Zegarek M.H., Kocherginsky M., Singh M., Davis E.M., Link P.A., Anastasi J., Le Beau M.M., Karpf A.R., Godley L.A. Dnmt3b is a haploinsufficient tumor suppressor gene in Myc-induced lymphomagenesis. Blood. 2013;121:2059–2063. doi: 10.1182/blood-2012-04-421065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Haney S.L., Upchurch G.M., Opavska J., Klinkebiel D., Hlady R.A., Roy S., Dutta S., Datta K., Opavsky R. Dnmt3a Is a Haploinsufficient Tumor Suppressor in CD8+ Peripheral T Cell Lymphoma. PLoS Genet. 2016;12 doi: 10.1371/journal.pgen.1006334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baccelli I., Gareau Y., Lehnertz B., Gingras S., Spinella J.F., Corneau S., Mayotte N., Girard S., Frechette M., Blouin-Chagnon V., et al. Mubritinib Targets the Electron Transport Chain Complex I and Reveals the Landscape of OXPHOS Dependency in Acute Myeloid Leukemia. Cancer Cell. 2019;36:84–99.e8. doi: 10.1016/j.ccell.2019.06.003. [DOI] [PubMed] [Google Scholar]
- 80.De Cecco M., Ito T., Petrashen A.P., Elias A.E., Skvir N.J., Criscione S.W., Caligiana A., Brocculi G., Adney E.M., Boeke J.D., et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature. 2019;566:73–78. doi: 10.1038/s41586-018-0784-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ku Y., Park J.H., Cho R., Lee Y., Park H.M., Kim M., Hur K., Byun S.Y., Liu J., Lee Y.S., et al. Noncanonical immune response to the inhibition of DNA methylation by Staufen1 via stabilization of endogenous retrovirus RNAs. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2016289118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chiappinelli K.B., Strissel P.L., Desrichard A., Li H., Henke C., Akman B., Hein A., Rote N.S., Cope L.M., Snyder A., et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162:974–986. doi: 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Roulois D., Loo Yau H., Singhania R., Wang Y., Danesh A., Shen S.Y., Han H., Liang G., Jones P.A., Pugh T.J., et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell. 2015;162:961–973. doi: 10.1016/j.cell.2015.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu M., Ohtani H., Zhou W., Ørskov A.D., Charlet J., Zhang Y.W., Shen H., Baylin S.B., Liang G., Grønbæk K., Jones P.A. Vitamin C increases viral mimicry induced by 5-aza-2′-deoxycytidine. Proc. Natl. Acad. Sci. USA. 2016;113:10238–10244. doi: 10.1073/pnas.1612262113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Maes K., De Smedt E., Lemaire M., De Raeve H., Menu E., Van Valckenborgh E., McClue S., Vanderkerken K., De Bruyne E. The role of DNA damage and repair in decitabine-mediated apoptosis in multiple myeloma. Oncotarget. 2014;5:3115–3129. doi: 10.18632/oncotarget.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shin D.Y., Sung Kang H., Kim G.Y., Kim W.J., Yoo Y.H., Choi Y.H. Decitabine, a DNA methyltransferases inhibitor, induces cell cycle arrest at G2/M phase through p53-independent pathway in human cancer cells. Biomed. Pharmacother. 2013;67:305–311. doi: 10.1016/j.biopha.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 87.Dreyling M., Doorduijn J.K., Gine E., Jerkeman M., Walewski J., Hutchings M., Mey U., Riise J., Trneny M., Vergote V.K., et al. Efficacy and Safety of Ibrutinib Combined with Standard First-Line Treatment or As Substitute for Autologous Stem Cell Transplantation in Younger Patients with Mantle Cell Lymphoma: Results from the Randomized Triangle Trial By the European MCL Network. Blood. 2022;140:1–3. [Google Scholar]
- 88.Ruan J., Leonard J.P., Chen G.Z., Chen Z., Allan J.N., Rutherford S.C., Hobbie B., Greig K., Rodriguez A., Tam W., et al. Phase 2 Trial of Acalabrutinib-Lenalidomide-Rituximab (ALR) with Real-Time Monitoring of MRD in Patients with Treatment-Naïve Mantle Cell Lymphoma. Blood. 2022;140:175–177. [Google Scholar]
- 89.Luo Y., Hitz B.C., Gabdank I., Hilton J.A., Kagda M.S., Lam B., Myers Z., Sud P., Jou J., Lin K., et al. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 2020;48:D882–D889. doi: 10.1093/nar/gkz1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ryan R.J.H., Petrovic J., Rausch D.M., Zhou Y., Lareau C.A., Kluk M.J., Christie A.L., Lee W.Y., Tarjan D.R., Guo B., et al. A B Cell Regulome Links Notch to Downstream Oncogenic Pathways in Small B Cell Lymphomas. Cell Rep. 2017;21:784–797. doi: 10.1016/j.celrep.2017.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Caeser R., Di Re M., Krupka J.A., Gao J., Lara-Chica M., Dias J.M.L., Cooke S.L., Fenner R., Usheva Z., Runge H.F.P., et al. Genetic modification of primary human B cells to model high-grade lymphoma. Nat. Commun. 2019;10:4543. doi: 10.1038/s41467-019-12494-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen Z.X., Mann J.R., Hsieh C.L., Riggs A.D., Chédin F. Physical and functional interactions between the human DNMT3L protein and members of the de novo methyltransferase family. J. Cell. Biochem. 2005;95:902–917. doi: 10.1002/jcb.20447. [DOI] [PubMed] [Google Scholar]
- 93.Li H., Rauch T., Chen Z.X., Szabó P.E., Riggs A.D., Pfeifer G.P. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J. Biol. Chem. 2006;281:19489–19500. doi: 10.1074/jbc.M513249200. [DOI] [PubMed] [Google Scholar]
- 94.Pertea M., Pertea G.M., Antonescu C.M., Chang T.C., Mendell J.T., Salzberg S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gaspar J. Genrich: Detecting Sites of Genomic Enrichment. 2018. https://github.com/jsh58/Genrich
- 97.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang Q., Li M., Wu T., Zhan L., Li L., Chen M., Xie W., Xie Z., Hu E., Xu S., Yu G. Exploring Epigenomic Datasets by ChIPseeker. Curr. Protoc. 2022;2:e585. doi: 10.1002/cpz1.585. [DOI] [PubMed] [Google Scholar]
- 99.Yu G., Wang L.G., He Q.Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–2383. doi: 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- 100.Benjamini Y., Speed T.P. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 2012;40:e72. doi: 10.1093/nar/gks001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pachitariu M., Stringer C. Cellpose 2.0: how to train your own model. Nat. Methods. 2022;19:1634–1641. doi: 10.1038/s41592-022-01663-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stringer C., Wang T., Michaelos M., Pachitariu M. Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods. 2021;18:100–106. doi: 10.1038/s41592-020-01018-x. [DOI] [PubMed] [Google Scholar]
- 103.Liao J., Karnik R., Gu H., Ziller M.J., Clement K., Tsankov A.M., Akopian V., Gifford C.A., Donaghey J., Galonska C., et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat. Genet. 2015;47:469–478. doi: 10.1038/ng.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rui L., Drennan A.C., Ceribelli M., Zhu F., Wright G.W., Huang D.W., Xiao W., Li Y., Grindle K.M., Lu L., et al. Epigenetic gene regulation by Janus kinase 1 in diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA. 2016;113:E7260–E7267. doi: 10.1073/pnas.1610970113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Huang X., Darzynkiewicz Z. Cytometric assessment of histone H2AX phosphorylation: a reporter of DNA damage. Methods Mol. Biol. 2006;314:73–80. doi: 10.1385/1-59259-973-7:073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Walsh A.J., Poole K.M., Duvall C.L., Skala M.C. Ex vivo optical metabolic measurements from cultured tissue reflect in vivo tissue status. J. Biomed. Opt. 2012;17 doi: 10.1117/1.JBO.17.11.116015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lakowicz J.R., Szmacinski H., Nowaczyk K., Johnson M.L. Fluorescence lifetime imaging of free and protein-bound NADH. Proc. Natl. Acad. Sci. USA. 1992;89:1271–1275. doi: 10.1073/pnas.89.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Georgakoudi I., Quinn K.P. Optical imaging using endogenous contrast to assess metabolic state. Annu. Rev. Biomed. Eng. 2012;14:351–367. doi: 10.1146/annurev-bioeng-071811-150108. [DOI] [PubMed] [Google Scholar]
- 111.Skala M.C., Riching K.M., Gendron-Fitzpatrick A., Eickhoff J., Eliceiri K.W., White J.G., Ramanujam N. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc. Natl. Acad. Sci. USA. 2007;104:19494–19499. doi: 10.1073/pnas.0708425104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chance B., Schoener B., Oshino R., Itshak F., Nakase Y. Oxidation-reduction ratio studies of mitochondria in freeze-trapped samples. NADH and flavoprotein fluorescence signals. J. Biol. Chem. 1979;254:4764–4771. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Mutations in DNMT, BTK, MEF2B, and NSD2. (B) Mutations in frequently mutated genes in MCL. (C) Mutations in metabolic-related genes, related to Figure 4
Data Availability Statement
-
•
RNA-seq, ChIP-seq from MCL cell lines, and WES data from MCL patients generated have been deposited at GEO and SRA, respectively, and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.