

Summary
Targeted therapies have improved outcomes for certain cancer subtypes, but cytotoxic chemotherapy remains a mainstay for triple-negative breast cancer (TNBC). The epithelial-to-mesenchymal transition (EMT) is a developmental program co-opted by cancer cells that promotes metastasis and chemoresistance. There are no therapeutic strategies specifically targeting mesenchymal-like cancer cells. We report that the US Food and Drug Administration (FDA)-approved chemotherapeutic eribulin induces ZEB1-SWI/SNF-directed chromatin remodeling to reverse EMT that curtails the metastatic propensity of TNBC preclinical models. Eribulin induces mesenchymal-to-epithelial transition (MET) in primary TNBC in patients, but conventional chemotherapy does not. In the treatment-naive setting, but not after acquired resistance to other agents, eribulin sensitizes TNBC cells to subsequent treatment with other chemotherapeutics. These findings provide an epigenetic mechanism of action of eribulin, supporting its use early in the disease process for MET induction to prevent metastatic progression and chemoresistance. These findings warrant prospective clinical evaluation of the chemosensitizing effects of eribulin in the treatment-naive setting.
Keywords: breast cancer, chemotherapy, EMT, epithelial-to-mesenchymal transition, mesenchymal-to-epithelial transition, drug resistance, metastasis, epigenetic
Graphical abstract
Highlights
-
•
FDA-approved drug eribulin induces ZEB1-SWI/SNF-directed chromatin remodeling
-
•
Remodeling drives mesenchymal-to-epithelial transition in triple-negative breast tumors
-
•
Eribulin sensitizes triple-negative breast cancer cells to other chemotherapeutics
Bagheri et al. demonstrate that the FDA-approved microtubule inhibitor eribulin, when applied in the treatment-naive setting, induces epigenetic remodeling to reverse the epithelial-to-mesenchymal transition, prevent metastasis, and sensitize breast cancer cells to other chemotherapeutics. These findings suggest that the order of exposure to chemotherapeutics can shape cancer cell biology.
Introduction
The epithelial-to-mesenchymal transition (EMT) is a cellular program that imparts plasticity to epithelial cells, enabling them to acquire traits such as motility and invasion.1 EMT is an integral step in developmental processes such as gastrulation and neural crest migration and normal processes such as wound healing. Cancer cells frequently co-opt EMT, and its roles in tumor progression are well studied, including in triple-negative breast cancers (TNBCs) that lack expression of estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2. In addition to contributing to the local invasion of primary tumor cells and enabling intravasation into blood/lymphatic vessels, EMT plays roles in tumor cell extravasation and chemoresistance.1,2 Despite our understanding of EMT and the signaling pathways and transcriptional networks that regulate this plasticity in diverse cellular contexts, the impact of EMT research on therapeutic targeting of cancers has been minimal. While there are US Food and Drug Administration (FDA)-approved therapies that may modulate EMT, there are no approved drugs administered specifically for their ability to modulate EMT. Indeed, there is a gap between (1) our biological understanding of EMT and (2) translation of that knowledge to improve clinical outcomes.
Eribulin (ERI) is a microtubule dynamics inhibitor administered to patients with advanced/metastatic breast cancer in the third-line setting or later.3 These patients have often received prior chemotherapy that included an anthracycline and a taxane. The proposed primary mechanism of ERI action is inhibition of tubulin addition to the plus end of microtubules, thereby inducing mitotic blockade.4 Through a seemingly unconnected molecular mechanism, ERI has been shown to induce mesenchymal-to-epithelial transition (MET) in some models5,6,7 and also in combination therapy,8 but the underlying molecular mechanism and therapeutic consequences remain unclear. We thus undertook studies using TNBC cell lines, murine and patient-derived xenograft (PDX) models, and patient-matched primary human tumor specimens acquired before and after neoadjuvant therapy to illuminate the MET-inducing mechanism of ERI action and identify therapeutic strategies to capitalize upon this mechanism.
Results
Therapeutic induction of MET in breast cancer cells
Given the previously demonstrated role of eribulin in the modulation of the EMT state,5 we sought to compare its effects with those of other chemotherapeutics within the same class of microtubule dynamics inhibitors. Paclitaxel (PAC) is used in neoadjuvant chemotherapy for breast cancer, and vinorelbine (VIN) and ERI are administered to treat advanced breast cancers. All 3 drugs inhibited the growth of PB3 cancer cells derived from a mouse mammary tumor virus promoter-driven polyoma virus middle T antigen (MMTV-PyMT) transgenic murine triple-negative mammary tumor (Figure 1A), which resides in a quasi-mesenchymal state.9 To mimic clinical treatment regimens in the development of drug-resistant cell lines, chemotherapeutics were applied intermittently to PB3 cells for 3 cycles, allowing for intervening drug holidays for cells to recover (Figure 1B). Treatment with eribulin at its half-maximal inhibitory concentration (IC50), but not with vinorelbine or paclitaxel at their IC50, led to the emergence of a drug-resistant cell population (PB3ERI-R) exhibiting upregulated levels of epithelial cell markers (i.e., epithelial cell adhesion marker [EpCAM] and E-cadherin), downregulated levels of mesenchymal markers (vimentin and Zeb1), and an epithelial cobblestone-like morphology compared with PB3 controls, vinorelbine-resistant derivatives (PB3VIN-R), and paclitaxel-resistant derivatives (PB3PAC-R) (Figures 1C and S1A–S1C). The altered phenotype of ERI-R cells was accompanied by decreased migration and invasion potential compared with PB3 controls (Figures 1D, 1E, and S1D). Chemotherapeutics were removed from drug-resistant cells 4 weeks prior to seeding for assays, indicating that these phenotypes were stable and not due to the continued presence of drugs. Similar effects of eribulin treatment were observed in human TNBC cells; ERI-R derivatives of MDA-MB-231 and SUM159PT cells showed increased E-cadherin and decreased VIM and ZEB1 (Figures 1F, 1G, and S1E–S1I).
Figure 1.

Eribulin induces MET in TNBC cells
(A) Dose-response curves in PB3 cells were generated to calculate IC50 values for eribulin, paclitaxel, and vinorelbine.
(B) Schematic of the multiround drug treatment procedure that resulted in the generation of drug-resistant cells.
(C–G) Estimation of EMT state was carried out by immunoblotting (C, F, and G), Transwell invasion assay 8 h post-seeding (D), and wound closure assay 16 h post seeding (E). Data are shown as mean of biological triplicates ± SD.
(H–K) PB3 parental and drug-resistant cells were implanted orthotopically. In (H), tumor volumes were serially measured. Tumor-initiating capacity (TIC) was assessed by limiting dilution transplantation (I). In (J), metastatic ability was assessed in lungs harvested from mice in (H) and stained with H&E. Metastases were enumerated in (K). Data in (H and K) are shown as mean of 10 mice ± SD. ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001; ns, not significant by Tukey-adjusted pairwise comparison.
Orthotopic implantation of parental PB3 cells or isogenic PB3VIN-R or PB3PAC-R cells into female non-obese diabetic (NOD)/severe combined immunodeficiency (SCID)/IL2Rγ−/− (NSG) mice resulted in robust tumor growth. However, PB3ERI-R cells did not readily form tumors and exhibited a more than 100,000-fold reduction in tumor initiation ability (Figures 1H and I). Furthermore, PB3ERI-R cells did not form spontaneous lung metastases despite the other cell lines yielding abundant lung lesions established from mammary fat pad implantation (Figures 1J and 1K), which may have resulted from differences in primary tumor sizes; however, PB3ERI-R cells were not able to initiate tumors. Cells that show CD44hi/CD104+ markers (such as MDA-MB-231, MDA-MB-231VIN-R, and MDA-MB-231PAC-R) are known to have a greater ability to initiate tumors compared with cells with CD44lo/CD104+ markers (like MDA-MB-231ERI-R). These cell groups can be classed as purely epithelial cells (CD44lo/CD104+, like MDA-MB-231ERI-R), quasi-mesenchymal to mesenchymal cells (CD44hi/CD104+, like MDA-MB-231VIN-R and MDA-MB-231PAC-R), and highly mesenchymal cells (CD44hi/CD104−).10 MDA-MB-231ERI-R cells also displayed a decrease in the cancer stem cell marker CD44 (Figure S1J), aligning with the reduced observed ability to start tumors in PB3ERI-R cells (Figures 1H and I). These results confirm the ability of eribulin to induce MET in breast cancer cells, driving cells toward an epithelial state accompanied by a significant reduction in tumor initiation potential.
Eribulin drives MET in human TNBC
We evaluated TNBC diagnostic core biopsy (pre-treatment) and surgical (post-treatment) specimens from patients treated with neoadjuvant chemotherapy. SOLTI1007 NeoEribulin specimens were obtained from 55 patients who received 4 cycles of neoadjuvant eribulin.11 Comparator specimens were from 15 patients who received standard-of-care neoadjuvant treatment with 4 cycles of adriamycin/cyclophosphamide followed by 4 cycles of paclitaxel (AC-T).
Specimens were multiplex immunostained for 6 EMT markers.12 To enumerate ratios of epithelial and mesenchymal cell types, cells were grouped based on marker expression (Figures 2A and B). In addition, we identified two new phenotypes (KRT8+, and ZEB1+/VIM+) that offer a more comprehensive representation of marker expression patterns. AC-T-treated tumors did not undergo major shifts in subtype proportions. In contrast, eribulin-treated tumors exhibited ∼20% and ∼30% increases in expression of E-cadherin [E-cad]+/KRT8+ and KRT8+ epithelial phenotypes, respectively, commensurate with decreases of ∼50% in VIM+ and ZEB1+/VIM+ mesenchymal phenotypes (Figure 2B).
Figure 2.

Eribulin promotes MET in human TNBC
Multiplexed, multiround TSA staining of EMT markers was performed in TNBC specimens acquired from patients before and after neoadjuvant treatment with eribulin or AC-T.
(A) Representative images.
(B) Proportions of tumor cells expressing markers of EMT phenotypes.
(C) EMT scores of tumors. ∗∗∗∗p < 0.0001; ns, not significant by Tukey-adjusted pairwise comparison.
EMT scores were generated for each tumor specimen, with “0” being most epithelial and “1” being most mesenchymal. Tumors treated with neoadjuvant eribulin showed a 41.7% mean decrease in EMT score compared with baseline (p < 0.0001), reflecting a shift toward an epithelial phenotype. In contrast, neoadjuvant AC-T did not significantly alter the EMT score (Figure 2C).
Clonal dynamics reveal induction of the epithelial-like phenotype in chemotherapy-resistant cells
Drug treatment of a heterogeneous cell population results in a diverse array of fates that are determined in part by the starting transcriptional and epigenetic states of each cell. To elucidate the cellular trajectories that result from drug treatment, we developed an approach to query indexed sequences and simultaneously measure epigenetic timelines (QISSMET). Similar approaches have been utilized to trace cell lineage through therapeutic bottlenecks.13,14 By analyzing changes in transcriptional and chromatin accessibility profiles induced by drug selection, QISSMET enables differentiation between pre-existing drug-resistant cells and emerging de novo cell types with acquired resistance. Changes that a given cell undergoes upon drug treatment would manifest as changes in gene expression and chromatin state, which would imply epigenetic reprogramming leading to the emergence of a de novo cell state. Alternatively, if no changes in gene expression or chromatin state are observed, this would suggest that a selection event had occurred, amplifying a pre-existing drug-resistant population.
To characterize the nature of drug-resistant cells, we stably transfected PB3 cells with an expressed barcode library. Barcoded cells were subject to 3–4 cycles of treatment with eribulin or paclitaxel (Figure 3A), and an aliquot of cells was harvested before each cycle for single-cell RNA sequencing (scRNA-seq). Eribulin treatment resulted in the emergence of cells (ERI3 and ERI4) that clustered distinctly from the untreated parental population by transcriptomic analysis, while paclitaxel did not (Figure 3B). Monocle pseudotime kinetics15,16,17 revealed that cells undergoing eribulin treatment took multiple trajectories from the most primitive (parental) to the most advanced (ERI4) transcriptional state (Figure 3C). In contrast, cells undergoing paclitaxel treatment took fewer distinct paths and remained more akin to parental controls without uniform directionality (Figure 3D).
Figure 3.

Clonal dynamics following drug treatment
(A) Schematic outlining genetic barcoding of cells and subsequent drug treatment strategy.
(B) UMAP projection of scRNA-seq data obtained from PB3 cells before and after 1–4 rounds of treatment with eribulin or paclitaxel.
(C and D) Monocle pseudotime projection showing the trajectory of cancer cell evolution upon drug treatment. The main lineage path or master path/node is indicated by “1” in a white circle. Sublineages (leaf nodes) from the master path are indicated by numbers in gray circles. Branches from leaf nodes are indicated by numbers in black circles. Left and right: cells colored according to pseudotime and sample, respectively.
(E–G) Euclidean distance between ERI4 vs. untreated cells (E) or PAC3 vs. untreated cells (F), with the same barcode are shown in swimmer plots, and UMAPs represent median points of ERI4 or PAC3 and untreated cells. ∗∗∗∗p < 0.0001 by Mann-Whitney U test.
(H) Proportions of induction vs. selection resistance for ERI and PAC treatments were compared by chi-square test.
(I and J) Muller plots outlining clonal diversity of the 10 most abundant barcodes following drug treatment.
We sought to determine whether eribulin treatment led to (1) an induction event that generated resistant cells with altered chromatin and transcriptional profiles or (2) the selection of pre-existing drug-resistant cells that were epigenetically/transcriptionally similar to the parental population. We selected the 28 most highly enriched barcodes that contained 1 or more cells in both the control and drug-treated clusters, and we calculated their Euclidean distances based on median positions on uniform manifold approximation and projection (UMAP) plots. Drug-resistant cells with barcodes that had larger Euclidean distances resided in the far left (“induction” region) of the UMAP, while drug-resistant cells with barcodes that had smaller Euclidean distances resided in the bottom island (“selection” region) of the UMAP (Figures 3E and 3F). Cells treated with eribulin had significantly larger Euclidian distances than cells treated with paclitaxel (median, 11.74 vs. 5.59; Figure 3G). Using a combination of Euclidian distance and Jaccard index, we calculated that 92% of barcodes in eribulin-resistant cells localized to the induction region, indicating that they underwent a shift in transcriptional profile as a result of eribulin treatment. In contrast, paclitaxel-resistant cells were transcriptionally more similar to the parental population, with 50% of barcodes localizing to the selection region (Figures 3H and S2A–S2E).
In addition to exhibiting altered transcriptional profiles, the epithelial-like cell subpopulations that emerged from eribulin treatment also exhibited altered chromatin landscapes, as observed by single-cell assay for transposase-accessible chromatin using sequencing (scATAC-seq; Figures S2F and S2G). Plotting the evolutionary trajectories of cells as they underwent drug treatments revealed that both agents imposed severe bottlenecks that only a few barcodes (i.e., individual cells) overcame. While eribulin treatment resulted in the emergence of a dominant clonal subpopulation, clonal frequencies were preserved with less variation upon paclitaxel treatment (Figures 3I and 3J). These results indicate that, in comparison with paclitaxel, eribulin treatment led to the evolution of resistant cells that were distinct from parental cells, consistent with their having undergone MET (Figure 1) and reprogramming of transcriptional and chromatin states.
Treatment with eribulin induces a shift in the chromatin profiles of cancer cells
In light of the observed functional properties of eribulin, we sought to further explore its mechanism of action that contributes to its ability to induce MET and therapeutic consequences. PB3ERI-R cells exhibited a significantly altered bulk transcriptomic profile relative to parental PB3, PB3PAC-R, and PB3VIN-R cells (Figures 4A, 4B, and S3A). Gene set enrichment analysis revealed significant enrichment for hallmark gene sets involved in EMT, the majority of which were downregulated in PB3ERI-R cells compared with parental controls (Figures 4C and D).
Figure 4.

Eribulin induces a shift in transcriptional and chromatin profiles of TNBC cells
(A–D) PB3 parental and drug-resistant cells were analyzed by bulk RNA-seq in biological duplicates. In (A), a heatmap shows the top 500 most differentially expressed genes. Genes significantly altered in drug-resistant cells vs. parental controls (log2-fold-change indicates mean expression level; Benjamini-Hochberg-corrected p value threshold = 0.01) are highlighted in volcano plots in (B). A dot plot outlining enrichment of hallmark gene sets in ERI-R vs. control cells is shown in (C); circle size depicts pathway significance, and red and blue dots indicate activated and suppressed pathways, respectively. Shown in (D) is a heatmap of EMT and chromatin organization genes in hallmark gene sets.
(E) Immunoblot of lysates from PB3 and MDA-MB-231 parental and drug-resistant cells.
(F) Top: average CUT&RUN enrichment profile of H3K4me3 in PB3 control and drug-resistant cells. Bottom: heatmap of CUT&RUN signal ±4 kb of peaks.
(G) H3K4me3 localization at genomic loci of canonical EMT genes as evaluated by CUT&RUN signal track analysis.
(H and I) Bulk ATAC-seq was performed in PB3 parental and drug-resistant cells. Peak accessibility surrounding all consensus regions (H, top), promoter-associated regions (H, bottom), and EMT marker genes (I) is shown.
(J) ATAC-seq and RNA-seq integration analysis. Shown are advanced volcano plots of highly significant transcription factors (TFs) as determined by DiffTF from ATAC-seq (x axis) and log2fold change in gene expression of TFs (y axis). TF classification is indicated by circle color. The number of TF binding sites used to determine TF activity is indicated by circle size.
We reasoned that transcriptomic changes may result from alterations in chromatin state. Indeed, the levels of the histone 3 lysine 4 trimethylation (H3K4me3) activation mark and the H3K27me3 repressive mark were both reduced in ERI-R derivatives, but not PAC-R or VIN-R derivatives, from PB3, MDA-MB-231, and SUM159 TNBC cells (Figures 4E, and S3B). H3K4me3 cistrome profiling revealed overall reduced representation of this activating histone mark in PB3ERI-R cells (Figure 4F). However, this reduction in H3K4me3-DNA binding was not universal, with some genomic loci coding for epithelial markers such as Cdh1, Grhl1, Ovol1, and Ovol2 gaining this mark in promoter regions in PB3ERI-R cells but not PB3PAC-R or PB3VIN-R cells (Figure 4G).
Assessing the chromatin accessibility landscape of cells by bulk ATAC-seq also revealed a more closed chromatin profile with fewer and different accessible regions of chromatin in PB3ERI-R cells compared with parental controls (Figures 4H, S3C and S3D). Consistent with H3K4me3 profiling, chromatin surrounding loci encoding epithelial markers (Cdh1, Ovol1, and Ovol2) was more accessible in PB3ERI-R cells (Figure 4I). Integrated analysis of bulk RNA-seq and ATAC-seq data with DiffTF18 inferred large-scale changes in the activity of key transcription factors in PB3ERI-R cells compared with parental controls but not in PB3VIN-R or PB3PAC-R cells (Figures 4J, S3E, and S3F). These data collectively point to eribulin treatment resulting in more pronounced reprogramming of the chromatin and transcriptional states of TNBC cells compared with other microtubule dynamics inhibitors, suggesting alternate mechanisms of drug action.
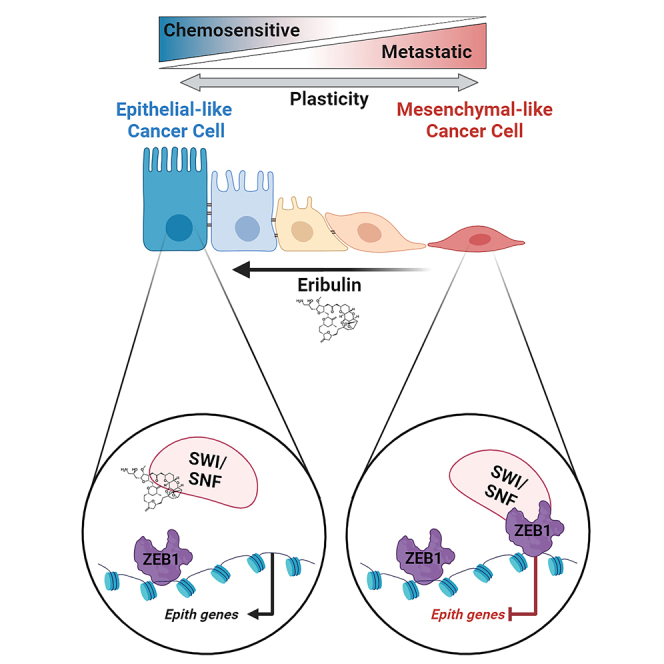
ZEB1-SWI/SNF interactions are required for maintenance of the mesenchymal state
To identify protein targets involved in chromatin/transcriptional responses to eribulin, we conducted an unbiased proteome integral solubility alteration (PISA) assay19 to identify proteins that exhibit altered thermal stability within 4 h in the presence of eribulin in PB3 cells. Among 6,527 proteins detected in all samples, 40 proteins had significant (p < 0.05) alterations in thermal stability (ΔSm ≥ 0.5) due to eribulin treatment (Tables S1 and S2). We focused validation studies on proteins with known roles in chromatin regulation. Smrd1 (Smarcd1) and Smrd3 (Smarcd3) are switch/sucrose non-fermentable (SWI/SNF)-family ATP-dependent chromatin remodelers that exhibited thermal stability shifts upon eribulin treatment, indicating that they may be bound by the drug (Figures 5A and S4A). CRISPR-Cas9-mediated knockout of Smarcd1, Smarcd2, or Smarcd3 (individually and all three together) in PB3 cells reduced the spindle-shaped (mesenchymal-like) morphology, reduced Zeb1 levels, and increased E-cad (Figures 5B, S4B, and S4C). Triple knockout of Smarcd1/2/3 phenocopied eribulin treatment, shifting PB3 and MDA-MB-231 cells toward an epithelial state (Figures 5B, S4D, and S4E) and phenocopied eribulin-induced decreases in H3K4me3 and H3K27me3 (Figure 5C). Rescue through the expression of exogenous human SMARCD1 in Smarcd1 knockout PB3 cells suppressed E-cad levels and restored Zeb1, indicating that SMARCD1/Smrd1 signaling drives a mesenchymal state (Figure S4F).
Figure 5.

ZEB1-SWI/SNF interactions are necessary for maintenance of a mesenchymal state
(A) PB3 cells treated with or without 300 nM eribulin for 4 h were analyzed by PISA assay. Proteins were plotted by ΔSm and −log10p (t test).
(B and C) Immunoblot analysis of cells with CRISPR-Cas9-mediated deletion of SmarcD1 (D1), SmarcD3 (D3), or SmarcD1/2/3.
(D) Zeb1 and Smarcc1 immunoprecipitates and lysates from cells treated with or without eribulin for 48 h.
(E–G) Genome-wide occupancy of Zeb1 in PB3 cells treated with or without eribulin for 24, 48, or 72 h was evaluated using CUT&RUN. In (E), average CUT&RUN enrichment profiles (top) and heatmap of the CUT&RUN signal ±2 kb of peaks (bottom) are shown. Shown in (F) are UpSet plots of differentially accessible peaks at the indicated time points as mean ± SEM. Signal tracks of Zeb1 localization at target gene loci are shown in (G).
Previous work has shown that members of the SWI/SNF family can interact with the transcription factor ZEB1, enabling repression of E-cad (CDH1) transcription and inducing EMT.20 We hypothesized that the specificity of SMARC proteins for EMT traits could be a result of interaction with ZEB1 to modulate ZEB1 function. Indeed, we observed Zeb1/ZEB1 interactions with Smarcd1/SMARCD1, Smarcd3/SMARCD3, and Smarcc1/SMARCC1 in PB3 and MDA-MB-231 cells (Figures 5D, S4G, and S4H). These interactions were altered by treatment with eribulin.
We tested the effects of interactions between Zeb1/Smarcd1 and Zeb1/Smarcc1 on DNA binding and transcriptional activity. In PB3 and MDA-MB-231 cells, eribulin progressively reduced Zeb1/ZEB1 binding to DNA across the genome (Figure 5E, 5F, S4I, and S4J). Eribulin elicited a time-dependent reduction in Zeb1 binding to the promoter regions of the epithelial target genes Cdh1 and Mmp2 and to other targets (Sox9, Vegfa, and Jun)21,22 (Figures 5G and S4K). These results point to roles of Smarcd1 and Smarcc1 as essential Zeb1 co-factors for the maintenance of a mesenchymal state.
Order of exposure to chemotherapeutics alters efficacy
Current treatment strategies for stage I-III TNBC often include the administration of neoadjuvant chemotherapy (NAC) consisting of several cycles of an anthracycline (e.g., adriamycin) with cyclophosphamide, followed by several cycles of a taxane (e.g., paclitaxel) prior to surgical resection of the tumor. Patients with advanced/metastatic TNBC are treated with agents such as vinorelbine or eribulin, where eribulin is approved for use following treatment with 3 or more lines of chemotherapy and prior exposure to an anthracycline and a taxane. We found that primary treatment of TNBC cells with eribulin resulted in the generation of epithelial-like ERI-R cells (Figures 1C–1F). It is generally accepted that epithelial-like cancer cells are more chemosensitive than mesenchymal-like cancer cells.1,2 We therefore sought to determine the MET-inducing effects of eribulin on sensitization to other chemotherapeutics. Treatment of PB3ERI-R cells with either paclitaxel or vinorelbine drastically increased senescence (50%–60%) and apoptosis (35%–50%) (Figures 6A, 6B, S5A, and S5B). Conversely, eribulin treatment of PB3PAC-R or PB3VIN-R cells induced modest senescence (<10%) and apoptosis (∼10%) (Figures 6C, S5C, and S5D). Similar effects were observed in growth assays with PB3 and MDA-MB-231 derivative cells. PB3 and PB3ERI-R cells showed respective vinorelbine IC50 values of 279.32 nM and 76 nM and respective paclitaxel IC50 values of 303.34 nM and 112.46 nM (Figure 6D), reflecting an ∼3-fold increase in chemosensitivity in ERI-R cells. Conversely, PB3PAC-R and PB3VIN-R cells showed relative resistance to eribulin (Figures 6E and 6F). In parallel, MDA-MB-231ERI-R cells showed increased sensitivity to paclitaxel and vinorelbine compared with parental MDA-MB-231 cells, while MDA-MB-231PAC-R and MDA-MB-231VIN-R cells showed relative resistance to eribulin (Figures 6G–6I). MDA-MB-231ERI-R cells also exhibited ∼70% senescence upon treatment with vinorelbine or paclitaxel, whereas MDA-MB-231PAC-R and MDA-MB-231VIN-R cells showed 3%–6% senescence in response to reciprocal drugs (Figure S5E). These results suggest that pharmacological MET induction with an agent such as eribulin efficiently sensitizes therapy-naive TNBC cells to other microtubule-targeted chemotherapeutics.
Figure 6.

Eribulin pre-treatment induces sensitization to subsequent chemotherapy
(A–C) Senescence-associated β-galactosidase assay and quantification of apoptotic cells. PB3 parental and drug-resistant cells were used as indicated and treated with paclitaxel (A), vinorelbine (B), or eribulin (C). Data are shown as mean of duplicates ± SEM.
(D–F) PB3 parental and drug-resistant cells were used to generate dose-response curves to determine IC50. (G–I) MDA-MB-231 parental and drug-resistant cells were treated as in (D)–(F). Data are shown as mean of triplicates ± SEM.
Pharmacological MET induction is associated with chemosensitization in vivo
To determine how MET induction via eribulin treatment affects tumor growth and metastatic progression, we first randomized mouse mammary tumor virus promoter-driven polyomavirus middle T antigen-expressing (MMTV-PyMT) transgenic female mice bearing late-stage triple-negative mammary tumors23 to drug treatments. While vinorelbine and paclitaxel each stunted tumor growth, eribulin led to robust tumor regression in all cases (Figure 7A). Furthermore, eribulin induced a near-complete inhibition of metastatic growth in the lungs that was significantly more effective than other drugs (Figure 7B). In contrast to vinorelbine or paclitaxel, which resulted in high-grade, poorly differentiated tumors containing an abundance of vimentin-expressing (i.e., mesenchymal-like) tumor cells, eribulin resulted in low-to moderate-grade, well-differentiated tumors containing islands of malignant cells primarily expressing the epithelial marker E-cad (Figures 7C, and S6A).
Figure 7.

MET induction is accompanied by robust tumor regression and reduced metastatic burden
(A–C) MMTV-PyMT tumor-bearing female mice were treated with vehicle, paclitaxel (20 mg/kg), vinorelbine (7 mg/kg), or eribulin (1.6 mg/kg) twice weekly to assess (A) tumor growth rate, (B) tumor metastasis to the lungs (red arrows), and (C) EMT status using immunofluorescence of E-cad (green) and vimentin (red). Tumors and lungs were harvested after 2 weeks of drug treatment and a 1-week drug holiday.
(D–F) MMTV-PyMT mice were treated as in (A), and palpable mammary tumors were surgically resected. After a 2-week drug holiday, mice were treated with a second round of therapy to assess tumor growth rate (D), EMT status (E), and tumor metastasis to lungs (F). Tumors and lungs were harvested after a 1-week drug holiday.
(G–N) NSG female mice bearing bilaterally implanted JAX-98naive or NCCC-470NAC orthotopic tumors were treated and assayed as in (A)–(F). After a 2-week treatment regimen (G and K), one tumor was resected from each mouse for molecular analysis (H and M). Mice were then maintained until an orthotopic tumor resumed growth and treated with a second round of therapy to assess tumor growth rate (I and L). Mice were maintained for an additional 3 months prior to tissue harvest to evaluate metastasis (J and N).
(O–Q) MDA-MB-231/Luc-ZsGreen cells were injected into the tail vein. Tumor burden was measured by bioluminescence imaging 2 weeks post injection (baseline) and after 2 weeks of drug treatment. Lung metastatic burden was quantified by counting metastases in H&E-stained sections.
Tumor volumes are shown as mean of 5 mice/group ± SEM. ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.0005, ∗∗∗∗p < 0.0001 by Tukey-adjusted pairwise comparison.
To determine the effects of prior chemotherapy exposure on sensitization to additional chemotherapeutics, tumor-bearing MMTV-PyMT mice received primary treatment with 5 doses of (1) paclitaxel or (2) vinorelbine over 2 weeks. Palpable tumors were surgically resected. After a 2-week drug holiday, mice received secondary treatment with eribulin for 2 weeks. Conversely, groups of mice that received primary treatment with eribulin for 2 weeks followed by resection and a 2-week drug holiday then received secondary treatment with (3) paclitaxel or (4) vinorelbine for 2 weeks. Tumors that emerged following primary eribulin treatment underwent robust regression in response to secondary paclitaxel or vinorelbine (Figure 7D). In contrast, secondary eribulin only induced stasis in tumors in mice that received primary vinorelbine or paclitaxel. Moreover, tumors harvested following primary eribulin plus secondary paclitaxel/vinorelbine (and a subsequent 1-week holiday) contained lower proportions of vimentin-expressing (mesenchymal-like) malignant cells and higher proportions of E-cad-expressing (epithelial-like) malignant cells compared with tumors exposed to secondary eribulin (Figure 7E). These observed differences in primary tumor growth and composition were reflected in differences in lung metastases; primary eribulin provided near-complete abrogation of metastatic propensity in lungs analyzed after secondary treatments (Figure 7F).
Chemotherapy exposure in patients directs drug sensitivity in xenografts
Our observations in cultured cells and MMTV-PyMT transgenic mice suggest that the order of exposure to chemotherapeutics dictates outcome; primary eribulin treatment may induce MET and sensitize TNBC cells to other chemotherapeutics, but primary treatment with other chemotherapeutics did not sensitize cells to eribulin. Such findings have clinical implications because patients with TNBC are commonly treated with an anthracycline, cyclophosphamide, and a taxane in the (neo)adjuvant setting long before the third-line treatment or more of recurrent/metastatic disease with eribulin. We thus tested drug effects in orthotopic PDX models established from primary TNBC tumors from patients who did or did not receive prior NAC. JAX-98naive and NCI-140naive PDX models were derived from treatment-naive patients. While 2 weeks of treatment with paclitaxel and vinorelbine inhibited the growth of JAX-98naive and NCI-140naive tumors, eribulin induced near-complete regression (Figures 7G and S6B). Tumors harvested after a 1-week drug holiday showed that eribulin induced differentiation with an abundance of E-cad-expressing cells (Figures 7H and S6C–S6E). In tumors that regrew following primary eribulin treatment, secondary vinorelbine and paclitaxel each provided robust regression (Figures 7I and S6F). Following secondary drug treatment, the remaining PDX tumors were resected, and mice were maintained for 3 months prior to organ harvest to assess metastasis. JAX-98naive engrafted mice showed metastases to lungs, liver, ovaries, and axillary lymph nodes. Primary eribulin followed by secondary paclitaxel or vinorelbine reduced metastatic occurrence, while the reverse order of treatments was less protective (Figures 7J and 6G).
The NCCC-470NAC and JAX-91NAC PDX models were derived from patients pre-treated with NAC. NCCC-470NAC tumors were resistant to all 3 chemotherapy drugs administered alone or in sequence (Figures 7K and 7L), tumors retained poor differentiation status without changes in the proportion of vimentin-expressing cells, and metastases (3 months post secondary drug treatment) were only marginally affected by primary eribulin treatment in sequence with paclitaxel/vinorelbine (Figures 7M, 7N, and S4H). While JAX-91NAC tumors behaved similarly to those of the treatment-naive models in response to single-agent drug treatment (Figure S6I–S6K), tumors that regrew following primary eribulin treatment were insensitive to secondary paclitaxel/vinorelbine (Figure S6L and S6M).
Metastasis and EMT are early events in the tumorigenic process, and patients presenting with early-stage breast cancer often already have disseminated cancer cells in other organs.24,25 It is thus imperative in preclinical studies to assess therapeutic effects on metastases. We injected luciferase-expressing MDA-MB-231 cells via the tail vein to establish lung metastases. Two weeks later, metastases were detected by bioluminescence imaging, and mice were treated with 4 rounds of vehicle, paclitaxel, vinorelbine, or eribulin. While vinorelbine and paclitaxel slowed (or did not affect) the growth of lung metastases, eribulin induced near-complete regression (Figures 7O–Q).
Eribulin reduces epithelial-mesenchymal heterogeneity in tumors
Primary eribulin treatment increased proportions of E-cad-expressing cells and decreased vimentin-expressing cells in TNBC PDX models (Figures 7H, 7M, S6E, and S6K). EMT scoring of PDX specimens showed that eribulin induced significant epithelial shifts in EMT score in NCI-140naive, JAX-98naive, or JAX-91NAC tumors. In contrast, NCCC-470NAC tumors did not exhibit a shift in EMT score upon eribulin treatment (Figures S7A–S7F), in line with resistance to eribulin-induced MET (Figure 7M). Paclitaxel did not significantly alter EMT score in any tumor model. Primary eribulin followed by secondary paclitaxel (E1P2) provided significantly lower EMT scores than the converse (P1E2) in NCI-140naive, JAX-98naive, and JAX-91NAC tumors (Figures S7A–S7F), affirming the contribution of drug order to EMT phenotype.
We also quantified tumor response to therapy based on changes in proportions of cancer vs. stromal cells. Eribulin significantly reduced proportions of cancer cells in all PDX models; such effects were most consistently observed in NCI-140naive and JAX-98naive tumors, while NCCC-470NAC and JAX-91NAC tumors showed more heterogeneity (Figures S7G–S7J). In contrast, paclitaxel had no significant effect on proportions of cancer cells within tumors. E1P2 caused significant decreases in cancer cell proportions in NCI-140naive and JAX-98naive tumors compared with P1E2—effects not observed in NCCC-470NAC and JAX-91NAC tumors. These data reinforce our observations that the effects of eribulin on MET induction and tumor regression are most profound when administered in the treatment-naive setting.
Discussion
Using a combination of TNBC cells, genetically engineered mouse models, and PDX, we uncovered a mechanism through which ZEB1 interacts with members of the SWI/SNF family of chromatin remodelers to maintain the mesenchymal state. This interaction, when disrupted using pharmacologically achievable concentrations of eribulin, induces MET and alters the differentiation state of tumor cells. This MET is accompanied by reduced tumor initiation capacity, reduced metastatic propensity, and increased chemosensitivity. Through a combination of lentiviral barcoding and single-cell analyses, we found that disrupting the ZEB1-SWI/SNF interaction using eribulin reprograms the chromatin and transcriptional profiles of TNBC cells, shifting their evolutionary trajectory as they progress through the bottlenecks of treatment. This work advances the understanding of how cancer cells utilize optimal chromatin remodeling machinery to maintain a mesenchymal state that imparts aggressive traits while unveiling a mechanism of action for eribulin to modulate cancer cell state, paving the way for optimal clinical use to exploit its multifaceted anti-tumor properties.
Therapeutic targeting of EMT has long been pursued as an attractive means of sensitizing tumors that exhibit mesenchymal-like traits to conventional chemotherapy.2,26,27,28 The prevailing understanding of the role of EMT in the invasion-metastasis cascade posits that cancer cells that acquire invasive traits by undergoing EMT often endure a reversal of this program via MET at a distant metastatic site, thereby enabling the colonization of foreign tissues by imparting proliferative traits.29,30 Such a notion would argue against the induction of MET as a possible therapeutic avenue, given the possibility of inadvertently promoting the colonization of cancer cells that have already embarked on the metastatic cascade and disseminated to distant sites. However, our data demonstrating the ability of eribulin to induce regression of established metastases provide evidence that MET induction could prove beneficial in countering metastatic progression. The regression of metastases induced by eribulin may be a result of its MET-inducing properties and/or anti-mitotic effects. The inability of paclitaxel or vinorelbine to eradicate metastases indicates that anti-mitotic effects alone are insufficient to achieve regression of established metastases.
A remaining question is whether cancer cells of both intermediate/hybrid and extreme mesenchymal-like states are equally susceptible to eribulin-induced MET. The PB3 and MDA-MB-231 TNBC cells used in our study reside in hybrid/quasi-mesenchymal states that can be reversed toward epithelial-like states through pharmacological or genetic means.9,31,32,33,34 In contrast, mesenchymal-like SUM159 cells showed more modest MET in response to eribulin (Figures 1C–1G and S1C–S1G), possibly due to their residence in a state that is less permissive to reversion. We previously observed a similar refractory response to MET induction by activation of PKA signaling in some cell systems,28 pointing to a more generalizable inability of cells that have advanced beyond a certain point along the epithelial-mesencymal (E-M) spectrum to regain epithelial traits. Ample evidence points to the aggressive nature of cells residing in an intermediate EMT state that exhibit enhanced metastatic ability.12,31,33,35,36 Whether targeting of extreme mesenchymal cell states holds therapeutic value is unknown, but recent work suggests that mesenchymal-like cancer cells may retain plasticity to acquire epithelial traits following treatment with chemotherapy.36
Drug treatment of cell populations typically results in a mixture of cell fate outcomes that are influenced in part by chromatin state. Using a combination of cellular barcoding and gene expression analyses (QISSMET), we developed a method to study the mode of resistance that drives drug resistance of populations, specifically grouping cells into two categories per prior definitions:37 Darwinian selection for cells where the terminal treatment UMAP state is similar to untreated cells and Lamarckian induction for cells where the terminal treatment UMAP state is transcriptionally distinct from that of untreated cells. QISSMET enables the uncovering of cellular trajectories as cells overcome treatment, and it became evident that the same drug can invoke different outcomes in a population of cells, likely determined by baseline chromatin and EMT states. The Muller plot in Figure 2I shows that eribulin treatment imposes a bottleneck that only a subpopulation of cells can overcome by undergoing a shift in their transcriptional and chromatin landscapes, reminiscent of previously described cancer stem cell subpopulations that drive relapse following therapy.26,38,39
We uncovered a mechanism that serves to maintain a mesenchymal state in TNBC cells by altering the transcriptional functionality of the key EMT factor ZEB1 through its interaction with the SMARCC and SMARCD members of the SWI/SNF chromatin remodeling complex. Several chromatin modulators have been implicated in the regulation of ZEB1, including CtBP, LSD1, HDAC1/2, and BRG1.20,40,41,42 Several of these interactions could constitute attractive therapeutic targets in EMT, such as work demonstrating that the histone deacetylase (HDAC) inhibitor mocetinostat reverses ZEB1-associated drug resistance.43 While HDAC inhibitors are FDA approved for certain indications and serve as proof-of-principle epigenetic therapeutics, the lack of a specific protein target has proven to be a hurdle in their success as widely used anti-neoplastic agents. We showed that eribulin, a microtubule dynamics inhibitor, alters the chromatin landscape of TNBC cells by interfering with interactions between ZEB1 and SWI/SNF complex members, providing a potential secondary mechanism of anti-cancer action.
Our findings have significant implications for the development of more effective treatment regimens for breast cancer patients. Analysis of tumors from the SOLTI1007 study revealed that treatment of patients with eribulin in the neoadjuvant setting can induce a reversion of tumor cells to a more epithelial-like state. We also observed in preclinical models that the chromatin-modulating and cellular reprogramming effects of eribulin enable pre-treatment with this agent to sensitize TNBC cells to other chemotherapeutics. Thus, there may be therapeutic value in using eribulin pre-treatment as an MET-inducing step, specifically for patients with tumors that exhibit EMT features. There are currently no clinical means to stratify patients by the extent of tumor EMT as a biomarker that could inform therapeutic decision-making, with the closest surrogate being a histopathological estimate of differentiation state. Our recently developed multiplexed immunostaining approach to quantify the extent of tumor epithelial-mesenchymal heterogeneity12 may enable the identification of patients that may benefit from pre-treatment with MET-inducing regimens with drugs such as eribulin prior to receiving standard NAC. In summary, we unveiled how a more refined understanding of the mechanism of action of an FDA-approved drug, including its effects on epithelial-mesenchymal plasticity, can open avenues to potentially improve clinical outcomes. Prospective clinical testing is warranted to evaluate the chemosensitizing effects of eribulin in the treatment-naive setting.
Limitations of the study
We temporally evaluated clonal dynamics to determine whether eribulin treatment yielded drug-resistant cell subpopulations due to selection (of pre-existing drug-resistant subpopulations) or induction (of an altered chromatin accessibility/transcriptional state). The use of expressed genetic barcodes enabled multiplexed analysis with scRNA-seq and scATAC-seq profiles via QISSMET. Our observation that eribulin-resistant cells exhibited newly acquired states of chromatin accessibility and gene expression supported the notion that eribulin resistance was caused by an induction event (Figures 3 and S2). A limitation of this analysis was the lack of replicate populations of barcoded cells at each time point, which can provide a means of distinguishing clones with pre-existing vs. acquired drug resistance based on the detection of the same vs. different barcodes across replicates. Bhang et al.44 used non-expressed genetic barcodes and found enrichment of overlapping subset of barcodes across replicate lung cancer cell populations following treatment with the EGFR inhibitor erlotinib, suggesting that such resistant cells were pre-existing and selected by drug treatment. Indeed, most erlotinib-resistant clones showed genomic amplification of MET,44 a known cause of resistance to EGFR inhibitors.45 Oren et al.46 improved upon lineage tracing technology through the development of the Watermelon reporter system that uses expressed genetic barcodes. They found that lung cancer cell clones surviving treatment with the EGFR inhibitor osimertinib had pre-defined cell fates prior to drug treatment, and duplicate experiments showed enrichment of overlapping sets of barcodes suggesting drug selection.46 Chang et al.13 used expressed barcodes to develop the method of tracking differential clonal responses by scRNA-seq (TraCe-seq) in lung cancer cells. Treatment of lung cancer cells with EGFR inhibitors yielded results analogous to those of Bhang et al.44 with enrichment of overlapping subsets of barcodes across replicate experiments.13,44 Notably, all 3 of those studies used EGFR-mutant lung cancer cell lines sensitive to EGFR inhibition, where known resistance mechanisms often involve upregulation of compensatory kinase pathways.45,47 In contrast, our study used TNBC models and the cytotoxic chemotherapeutic eribulin. Thus, the type of drug and mechanism(s) of action likely dictates the path to resistance.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC anti-mouse CD326 (Ep-CAM) | BioLegend | Cat #118214; RRID:AB_1134102 |
| anti-E-cadherin | BD Biosci. | Cat #610182; RRID:AB_397581 |
| anti-E-cadherin | Cell Sig. Tech. | Cat #3195; RRID:AB_2291471 |
| anti-Vimentin | Cell Sig. Tech. | Cat #5741; RRID:AB_10695459 |
| anti-Snail | Cell Sig. Tech. | Cat #3879; RRID:AB_2255011 |
| anti-Snail | Cell Sig. Tech. | Cat #3895; RRID:AB_2191759 |
| anti-Tubulin | Cell Sig. Tech. | Cat #5666; RRID:AB_10691594 |
| anti-Gapdh | Cell Sig. Tech. | Cat #2118; RRID:AB_561053 |
| anti-Zeb1 | Bethyl | Cat #A301-922A; RRID:AB_1524126 |
| anti-Smarcd1 | ThermoFisher | Cat #PA5-52049; RRID:AB_2647616 |
| anti-Smarcc1 | Bethyl Laborat. | Cat #A301-020A; RRID:AB_817978 |
| anti-Smarcc1 | Cell Sig. Tech. | Cat #11956; RRID:AB_2797776 |
| anti-Smarcd3 | Cell Sig. Tech. | Cat #62265; RRID:AB_2799624 |
| anti-H3K4me3 | ThermoFisher | Cat #MA5-11199; RRID:AB_10977872 |
| anti-H3K27me3 | ThermoFisher | Cat #MA5-11198; RRID:AB_11000749 |
| anti-CD104APC | BioLegend | Cat #123612; RRID:AB_2734181 |
| anti-CD44PECy7 | BioLegend | Cat #858802; RRID:AB_2801225 |
| anti-KRT8 | ThermoFisher | Cat #PA5-29607; RRID:AB_2547083 |
| anti-KRT14 | ThermoFisher | Cat #MA5-11599; RRID:AB_10982092 |
| Biological samples | ||
| Human FFPE TNBC tissue specimens | SOLTI-1007-NeoEribulin study | N/A |
| Human FFPE TNBC tissue specimens | Dartmouth-Hitchcock Medical Center Dept. of Pathology | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| VivoGlo Luciferin | Promega | Cat #P1043 |
| Eribulin | Eisai Pharm. Inc. | (via Material Transfer Agreem.) |
| Vinorelbine | SAGENT | NDC 25021-204-01 |
| Paclitaxel | Hospira | NDC 61703-342-09 |
| Critical commercial assays | ||
| SA-bGal kit | Cell Sig. Tech. | Cat #23833 |
| LIVE/DEAD Viability/Cytotoxicity Kit | Invitrogen | Cat #L3224 |
| Universal Magnetic Co-Ip kit | Active Motif | Cat #54002 |
| Deposited data | ||
| RNA-seq, ATAC-seq, CUT&RUN, single-cell RNA-seq data | This paper | GEO: GSE207482 and GSE207773 |
| Experimental models: Cell lines | ||
| PB3 | Robert Weinberg Lab | N/A |
| MDA-MB-231 | Robert Weinberg Lab | RRID:CVCL_0062 |
| SUM-159 | Robert Weinberg Lab | RRID:CVCL_5423 |
| Experimental models: Organisms/strains | ||
| MMTV-PyMT transgenic mice | Jackson Laboratory | Cat #022974; RRID:IMSR_JAX:022974 |
| NOD/SCID/IL2Rg−/− (NSG) mice | Jackson Laboratory | Cat #005557; RRID:IMSR_JAX:005557 |
| Recombinant DNA | ||
| Plasmid psPAX2 | Addgene | Cat #12260; RRID:Addgene_12260 |
| Plasmid pMD2.G | Addgene | Cat #12259; RRID:Addgene_12259 |
| Plasmid, lentiviral transfer, encoding SMARCD1 | Genecopoeia | Cat #EX-A1760-LX304 |
| Plasmid Tet-pLKO-puro | Addgene | Cat #21915; RRID:Addgene_21915 |
| Plasmid Lenti-CRISPR V2 | Addgene | Cat #52961; RRID:Addgene_52961 |
| Plasmid pHIV-Luc-ZsGreen | Addgene | Cat #39196; RRID:Addgene_39196 |
| Software and algorithms | ||
| Tutorial and original code | This paper | https://github.com/lifebytes/bagheri-et-al_scRNA-ATAC_analysis |
| Other | ||
| CUTANA CUT&RUN Kit | EpiCypher | Cat #14-1048 |
| RNeasy plus kit | Qiagen | Cat #74134 |
| QIAamp DNA isolation kit | Qiagen | Cat #51304 |
| MinElute Purification Kit | Qiagen | Cat #28004 |
| ProLong™ Diamond Antifade Mountant | ThermoFisher | Cat #P36961 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Todd Miller (tomiller@mcw.edu).
Materials availability
Plasmids and cell lines generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
The raw and analyzed RNA-seq, ATAC-seq, CUT&RUN, and single-cell RNA-seq data were deposited in the NCBI Gene Expression Omnibus at accession numbers GSE207482 and GSE207773, which are publicly available. The tutorial and original code are available at:
https://github.com/lifebytes/bagheri-et-al_scRNA-ATAC_analysis. Original immunoblot and microscopy data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact.
Experimental model and study participant details
Cell lines
MMTV-PyMT murine mammary tumor-derived PB3 cells, and human MDA-MB-231 and SUM-159 breast cancer cells were gifts from Dr. Bob Weinberg (Whitehead Institute for Biomedical Research). PB3 cells were maintained in DMEM/F12 medium supplemented with 5% adult bovine serum (ABS), 2 mM L-Glutamine, and 1X MEM Non-Essential Amino Acids Solution (ThermoFisher Scientific cat. #11140050). MDA-MB-231 and SUM-159 cells were maintained in DMEM and F12 medium, respectively, supplemented with 10% fetal bovine serum (FBS). Cell lines were propagated for <4 months following authentication by STR genotyping, and were confirmed to be mycoplasma-negative.
Mouse models
Animal studies were approved by the Dartmouth College Institutional Animal Care and Use Committee. MMTV-PyMT transgenic mice [(Tg(MMTV-PyVT)634Mul/LellJ mice on a C57Bl/6 J background, strain #022974, ref. 48] were obtained from the Jackson Laboratory and bred in-house. Female 8-to-10-week-old NOD/SCID/IL2Rγ−/− (NSG) mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, strain #005557) were obtained from the Dartmouth Cancer Center Mouse Modeling Shared Resource.
Human tissue specimens
Human FFPE TNBC tissue specimens were obtained from two sources: the SOLTI-1007-NeoEribulin study11; Dartmouth-Hitchcock Medical Center pathology archives. SOLTI-1007-NeoEribulin was done in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. That study protocol was approved by independent ethics committees at each center and the Agencia Española de Medicamentos y Productos Sanitarios. All patients provided written informed consent. Specimens from Dartmouth-Hitchcock Medical Center were obtained through clinically indicated procedures for diagnostic purposes. Use of these specimens for this research was approved the Dartmouth Health Human Research Protection Program (STUDY02000731).
Method details
Genetic constructs
Lentiviral particles were produced using standard protocols. Briefly, HEK293T cells were transfected with transfer plasmid and packaging plasmids psPAX2 (Addgene #12260) and pMD2.G (Addgene #12259) using Lipofectamine 3000 (ThermoFisher Scientific cat. #L3000015). Lentiviral particles were concentrated using Lenti-X Concentrator (Takara Bio, cat. #631232), and cancer cells were transduced with lentivirus in the presence of polybrene (8–30 μg/mL).
A lentiviral transfer plasmid encoding SMARCD1 cDNA was purchased from GeneCopoeia (cat. #EX-A1760-LX304) and used to generate lentivirus to transduce PB3 and MDA-MB-231 cells; stably transfected cells were selected with 10ug/mL blasticidin for 7 days shRNA constructs were designed and synthesized by IDT and cloned into the Tet-pLKO-puro transfer plasmid (Addgene plasmid #21915) as described.49 CRISPR/Cas9 sgRNAs were designed and synthesized by IDT and cloned into the pLenti-CRISPR V2 transfer plasmid (Addgene plasmid #52961) as described.50 To trace metastasis in vivo, MDA-MB-231 cells were stably transfected with lentivirus generated using the pHIV-Luc-ZsGreen transfer plasmid encoding fluorescent ZsGreen and firefly luciferase (Addgene #39196); stably expressing cells were collected by FACS for ZsGreen fluorescence.
In vitro drug treatment and IC50 assays
Stock solutions for eribulin and vinorelbine (10 mM) were prepared using water, and paclitaxel was prepared using DMSO. Cells were plated in 96-well white-walled, clear, flat-bottom microplates at a density of 7x103 cells/well in 100 μl growth medium. Twenty-four hours later, cells were treated with dose ranges of drugs. Ten ul resazurin (Alamar blue, 0.2 mg/mL, ThermoFisher Scientific cat. #R12204) was added to each well for 4 h at 37°C, and Abs590 was measured to reflect cell viability. IC50 was determined for each drug using nonlinear regression in Prism software (Graphpad).
Cell senescence, death, and apoptosis assays
Senescence-associated β-galactosidase activity was evaluated using CellEvent Senescence Green Flow Cytometry Assay Kit (ThermoFisher Scientific cat. #C10841). Cells were treated with drugs as indicated, trypsinized, washed with 1X PBS, resuspended in 100 μL fixation solution, and incubated for 10 min at room temp. Then cells were washed in 1% BSA in PBS to remove fixation solution, resuspended in 100 μL working solution, and incubated for 2 h at 37°C (atmospheric CO2). Cells were resuspended in 1% BSA in PBS and analyzed by flow cytometry.
Cell death and apoptosis were measured using the Dead Cell Apoptosis Kit (ThermoFisher Scientific cat. #V35114). Cells were treated with drugs as indicated for 72 h, harvested, and washed in 1X Annexin binding buffer. Cells were collected by centrifugation, resuspended in 1X Annexin binding buffer, and stained with SYTOX Green, resazurin, and APC-Annexin V. Cells were incubated at 37°C with 5% CO2 for 15 min. Then 400 μL of 1X annexin-binding buffer was added to samples, and cells were analyzed by flow cytometry. Data were analyzed with FlowJo software (BD Biosciences).
Transwell cell migration assay
Invasion capabilities of cells were measured using 24-well Transwell permeable polycarbonate membrane supports with 8-μm pores. The tops of Transwell membranes were treated with Matrigel (Corning cat. #CLS354234) for 3 h at room temperature. Cells suspended in serum-free medium (105 cells/mL) were seeded on top of Transwell chambers (0.4 mL/chamber). The lower chamber contained growth medium with 10% FBS. At 8 h after cell seeding, non-invaded cells were removed from the top of the membrane using a cotton swab. Cells were fixed using 4% paraformaldehyde and stained with 0.1% crystal violet. Cells that had invaded through the membrane to the bottom were counted manually by microscopy.
Cell monolayer scratch (wound closure) assay
Migration capabilities of cells were assessed using a scratch assay. Cells were seeded into 6-well tissue culture plates (5×105 cells/well to provide near-confluent monolayers) and incubated overnight at 37°C. Scratch wounds were created in monolayers by dragging a sterile 100-μL plastic pipette tip across the plate. Wells were washed with PBS to remove debris. Cells were incubated in growth medium at 37°C with 5% CO2 and imaged after 2, 4, 8, and 16 h. Cell migration was measured as the relative amount of closure of the scratch/wound.
Immunoblotting
Cells were lysed in cold RIPA buffer (ThermoFisher Scientific cat. #89901) supplemented with phosphatase/protease inhibitors (ThermoFisher Scientific cat. #78446) for 1 h on ice. Lysates were centrifuged at 20,000 x g for 10 min at 4°C, and supernatant was analyzed by Quick Start Bradford Protein Assay kit (BIO-RAD cat. #5000202). Lysates were diluted with NuPAGE LDS sample buffer (Life Technologies cat. #NP0007) and NuPAGE Sample Reducing Agent (Life Technologies cat. #NP0009). Samples were heated at 72°C before resolving on 4–12% Bis-Tris gradient gels. Protein was transferred to nitrocellulose using either MiniBlot module or iBlot2 Gel Transfer Device (ThermoFisher Scientific cat. #B1000 and #IB21001). Membranes were blocked with 2.5% BSA or 2.5% non-fat dry milk, and then incubated overnight with primary antibodies in TBST (TBS supplemented with 0.1% Tween 20). HRP-conjugated secondary antibodies were incubated with membranes at room temp. with agitation for 1 h in blocking buffer. Membranes were developed using SuperSignal Western Blot Substrate (ThermoFisher Scientific cat. # A45915).
Immunoprecipitation
Cells were seeded in 150-mm dishes and treated +/− eribulin for 48 h. Nuclear fractions were extracted with the Universal Magnetic co-IP kit (Active Motif cat #54002). One mg of protein in lysate was mixed with 5 μg antibody and protein G magnetic beads. Immunoprecipitates were analyzed by western blot.
Immunocytochemistry
Eight-chamber cell culture slides (BD Falcon) with pretreated with poly-L-lysine (Gibco cat. #A3890401). Cells (5x103 per chamber) were seeded onto slides and treated as indicated. Cells were rinsed with cold PBS and fixed with 4% paraformaldehyde or cold acetone-methanol (1:1) for 10 min at room temp., followed by permeabilization with 0.1% Triton X-100. Cells were treated with blocking buffer recommended by the manufacturer of each primary antibody for 1 h at room temp. followed by primary antibody incubation overnight at 4°C. Cells were washed with cold PBS three times for 3 min each, and then incubated with Alexa 555-labeled anti-rabbit and Alexa 488-labeled anti-mouse secondary antibodies (ThermoFisher Scientific cat. # A-11001) at room temp. for 1 h. Cells were washed three times with cold PBS. Cells were analyzed by fluorescence confocal microscopy.
Proteome integral solubility alteration (PISA) assay
PISA assay was performed as described19 with the following modifications. PB3 cells were cultured in 10-cm dishes in triplicate and treated with DMSO (vehicle), 300 nM eribulin, or 600 nM eribulin for 4 h. Cells were harvested by trypsinization, washed with PBS, and resuspended in PBS to a cell density equivalent to 0.4 mg protein/mL. One-hundred uL of each cell suspension was split among nine replicate 0.2-mL tubes, followed by incubation at 9 different temperatures across a gradient of 34°C–42°C for 3 min in a thermocycler. Samples were snap-frozen in liquid nitrogen, and then thawed at room temperature for 5 min in a water bath. This freeze/thaw process was repeated twice, followed by centrifugation at 21,000 x g at 4°C for 10 min. Fifty uL of supernatant from each temperature condition was transferred to a new 1.5-mL tube, followed by the addition of 1 mL of a solution containing 9 M urea/50 mM Tris pH 8.1/150 mM NaCl/2 mM DTT. Mixtures were heated at 45°C for 15 min, followed by alkylation with 7 mM iodoacetamide for 1 h in the dark. The alkylation reaction was quenched with DTT (2 mM final conc.) for 15 min, followed by dilution in 1.5 mL 50 mM Tris pH 8.1/150 mM NaCl and digestion with 1 mg sequencing-grade trypsin overnight at 30°C. Each sample was desalted on an OASIS mHLB plate (2 mg sorbent). Peptide concentrations were quantified by BCA assay (ThermoFisher Scientific cat. #23225). The equivalent of 40 mg of peptide from each replicate of each temperature condition was labeled using unique isobaric labeling tags (TMT 10-plex reagents, ThermoFisher Scientific cat. # EPX140-40040-901) by resuspension in 30 μg of 166 mM EPPS pH 9 and addition of TMT reagent (80 μg reagent in 8 μL dry acetonitrile) for 1 h, followed by quenching with 1% hydroxylamine for 20 min at room temperature. Each multiplex sample was combined in a single 1.5-mL tube, followed by the addition of 600 μL 1% TFA and desalting on an OASIS C18 plate (10 mg sorbent). The desalted multiplex was dried by vacuum centrifugation and separated on a pentafluorophenyl (PFP) reverse-phase column (Waters) into 48 distinct fractions,51 concatenated into 16 combined fractions by sequential addition, dried by vacuum centrifugation, and analyzed by LC-MS/MS on a Proxeon Easy-nLC 1200-Orbitrap Fusion Lumos mass spectrometer platform using SPS-MS3-based quantification. The resulting tandem mass spectra were data-searched using the Comet algorithm52 against the human UniProt database (www.uniprot.org), filtered to a 1% false discovery rate using the target-decoy strategy.53 Quantification was accomplished using in-house software to extract reporter ion intensities per-peptide and summed across all peptides for each unique UniProt identifier. Each channel was normalized across all proteins to the total intensity of the least abundant channel, and protein intensities were adjusted accordingly. Final protein stability ratios were reported as ΔSm values as described.19
RNA-seq
Cells were washed twice and harvested in PBS. Total RNA was extracted using RNeasy Plus Mini Kit (Qiagen cat. #74136). Purified RNA was used for reverse transcription with High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems cat. #4374967): 1.5 μg RNA was used in a 20-μL reaction and incubated at 25°C for 10 min, and then at 37°C for 120 min. Reverse transcriptase was inactivated by heating to 85°C for 5 min cDNA was used for library preparation (Roche, Kapa mRNA HyperPrep Kit cat. #KR1351) and massively parallel sequencing on an Illumina NextSeq500. RNA-seq data were analyzed as described previously.12
ATAC-seq
Methodology was adapted from ref. 1. Cells were washed with cold PBS and trypsinized. Cells (105) were resuspended in cold PBS and centrifuged at 500 x g for 5 min. Cells were resuspended in 1 mL cold ATAC-seq resuspension buffer (RSB: 10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2 in water) and centrifuged at 4 °C at 500 x g for 5 min. Nuclei were isolated by resuspending cells in 100 μL ATAC-seq lysis buffer (RSB plus 0.1% NP40, 0.1% Tween 20, 0.01% digitonin) by pipetting up and down 5 times. Cells suspensions were incubated on ice for 3 min and washed with 1 mL ATAC-seq RSB containing 0.1% Tween 20. Suspensions were centrifuged at 4 °C at 500 x g for 10 min. Supernatant was removed and discarded, and pelleted nuclei were resuspended in 100 μL Transposition Mix (50 μL 2x TD buffer, 5 μL transposase (100 nM final conc.), 33 μL PBS, 1 μL 1% digitonin, 1 μL 10% Tween 20, 10 μL water). Transposition reactions were incubated at 37°C for 30 min in a thermomixer with shaking at 1,000 rpm. DNA was analyzed by massively parallel sequencing on an Illumina NextSeq500. ATAC-seq data were analyzed as described.12
CUT&RUN
CUT&RUN was performed using CUTANA Kit (EpiCypher cat. #14–1048) per manufacturer’s recommendations with minor modifications. Cells (5x105) were washed with PBS and bound to concanavalin A-coated magnetic beads. Cells were permeabilized with Wash Buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM spermidine, and 1x Roche EDTA-free Complete Protease Inhibitor) containing 0.025% digitonin (Digitonin Buffer) and 2 mM EDTA, and incubated with primary antibody (anti-ZEB1, anti-H3K4me3, or IgG isotype control) overnight at 4°C. The cell-bead slurry was washed twice with Digitonin Buffer and incubated with 1x Protein-A/G-MNase (pAG-MNase) in Digitonin Buffer for 10 min at room temp. The slurry was washed twice with Digitonin Buffer, and incubated in Digitonin Buffer containing 2 mM CaCl2 for 2 h at 4°C to activate pAG-MNase digestion. Digestion was stopped by addition of 2x Stop Buffer (340 mM NaCl, 20 mM EDTA, 4 mM EGTA, 50 μg/mL RNase A, 50 μg/mL glycogen), and sample was incubated for 10 min at 37°C to release chromatin into supernatant and degrade RNA. Supernatant was recovered, and DNA was isolated using MinElute Reaction Cleanup Kit (Qiagen cat. #28206). Isolated CUT&RUN DNA fragments were quantified by Qubit, and 5–10 ng DNA was used for library preparation with the NEB Ultra II DNA Kit. Library amplification was performed using the modified PCR cycling conditions described in Step 39 of the Epicypher CUTANA (EpiCypher) protocol. Barcoded libraries were pooled for sequencing on a NextSeq500 (Mid Output flow cell,10 million 50-bp paired-end reads per sample).
Massively parallel sequencing and bioinformatic analyses
Raw sequence quality of reads was determined using FastQC (v0.11.9). Reads were trimmed using Cutadapt (v2.4) for adapter sequences with parameters “--nextseq-trim 20 --max-n 0.8 --trim-n -m 1”. Reads were mapped to hg38 genome (for human samples) or mm10 genome (for murine samples) using Bowtie2 (v2.4.2)54 with parameters “--local --no-mixed --no-discordant.” Alignments sorted with samtools (v1.11),55 and unmapped or multi-mapping reads were filtered using sambamba (v0.8.0).56 MarkDuplicates (Picard Tools) was used to identify and remove duplicate reads. Normalized signal tracks were generated using deepTools (v 3.3.0) command BamCoverage with parameters “--binSize 20 --smoothLength 60 --normalizeUsing CPM --centerReads –extendReads." Command “bamPEFragmentSize” (deepTools) was used to confirm expected fragment length distributions were present in each sample, and plotFingerprint was used to confirm genomic enrichment in target-captured samples over corresponding IgG controls. Peaks were called using the MACS2 (v2.2.7.1) “call peak” command in narrowpeak mode with parameters “-f BAMPE --keep-dup all -q 0.05.” Corresponding target-captured controls were specified using option -c. Effective genome size was provided using option -g with a value of 2913022398 for human data, and 2652783500 for mouse data. Target capture signal was assessed using Fraction of Reads in Peaks (FRiP) calculated for each sample. Called peaks were filtered against the ENCODE human blacklist (human: ENCFF356LFX, mouse: ENCFF547MET) and further restricted to peaks demonstrating a 2-fold or greater signal increase relative to IgG controls. Reproducible peak sets were identified for replicate pairs using BEDTools (v 2.30.0) command “intersect” with parameter “-a.” Peak sets were annotated using the annotatePeak() function (ChIPseeker package in R software). Promoters were defined as transcriptional start sites (TSS) +/− 1 kb. The R packages “TxDb.Hsapiens. UCSC.hg38. knownGene” and “TxDb.Mmusculus. UCSC.mm10. knownGene” were used to define genomic features for human and mouse peaks, respectively. Any peaks >10 kb from the closest gene were annotated as “intergenic” peaks. A consensus peak set was generated by merging overlapping peaks called across all samples. Read counts in each consensus peak region were counted from BAM files using the featureCounts() function (Rsubread package) with options “isPairedEnd = TRUE, countMultiMappingReads = FALSE”. DESeq257 was used to perform differential binding analysis on the matrix of read counts from consensus peak regions. Adequate dispersion trends were confirmed during model fitting for all analyses. A Benjamini-Hochberg adjusted p value threshold of <0.05 (Wald-test) was used to denote statistically significant peaks.
Mouse tumor growth studies
JAX-98 and JAX-91 PDX models were purchased from The Jackson Laboratory (cat. # TM00098 and TM00091). NCI-140-R was obtained from the NCI Patient-Derived Models Repository (cat. # 994819). NCCC-470 was obtained from the Dartmouth Cancer Center Mouse Modeling Shared Resource. All PDX models were derived from primary tumor surgical specimens. JAX-98 and NCI-140-R were derived from treatment-naïve patients. NCCC-470 and JAX-91 were derived from patients following treatment with neoadjuvant chemotherapy.
PDX tumors were subcutaneously engrafted into female 8-to-10-week-old NSG mice. When tumor volumes reached ∼1500 mm3, tumors were harvested and washed with cold PBS containing antibiotics [penicillin (100 U/mL), streptomycin (100 μg/mL), amphotericin B (0.25 μg/mL)]. Tumors were sliced into 5-mm3 fragments and either cryopreserved or serially engrafted into the inguinal mammary fat pads of NSG female mice for experimentation.
Female heterozygous MMTV-PyMT mice were monitored for mammary tumor occurrence by palpation. Tumor dimensions were measured twice weekly with a digital caliper, and volume was calculated as Length × Width2/2. MMTV-PyMT mice with a tumor ≥800 mm3, and mice bearing bilateral PDX tumors with one tumor that reached 500 mm3, were randomized to treatment with drug/vehicle. Vinorelbine and paclitaxel solutions were purchased from the Dartmouth-Hitchcock Medical Center pharmacy. Eribulin was supplied by Eisai Pharmaceuticals, Inc. and solubilized in water. Solutions were protected from light and made fresh daily. Vinorelbine (7 mg/kg), paclitaxel (20 mg/kg), and eribulin (1.6 mg/kg) were administered by i.p. injection of 200 μL twice per wk for 2 weeks. In MMTV-PyMT mice, palpable tumors were surgically resected after a 1-wk drug holiday for downstream molecular analyses. In mice bearing bilateral orthotopic PDXs, one tumor was surgically after a 1-wk drug holiday for downstream molecular analyses. After a 2-week holiday in MMTV-PyMT mice, and when remaining PDX tumors resumed growth, mice were administered a second round of drug treatment for 2 weeks. In MMTV-PyMT mice, tissues were harvested after a 1-wk drug holiday following secondary drug treatment. In mice bearing PDXs, remaining tumors were surgically resected, and mice were maintained for 3 months prior to tissue harvest to assess metastasis.
MDA-MB-231/Luc-ZsGreen cells were injected i.v. via tail vein into NSG female mice at 8 weeks of age. Tumor burden was measured bioluminescence imaging. Mice were injected i.p. with 100 μL D-luciferin (2 mg/mL). After a 10-min uptake period, mice were imaged for bioluminescence using an IVIS Lumina III In Vivo Imaging System (PerkinElmer).
Immunofluorescence staining of tissue sections
Slide-mounted sections of FFPE tissue were deparaffinized using xylene and rehydrated through a graded ethanol series to water. Antigen retrieval was performed by incubation in sodium citrate buffer (pH 6.0) for 20 min in a pressure cooker, followed by 20 min of cooling at room temp. Sections were then incubated in blocking buffer (1% goat serum and 1% BSA in PBS) for 1 h at room temp. Slides were incubated at 4°C overnight with primary antibodies. After rinsing slides in TBST, slides were incubated in fluorophore-conjugated secondary antibodies (as above) and DAPI for 1 h at room temp. Slides were washed with TBST and mounted using Prolong Diamond Antifade mountant (ThermoFisher Scientific cat. #P36961). Tissues were imaged by fluorescence confocal microscopy.
Multiplexed Tyramide signal amplification (TSA) staining
Methodology was adapted from ref. 12. PDX tumors were harvested from mice treated as indicated and FFPE. Five representative cores were selected from each specimen to make a tissue microarray. Human TNBC specimens were obtained from SOLTI-1007-NeoEribulin study11 and Dartmouth-Hitchcock Medical Center pathology archives. Human FFPE diagnostic core biopsy specimens obtained at baseline were used directly for TSA staining. Human FFPE surgical tumor tissue specimens obtained after neoadjuvant chemotherapy were used to make a tissue microarray with 5 cores per specimen. Tissue sections were immunostained with antibodies targeting a panel of EMT markers: anti-Snail (Cell Signaling Technology cat. #3895, dilution 1:400); anti-KRT8 (Invitrogen cat. #PA5-29607, dilution 1:300); anti-KRT14 (Invitrogen cat. #MA5-11599, dilution 1:1000); anti-Vimentin (Cell Signaling Technology cat. #5741, dilution 1:500); anti-E-cadherin (BD Bioscience cat. #610182, dilution 1:500); anti-ZEB1 (Invitrogen cat. #PA5-82982, dilution 1:1000). The PerkinElmer OPAL Assay Development Guide was used to optimize antibodies and multiplexed staining procedures. Slides were microscopically scanned with the PerkinElmer Vectra 3 automated multispectral imaging system. Images were imported into InForm software (PerkinElmer), and analysis was performed per manufacturer’s instructions and methods adapted from ref. 12. Cells were phenotyped based on expression of one or multiple EMT markers (E-cadherin only; KRT8/E-cadherin; KRT14 only; KRT8/E-cadherin/vimentin; snail only; vimentin only; vimentin/ZEB1) and validated by marker distribution. Cell Mean Fluorescent units were extracted for each marker and normalized as a percentage of maximum and minimum fluorescence across all cells in all images. EMT was scored as in ref. 12.
Lentivirus-mediated genetic barcoding of cells
PB3 cells (2x105) were seeded in a 6-cm dish and treated with 6 μg/mL polybrene transfection reagent (Millipore cat. #TR-1003-G) for 1 h. A packaged lentiviral barcode library (CloneTracker XPTM 10M Barcode-3′ Library with RFP-puro, Cellecta cat. #BCXP10M3RP-V) was added onto cells at a multiplicity of infection of 0.5, and cells were incubated at 37°C for 24 h. Medium was refreshed and cells were maintained for 2 days. Cells were then trypsinized to obtain a single-cell suspension, and sorted by flow cytometry to collect the RFP-expressing subpopulation. RFP+ (barcoded) cells were re-plated and expanded for experimentation or archiving in liquid nitrogen.
Drug treatment of genetically barcoded cells
Since the 10x genomics platform could only analyze a maximum of 103 cells per sample, a founder population of 103 genetically barcoded PB3 cells was obtained to ensure consistency of barcode distribution across samples; this founder population was expanded for experimentation. Cells (8x105) were seeded into each well of a 12-well plate. The next day, cells were treated +/− 500 nM eribulin or 100 nM paclitaxel. Cells were incubated with drug for 72 h before drug washout. Cells were allowed to recover for 48 h, and then re-plated into 6-well plates to provide more surface area for expansion. Since cells seemed resistant to 100 nM paclitaxel, the dosage was increased to 250 nM for subsequent rounds of treatment. Drug treatments were repeated as above for 2-3 additional cycles, and a sampling of cells was removed and snap-frozen after each cycle.
Single-cell/single-nucleus RNA-seq and ATAC-seq of genetically barcoded cells
Cells were treated as indicated, trypsinized, washed in PBS with 0.04% BSA, and counted. Single-cell suspensions were processed using the 10x Genomics NextGen Single-Cell RNA-seq v3.1 and Single-Cell ATAC-seq v1 kits with or without multiplexing using lipid-oligo hashtags using manufacturer’s instructions for nuclei isolation (scATAC), library preparation, and sequencing. Second batches of control-treated and eribulin-treated (Eri4) cells were analyzed on the 10x Genomics Multiome ATAC + Gene Expression assay. For retrieval of genetic barcodes, 10 μL of amplified cDNA was used for 10 cycles of PCR using the 10x Genomics SI-PCR primer (5′AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGC∗T∗C) and BC_RPI_X_Fwd primer (CAAGCAGAAGACGGCATACGAGATNNNNNNGTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGACCGAACGCAACGCACGCA), where ‘N’ denotes sample-specific index sequences. Amplicons were purified with SPRI Select beads (Beckman Coulter), pooled with scRNA-seq libraries, and sequenced to a target depth of 5,000 reads/cell.
Genetic barcode whitelist preparation
We prepared a whitelist of barcodes by combining BC14 and BC30 (BC14sequence+BC30sequence) barcodes. In custom barcode-only sequencing, barcodes are expected in the following format: Custom barcode = 19bp constant + BC14 sequence + TGGT + BC30 sequence. We prepared barcodes in various formats as per the requirements of downstream tools using in-house scripts. The reference files play a crucial role in downstream analyses of custom barcodes FASTQ files by facilitating feature barcode demultiplexing.
Single-cell/single-nucleus RNA-seq data analysis
Eight libraries for single-cell/single-nuclei transcriptome data were obtained in 3 batches (Run1, Run2, Run3). Each batch contains samples from different treatments and dosages sequenced. While Run1 and Run2 data were from single-cell 3′ V3 sequencing (scRNA) protocol, Run3 used the single-nuclei RNA+ATAC V1 sequencing (snRNA multiome) protocol. Hence, FASTQ and count matrix files were generated using 10x Genomics Cellranger-3.1.0, 10x Genomics Cellranger-4.0.0,58 and 10x Genomics Cellranger-arc-1.0.059 for Run1, Run2, and Run3, respectively. Cellranger genome reference mm10 (mouse) was used as the reference for all runs. For Run1 and Run2, whitelist custom barcodes (described above) were used in Cellranger “--feature-ref” parameter for custom barcodes FASTQ. As Cellranger-arc pipeline doesn’t support “--feature-ref” barcoding, custom barcode FASTQ files were processed using CITE-seq-Count v1.4.3 tool.
Count matrices were analyzed using the Seurat v3.1.4 pipeline.60 Low-quality cells (<200 genes expressed; >50% UMIs in mitochondrial or ribosomal genes) were removed. Using selected variably expressing genes and significant principal components from PCA, clustering and UMAP projections were generated. All the samples were merged into one object to illustrate differences due to drug treatment and dosage. After merging, batch effects due to run and platform differences were regressed out. Pseudotime analysis was performed against the all-sample merged dataset using Monocle3 v0.2.115,16,17 while considering “Untreated” cells as the root cells. To represent the relative frequency of each barcode over pseudotime, Muller plot representation was used.61 Relative frequencies were calculated as the ratio between number of cells associated with a barcode at a given pseudotime (binned) and the sum of all cells (from all barcodes) at the same pseudotime bin. Muller plot is accompanied with line chart illustrating the total number of cells at any given pseudotime.
Single-cell/single-nucleus ATAC-seq data analysis
Four libraries for single-cell/nucleus ATAC-seq data were obtained in 2 batches: Run1 with Run3 using scATAC V1 and snATAC (multiome) V1 protocols. FASTQ and peak count matrix files were generated using 10x Genomics Cellranger-atac-1.2.0 for Run1, and 10x Genomics Cellranger-arc-1.0.0 for Run3. Cellranger reference genome mm10 (mouse) was used as the reference. Custom barcode FASTQ files were processed using CITE-seq-Count v1.4.3 tool. Peak count matrices were analyzed using the Signac v1.1.1 pipeline. Low-quality cells (fragments in peak regions <1000 and >75000; percentage of UMIs in peaks >20%; ratio of UMIs in blacklist regions vs. peak regions >0.05; nucleosome signal >10; TSS enrichment <2) were removed. Using selected top features and 2-to-30 principal components from LSI, clustering and UMAP projection were generated. All samples were also integrated into one object using Harmony R package62 to illustrate differences due to drug treatment and dosage. In Harmony, batch effects due to run and technology differences were regressed out.
Integration and genetic barcode classification in single-cell/single-nucleus seq
To assign each cell to a genetic barcode, we applied the MULTIseqDemux method63 from the Seurat pipeline. Though the method differentiated barcodes, this method was originally designed for demultiplexing samples where background estimation is appropriated for each cell from all barcodes. Herein, the enrichment level for each barcode could vary, and it was appropriate to calculate the background for each barcode from all cells. We deployed an in-house-developed R script using binomial distribution that estimates background for each barcode from all cells. We first annotated doublets (cells with ≥2 barcodes) and barcode-negative cells using MULTIseqDemux. Using this approach, we were able to re-annotate and increase the number of singlets (cells with 1 barcode), and remove doublets and barcode-negative cells. scRNA-seq and scATAC-seq datasets were then integrated with the TransferData module in the Seurat pipeline.
Selection vs. Induction identification by QISSMET: To identify and illustrate the cells that undergo bioinformatic Selection (i.e., drug-treated and untreated cells that cluster together) or Induction (i.e., drug-treated and untreated cells that clustering far apart), all cells in each barcode were analyzed using Jaccard index- and Euclidean distance-based methods. Euclidean distance was calculated between median points of cells from a sample per barcode in their UMAP space. For differences within samples, we used the Jaccard index method from scclusteval v1.0,64 where cells of each barcode were re-clustered using a density clustering method (with same variable genes from original integrated dataset). Based on the number of cells for each sample in various clusters, the Jaccard index was calculated. In density clustering, proximal cells were assigned to one cluster. Therein, if all the cells are similar in a given scenario, then they will cluster closely and be assigned to one cluster.
Quantification and statistical analysis
Datasets were analyzed by unpaired t-test (for two-sample experiments) or multiple comparison-adjusted one-way ANOVA or two-way ANOVA with Tukey-adjusted posthoc pairwise comparisons according using Prism software (GraphPad).
Acknowledgments
We thank the following Dartmouth Cancer Center Shared Resources: Genomics and Molecular Biology, DartLab Flow Cytometry, Pathology, and Mouse Modeling. We thank Dr. Meredith Brown, Jennifer Fields, and Dr. Radu Stan for technical assistance and helpful discussions and Dr. Robert A. Weinberg (Whitehead Institute) for critical reading of the manuscript. Funding was provided by a grant from METAvivor (to D.R.P.), a Prouty Pilot Grant from Friends of the Dartmouth Cancer Center (to D.R.P. and S.A.G.), The Elmer R. Pfefferkorn & Allan U. Munck Education and Research Fund (to D.R.P.), a Sponsored Research Agreement with Eisai, Inc. (D.R.P.), and NIH (P30CA023108, a Dartmouth Center for Quantitative Biology pilot grant from P20GM130454, S10OD025235, and R01GM122846 [to S.A.G.]; R00CA201574 [to D.R.P.]; R01CA267691 [to D.R.P. and T.W.M.]; and R01CA262232 [to T.W.M.]).
Author contributions
Conception and design, M.B. and D.R.P.; development of methodology, M.B., N.B.O., G.A.M., H.L., M.A.M.S., F.W.K., S.H.N., S.A.G., and D.R.P.; acquisition of data, M.B., G.A.M., N.B.O., H.L., I.S.L., S.A.G., F.W.K., and D.R.P.; analysis and interpretation of data M.B., N.B.O., G.A.M., M.A.M.S., S.D., H.L., F.W.K., O.M.W., I.L.C., S.H.N., K.E.M., S.A.G., T.W.M., and D.R.P.; writing, review, and/or editing of the manuscript, M.B., G.A.M., T.W.M., and D.R.P.; study supervision, D.R.P. and T.W.M.
Declaration of interests
M.A.M.S. is an employee of Lifebytes India Pvt. Ltd. D.R.P. received research funding from Eisai Pharmaceutical Ltd.
Published: April 8, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101504.
Contributor Information
Todd W. Miller, Email: tomiller@mcw.edu.
Diwakar R. Pattabiraman, Email: diwakar.r.pattabiraman@dartmouth.edu.
Supplemental information
References
- 1.Nieto M.A., Huang R.Y.J., Jackson R.A., Thiery J.P. Emt: 2016. Cell. 2016;166:21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 2.Shibue T., Weinberg R.A. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017;14:611–629. doi: 10.1038/nrclinonc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cortes J., O'Shaughnessy J., Loesch D., Blum J.L., Vahdat L.T., Petrakova K., Chollet P., Manikas A., Diéras V., Delozier T., et al. Eribulin monotherapy versus treatment of physician's choice in patients with metastatic breast cancer (EMBRACE): a phase 3 open-label randomised study. Lancet. 2011;377:914–923. doi: 10.1016/S0140-6736(11)60070-6. [DOI] [PubMed] [Google Scholar]
- 4.Smith J.A., Wilson L., Azarenko O., Zhu X., Lewis B.M., Littlefield B.A., Jordan M.A. Eribulin binds at microtubule ends to a single site on tubulin to suppress dynamic instability. Biochemistry. 2010;49:1331–1337. doi: 10.1021/bi901810u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida T., Ozawa Y., Kimura T., Sato Y., Kuznetsov G., Xu S., Uesugi M., Agoulnik S., Taylor N., Funahashi Y., Matsui J. Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial-mesenchymal transition (EMT) to mesenchymal-epithelial transition (MET) states. Br. J. Cancer. 2014;110:1497–1505. doi: 10.1038/bjc.2014.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dybdal-Hargreaves N.F., Risinger A.L., Mooberry S.L. Regulation of E-cadherin localization by microtubule targeting agents: rapid promotion of cortical E-cadherin through p130Cas/Src inhibition by eribulin. Oncotarget. 2018;9:5545–5561. doi: 10.18632/oncotarget.23798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaul R., Risinger A.L., Mooberry S.L. Eribulin rapidly inhibits TGF-beta-induced Snail expression and can induce Slug expression in a Smad4-dependent manner. Br. J. Cancer. 2019;121:611–621. doi: 10.1038/s41416-019-0556-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oba T., Ito K.I. Combination of two anti-tubulin agents, eribulin and paclitaxel, enhances anti-tumor effects on triple-negative breast cancer through mesenchymal-epithelial transition. Oncotarget. 2018;9:22986–23002. doi: 10.18632/oncotarget.25184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dongre A., Rashidian M., Reinhardt F., Bagnato A., Keckesova Z., Ploegh H.L., Weinberg R.A. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017;77:3982–3989. doi: 10.1158/0008-5472.CAN-16-3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kröger C., Afeyan A., Mraz J., Eaton E.N., Reinhardt F., Khodor Y.L., Thiru P., Bierie B., Ye X., Burge C.B., Weinberg R.A. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl. Acad. Sci. USA. 2019;116:7353–7362. doi: 10.1073/pnas.1812876116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pascual T., Oliveira M., Villagrasa P., Ortega V., Paré L., Bermejo B., Morales S., Amillano K., López R., Galván P., et al. Neoadjuvant eribulin in HER2-negative early-stage breast cancer (SOLTI-1007-NeoEribulin): a multicenter, two-cohort, non-randomized phase II trial. NPJ Breast Cancer. 2021;7:145. doi: 10.1038/s41523-021-00351-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown M.S., Abdollahi B., Wilkins O.M., Chakraborty P., Ognjenovic N.B., Muller K.E., Jolly M.K., Hassanpour S., Pattabiraman D.R. Dynamic plasticity within the EMT spectrum, rather than static mesenchymal traits, drives tumor heterogeneity and metastatic progression of breast cancers. bioRxiv. 2021;2021 doi: 10.1101/2021.03.17.434993. Preprint at. [DOI] [Google Scholar]
- 13.Chang M.T., Shanahan F., Nguyen T.T.T., Staben S.T., Gazzard L., Yamazoe S., Wertz I.E., Piskol R., Yang Y.A., Modrusan Z., et al. Identifying transcriptional programs underlying cancer drug response with TraCe-seq. Nat. Biotechnol. 2022;40:86–93. doi: 10.1038/s41587-021-01005-3. [DOI] [PubMed] [Google Scholar]
- 14.Emert B.L., Cote C.J., Torre E.A., Dardani I.P., Jiang C.L., Jain N., Shaffer S.M., Raj A. Variability within rare cell states enables multiple paths toward drug resistance. Nat. Biotechnol. 2021;39:865–876. doi: 10.1038/s41587-021-00837-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao J., Spielmann M., Qiu X., Huang X., Ibrahim D.M., Hill A.J., Zhang F., Mundlos S., Christiansen L., Steemers F.J., et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566:496–502. doi: 10.1038/s41586-019-0969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qiu X., Mao Q., Tang Y., Wang L., Chawla R., Pliner H.A., Trapnell C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods. 2017;14:979–982. doi: 10.1038/nmeth.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berest I., Arnold C., Reyes-Palomares A., Palla G., Rasmussen K.D., Giles H., Bruch P.M., Huber W., Dietrich S., Helin K., Zaugg J.B. Quantification of Differential Transcription Factor Activity and Multiomics-Based Classification into Activators and Repressors: diffTF. Cell Rep. 2019;29:3147–3159.e12. doi: 10.1016/j.celrep.2019.10.106. [DOI] [PubMed] [Google Scholar]
- 19.Gaetani M., Sabatier P., Saei A.A., Beusch C.M., Yang Z., Lundström S.L., Zubarev R.A. Proteome Integral Solubility Alteration: A High-Throughput Proteomics Assay for Target Deconvolution. J. Proteome Res. 2019;18:4027–4037. doi: 10.1021/acs.jproteome.9b00500. [DOI] [PubMed] [Google Scholar]
- 20.Sánchez-Tilló E., Lázaro A., Torrent R., Cuatrecasas M., Vaquero E.C., Castells A., Engel P., Postigo A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene. 2010;29:3490–3500. doi: 10.1038/onc.2010.102. [DOI] [PubMed] [Google Scholar]
- 21.Liu L., Tong Q., Liu S., Cui J., Zhang Q., Sun W., Yang S. ZEB1 Upregulates VEGF Expression and Stimulates Angiogenesis in Breast Cancer. PLoS One. 2016;11 doi: 10.1371/journal.pone.0148774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Debnath P., Huirem R.S., Dutta P., Palchaudhuri S. Epithelial-mesenchymal transition and its transcription factors. Biosci. Rep. 2022;42 doi: 10.1042/BSR20211754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christenson J.L., Butterfield K.T., Spoelstra N.S., Norris J.D., Josan J.S., Pollock J.A., McDonnell D.P., Katzenellenbogen B.S., Katzenellenbogen J.A., Richer J.K. MMTV-PyMT and Derived Met-1 Mouse Mammary Tumor Cells as Models for Studying the Role of the Androgen Receptor in Triple-Negative Breast Cancer Progression. Horm. Cancer. 2017;8:69–77. doi: 10.1007/s12672-017-0285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hüsemann Y., Geigl J.B., Schubert F., Musiani P., Meyer M., Burghart E., Forni G., Eils R., Fehm T., Riethmüller G., Klein C.A. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13:58–68. doi: 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 25.Rhim A.D., Mirek E.T., Aiello N.M., Maitra A., Bailey J.M., McAllister F., Reichert M., Beatty G.L., Rustgi A.K., Vonderheide R.H., et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta P.B., Onder T.T., Jiang G., Tao K., Kuperwasser C., Weinberg R.A., Lander E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ognjenovic N.B., Bagheri M., Mohamed G.A., Xu K., Chen Y., Mohamed Saleem M.A., Brown M.S., Nagaraj S.H., Muller K.E., Gerber S.A., et al. Limiting Self-Renewal of the Basal Compartment by PKA Activation Induces Differentiation and Alters the Evolution of Mammary Tumors. Dev. Cell. 2020;55:544–557.e6. doi: 10.1016/j.devcel.2020.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pattabiraman D.R., Bierie B., Kober K.I., Thiru P., Krall J.A., Zill C., Reinhardt F., Tam W.L., Weinberg R.A. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science. 2016;351 doi: 10.1126/science.aad3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ocaña O.H., Córcoles R., Fabra A., Moreno-Bueno G., Acloque H., Vega S., Barrallo-Gimeno A., Cano A., Nieto M.A. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell. 2012;22:709–724. doi: 10.1016/j.ccr.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 30.Tsai J.H., Donaher J.L., Murphy D.A., Chau S., Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22:725–736. doi: 10.1016/j.ccr.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bierie B., Pierce S.E., Kroeger C., Stover D.G., Pattabiraman D.R., Thiru P., Liu Donaher J., Reinhardt F., Chaffer C.L., Keckesova Z., Weinberg R.A. Integrin-beta4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc. Natl. Acad. Sci. USA. 2017;114:E2337–E2346. doi: 10.1073/pnas.1618298114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dongre A., Rashidian M., Eaton E.N., Reinhardt F., Thiru P., Zagorulya M., Nepal S., Banaz T., Martner A., Spranger S., Weinberg R.A. Direct and Indirect Regulators of Epithelial-Mesenchymal Transition-Mediated Immunosuppression in Breast Carcinomas. Cancer Discov. 2021;11:1286–1305. doi: 10.1158/2159-8290.CD-20-0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pastushenko I., Brisebarre A., Sifrim A., Fioramonti M., Revenco T., Boumahdi S., Van Keymeulen A., Brown D., Moers V., Lemaire S., et al. Identification of the tumour transition states occurring during EMT. Nature. 2018;556:463–468. doi: 10.1038/s41586-018-0040-3. [DOI] [PubMed] [Google Scholar]
- 34.Vijay G.V., Zhao N., Den Hollander P., Toneff M.J., Joseph R., Pietila M., Taube J.H., Sarkar T.R., Ramirez-Pena E., Werden S.J., et al. GSK3beta regulates epithelial-mesenchymal transition and cancer stem cell properties in triple-negative breast cancer. Breast Cancer Res. 2019;21:37. doi: 10.1186/s13058-019-1125-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jolly M.K., Boareto M., Huang B., Jia D., Lu M., Ben-Jacob E., Onuchic J.N., Levine H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015;5:155. doi: 10.3389/fonc.2015.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lüönd F., Sugiyama N., Bill R., Bornes L., Hager C., Tang F., Santacroce N., Beisel C., Ivanek R., Bürglin T., et al. Distinct contributions of partial and full EMT to breast cancer malignancy. Dev. Cell. 2021;56:3203–3221.e11. doi: 10.1016/j.devcel.2021.11.006. [DOI] [PubMed] [Google Scholar]
- 37.Boumahdi S., de Sauvage F.J. The great escape: tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 2020;19:39–56. doi: 10.1038/s41573-019-0044-1. [DOI] [PubMed] [Google Scholar]
- 38.Chen J., Li Y., Yu T.S., McKay R.M., Burns D.K., Kernie S.G., Parada L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diehn M., Cho R.W., Lobo N.A., Kalisky T., Dorie M.J., Kulp A.N., Qian D., Lam J.S., Ailles L.E., Wong M., et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Postigo A.A., Dean D.C. ZEB represses transcription through interaction with the corepressor CtBP. Proc. Natl. Acad. Sci. USA. 1999;96:6683–6688. doi: 10.1073/pnas.96.12.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J., Scully K., Zhu X., Cai L., Zhang J., Prefontaine G.G., Krones A., Ohgi K.A., Zhu P., Garcia-Bassets I., et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature. 2007;446:882–887. doi: 10.1038/nature05671. [DOI] [PubMed] [Google Scholar]
- 42.Aghdassi A., Sendler M., Guenther A., Mayerle J., Behn C.O., Heidecke C.D., Friess H., Büchler M., Evert M., Lerch M.M., Weiss F.U. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut. 2012;61:439–448. doi: 10.1136/gutjnl-2011-300060. [DOI] [PubMed] [Google Scholar]
- 43.Meidhof S., Brabletz S., Lehmann W., Preca B.T., Mock K., Ruh M., Schüler J., Berthold M., Weber A., Burk U., et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol. Med. 2015;7:831–847. doi: 10.15252/emmm.201404396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhang H.e.C., Ruddy D.A., Krishnamurthy Radhakrishna V., Caushi J.X., Zhao R., Hims M.M., Singh A.P., Kao I., Rakiec D., Shaw P., et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat. Med. 2015;21:440–448. doi: 10.1038/nm.3841. [DOI] [PubMed] [Google Scholar]
- 45.Engelman J.A., Zejnullahu K., Mitsudomi T., Song Y., Hyland C., Park J.O., Lindeman N., Gale C.M., Zhao X., Christensen J., et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 46.Oren Y., Tsabar M., Cuoco M.S., Amir-Zilberstein L., Cabanos H.F., Hütter J.C., Hu B., Thakore P.I., Tabaka M., Fulco C.P., et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature. 2021;596:576–582. doi: 10.1038/s41586-021-03796-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sattler M., Mambetsariev I., Fricke J., Tan T., Liu S., Vaidehi N., Pisick E., Mirzapoiazova T., Rock A.G., Merla A., et al. A Closer Look at EGFR Inhibitor Resistance in Non-Small Cell Lung Cancer through the Lens of Precision Medicine. J. Clin. Med. 2023;12 doi: 10.3390/jcm12051936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guy C.T., Cardiff R.D., Muller W.J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol. Cell Biol. 1992;12:954–961. doi: 10.1128/mcb.12.3.954-961.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wiederschain D., Wee S., Chen L., Loo A., Yang G., Huang A., Chen Y., Caponigro G., Yao Y.M., Lengauer C., et al. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle. 2009;8:498–504. doi: 10.4161/cc.8.3.7701. [DOI] [PubMed] [Google Scholar]
- 50.Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelson T., Heckl D., Ebert B.L., Root D.E., Doench J.G., Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grassetti A.V., Hards R., Gerber S.A. Offline pentafluorophenyl (PFP)-RP prefractionation as an alternative to high-pH RP for comprehensive LC-MS/MS proteomics and phosphoproteomics. Anal. Bioanal. Chem. 2017;409:4615–4625. doi: 10.1007/s00216-017-0407-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eng J.K., Jahan T.A., Hoopmann M.R. Comet: an open-source MS/MS sequence database search tool. Proteomics. 2013;13:22–24. doi: 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- 53.Elias J.E., Gygi S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 54.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tarasov A., Vilella A.J., Cuppen E., Nijman I.J., Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31:2032–2034. doi: 10.1093/bioinformatics/btv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8 doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Satpathy A.T., Granja J.M., Yost K.E., Qi Y., Meschi F., McDermott G.P., Olsen B.N., Mumbach M.R., Pierce S.E., Corces M.R., et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 2019;37:925–936. doi: 10.1038/s41587-019-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Farahpour F., Saeedghalati M., Hoffmann D. 2016. MullerPlot: Generates Muller Plot from Population/Abundance/Frequency Dynamics Data.https://cran.r-project.org/package=MullerPlot [Google Scholar]
- 62.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McGinnis C.S., Patterson D.M., Winkler J., Conrad D.N., Hein M.Y., Srivastava V., Hu J.L., Murrow L.M., Weissman J.S., Werb Z., et al. MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nat. Methods. 2019;16:619–626. doi: 10.1038/s41592-019-0433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tang M., Kaymaz Y., Logeman B.L., Eichhorn S., Liang Z.S., Dulac C., Sackton T.B. Evaluating single-cell cluster stability using the Jaccard similarity index. Bioinformatics. 2021;37:2212–2214. doi: 10.1093/bioinformatics/btaa956. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw and analyzed RNA-seq, ATAC-seq, CUT&RUN, and single-cell RNA-seq data were deposited in the NCBI Gene Expression Omnibus at accession numbers GSE207482 and GSE207773, which are publicly available. The tutorial and original code are available at:
https://github.com/lifebytes/bagheri-et-al_scRNA-ATAC_analysis. Original immunoblot and microscopy data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact.