

SUMMARY
There is a broad diversity among Cas12a endonucleases that possess nucleic acid detection and gene-editing capabilities, but few are studied extensively. Here, we present an exhaustive investigation of 23 Cas12a orthologs, with a focus on their cis- and trans-cleavage activities in combination with noncanonical crRNAs. Through biochemical assays, we observe that some noncanonical crRNA:Cas12a effector complexes outperform their corresponding wild-type crRNA:Cas12a. Cas12a can recruit crRNA with modifications such as loop extensions and split scaffolds. Moreover, the tolerance of Cas12a to noncanonical crRNA is also observed in mammalian cells through the formation of indels. We apply the adaptability of Cas12a:crRNA complexes to detect SARS-CoV-2 in clinical nasopharyngeal swabs, saliva samples, and tracheal aspirates. Our findings further expand the toolbox for next-generation CRISPR-based diagnostics and gene editing.
In brief
Nguyen et al. explore the diversity and adaptability of 23 Cas12a orthologs in combination with their canonical and 8 noncanonical crRNAs. A total of 207 combinations of Cas12a and crRNAs were applied for clinical nucleic acid detection and gene editing in cells. The data expand the toolbox for next-generation CRISPR-based diagnostics and genome engineering.
Graphical Abstract

INTRODUCTION
Since their discovery, CRISPR-Cas12a systems have been harnessed extensively for gene editing in plants, insects, and mammalian cells due to their comparable editing efficiency to Cas9.1–6 Unlike Cas9, Cas12a cleaves single-stranded DNA (ssDNA) nonspecifically upon binding to the double-stranded DNA (dsDNA) target (cis-cleavage). The additional collateral cleavage activity (referred to as trans-cleavage) of these systems has successfully been repurposed for the detection of nucleic acids for the diagnosis of cancer and infectious diseases.7–14 However, despite the identification of a large diversity of Cas12a orthologs, few have been purified and studied biochemically, with most applications using the prominent orthologs such as LbCas12a, AsCas12a, and FnCas12a.4,7,13,15–20 Therefore, a more expansive investigation is necessary to uncover key properties that inform crRNA and protein engineering to yield highly efficient Cas12a systems with broad applications.
Here, we present a study of 23 Cas12a effectors,3,15,17,20 specifically their cis-cleavage, trans-cleavage, and thermal stability in combination with their 9 distinct CRISPR RNAs (crRNAs). We observed that when these Cas12a proteins complex with some noncanonical crRNAs, their cis- and trans-cleavage activities outperformed their activities when paired with the wild-type (WT) crRNAs. Furthermore, various orthologs showed an increased thermal stability when complexed with noncanonical crRNAs compared to their WT ribonucleoprotein (RNP) complex and apo form.
A combinatorial genome editing screen of the 23 Cas12a proteins and 9 crRNAs revealed that many Cas12a paired with crRNAs from different orthologs had comparable activity to the WT. Specifically, ErCas12a maintained the highest gene-editing activity even when paired with diverse crRNAs. These findings identified an engineerable region in the hairpin loop and indicated that modified guide RNAs (gRNAs) could be designed for enhanced nucleic acid detection and gene-editing applications. Additional noncanonical crRNAs, including loop-extended scaffolds, were tested for insertions and deletions (indels) formation in mammalian cells and found to be tolerated by ErCas12a.
The highly adaptable properties of Cas12a systems were also beneficial for nucleic acid detection. We show that when complexed with noncanonical crRNAs, some Cas12a have improved diagnostic capabilities. As a proof of concept, we applied these combinations to detect severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) in clinical nasopharyngeal swabs, saliva samples, and tracheal aspirates. Along with crRNA manipulation, biodiverse Cas12a orthologs expand the toolbox for next-generation diagnostics and genome engineering. We call our approach of combining non-canonical crRNA with Cas12a cCRISPR (combinatorial CRISPR).
RESULTS
crRNA scaffold has a solvent accessible stem-loop in the RNP complex
Inspired by previous studies on Cas12a orthologs with diverse applications, we sought to further investigate a collection of these Cas12a proteins and analyze their phylogenetic relevance and direct repeat sequences.3,17,20 We were interested in the conformational adaptability of crRNA and Cas12a, particularly how non-canonical binary complexation affects their functionality. A total of 23 Cas12a orthologs with highly similar corresponding mature crRNAs were selected for this study. As reported by Teng et al. and Zetsche et al., many of these functional Cas12a proteins have very similar mature crRNA but do not necessarily have significant percent identity in their amino acid sequences, confirmed by phylogenetic analyses (Figures 1A and S1).17,20 Interestingly, these mature crRNAs differ in sequences between the stem left and stem right (Figure 1B). After analyzing the mature crRNA sequences resulting from protein-mediated pre-crRNA processing,21 9 groups of distinct crRNAs were identified. We expressed and purified 23 Cas12a proteins by subcloning human codon-optimized gene fragments into an expression plasmid and transforming them into Escherichia coli (Figure S2).
Figure 1. A survey of in vitro performance of Cas12a proteins.

(A) Phylogenetic analysis of 23 Cas12a orthologs. Sequence alignment of Cas12a shows conserved regions of RuvC and Nuc domains.
(B) Sequence alignment of mature crRNA from CRISPR direct repeats across all 23 Cas12a orthologs display high similarity and conserved stem-loops. To minimize the number of combinations, identical and/or similar crRNA sequences were clustered together into 9 groups.
(C) Percentage of cis-cleavage. Double-stranded GFP fragment was added to the precomplexed Cas12a:crRNA and incubated at 37°C for 30 min followed by gel electrophoresis. The percentage of cleavage of the dsDNA target was analyzed by ImageJ (mean, n = 2 independent experiments).
(D) Background corrected fluorescence of 22 functional Cas12a against 9 crRNAs in a combinatorial fashion with TTTTT reporters. The fluorescence signal was collected using the trans-cleavage reporter assay at t = 20 min (mean, n = 3 independent experiments). Canonical crRNAs are highlighted in the boxes with borders.
(E) Binary structure of LbCas12a (yellow) in complex with its canonical crRNA (red) (PDB: 5ID6).18,22
(F) Ternary structure of LbCas12a (yellow) in complex with its canonical crRNA (red) and target DNA (teal) (PDB: 5XUS).
Arrow indicates solvent-accessible region in the stem-loop of crRNA (E and F).
We proceeded to investigate the performance of these Cas12a proteins by carrying out cis-cleavage and trans-cleavage reporter assays via complexing 9 distinct crRNAs to 23 Cas12a proteins. Among 23 Cas12a proteins, many of them prefer T-rich protospacer adjacent motif (PAM) sequences, although some appeared to have been studied without an identified PAM. We designed gRNAs targeting a dsDNA region within the gene encoding GFP with a PAM sequence TTTN (Table S1). Through in vitro cis-cleavage assays, we discovered that many of the Cas12a effector proteins can adopt crRNA orthologs derived from 23 examined species, except for LpCas12a, whose function we failed to verify with these assays. Interestingly, we found that the cis-cleavage activity of the majority of Cas12a proteins was enhanced when complexed with select noncanonical crRNAs (Figures 1C and S5).
We performed trans-cleavage assays with a variety of reporters (TTTTT, TTATT, CCGCC, rUrUrUrUrU, rUrArUrArU), and unanimously found the highest fluorescence signal with the poly-T sequence reporter. Consistent with the literature, we observed higher cleavage rates with DNA reporters when compared with RNA reporters (Figures 1D, S3, and S4). Some orthologs demonstrated noticeable RNA cleavage activity (Figure S4). Of the examined effectors, BoCas12a, BsCas12a, ErCas12a, and TsCas12a exhibited high trans-cleavage activity comparable to the widely used LbCas12a and AsCas12a. Notably, the majority of Cas12a trans-cleavage activity was enhanced when complexed with noncanonical crRNAs (Figures 1D, S3, and S4). Additionally, some Cas12a showed efficient cis-cleavage but poor trans-cleavage activity (FnCas12a, Mb2Cas12a, PrCas12a), and vice versa (BoCas12a, BsCas12a) (Figures 1C and 1D). These comprehensive results unveiled several new combinations of Cas12a:crRNA orthologs for developing robust tools for diagnostics and gene editing.
Previous studies on Cas12a structures and Cas12a-crRNA binding interactions showed that some nucleotides within the hairpin loops do not interact with amino acids in Cas12a.18,22–25 We studied the solved structures of Cas12a:crRNA binary and Cas12a:crRNA:dsDNA ternary complexes to further understand how Cas12a maintains functionality with noncanonical crRNAs.18,22,23 We noticed that the loop region between the stem left and stem right of the crRNA is solvent accessible (Figures 1E and 1F). We hypothesized that because this region makes minimal interactions with the Cas12a effector, noncanonical crRNA with changes in the loop region can preserve the activity of the protein. Additionally, we predicted that modifications such as nucleotide extensions and splitting may also be tolerated.
Cas12a can recruit segments of crRNA
By using trans-cleavage reporter assays to monitor Cas12a performance, we designed multiple truncated versions of a crRNA (referred to as α, β, γ, δ, ε, ζ, and Ψ) targeting a GFP gene fragment. Truncated crRNAs either missing the stem left or stem right exhibited no observable fluorescence signal for AsCas12a, ErCas12a, and LbCas12a (Figures 2A–2C and 2E). Interestingly, AsCas12a showed a substantial fluorescence signal when combined with crRNA(ζ) and crRNA(Ψ) (Figure 2A), which do not contain the first 5 nucleotides and the loop, respectively.
Figure 2. Part of the hairpin loop of the crRNA scaffold is not crucial for cleavage, but sequence changes can enhance the overall performance of Cas12a.

RNAfold was used to predict some of the crRNA structures.26
(A, C, and E) Fluorescence signal of individual truncated crRNAs.
(B, D, and F) Fluorescence signal of combined truncated crRNAs. The fluorescence was collected using the trans-cleavage reporter assay at t = 20 min (mean ± SD, n = 3 independent experiments).
(G and H) cis-Cleavage assay of all truncated crRNAs from (A) to (F). The cis-cleavage experiment has been repeated with similar results.
We speculated that sequence changes in the canonical loop region are not necessarily crucial for the protein’s functionality and can be engineered for enhanced collateral cleavage efficiency. To investigate this hypothesis and to test the protein’s ability to recruit segments of the crRNA, combinations of truncated crRNAs were added in increasing concentration (50, 100, and 500 nM) to complex with a constant concentration of Cas12a (50 nM) (Figures 2B–2D and 2F). AsCas12a exhibited robust trans-cleavage activity in which the fluorescence signal was comparable to the WT crRNAs when a high concentration of truncated crRNA was supplied (Figure 2B). Notably, crRNA(α+ε):ErCas12a and crRNA(α+β):ErCas12a mirrored the cleavage activity of the WT complex, even at the low concentrations (Figure 2D). These results corroborate a previous study in which crRNA with a minimized scaffold region displayed protein binding and led to efficient cis-cleavage.27 However, the findings somewhat contradicted our study, which showed that AsCas12a maintained robust cis-cleavage with split crRNAs that were truncated at least 10 nt on the 5′-end of the crRNA. Although the trans-cleavage activity was not investigated in that study, we speculated that these truncated versions of crRNA may work in a target sequence-specific manner or that high concentrations (3–5 μM) of split crRNA may shift the equilibrium for higher binding to the Cas protein. Regarding LbCas12a, some signal was observed with high concentrations of the crRNA(α+ε) or crRNA(α+β), indicating minimal activation of the protein collateral activity (Figure 2F).
We also observed that the crRNA(α+ε) lacking the loop segment outperformed those which, when combined, form a complete mature crRNA (α+β and γ+δ) (Figures 2B–2D and 2F). These phenomena indicate that this loop region can be engineered to increase the overall functionality of the Cas12a proteins. Regarding the cis-cleavage activity of these truncated crRNAs, we found that they induced cutting of dsDNA for crRNA(α+ε) or crRNA(α+β), but their efficiency was reduced compared to the WT complex (Figures 2G and 2H). The discrepancy between the cis- and trans-cleavage activities, for example, in the case of ErCas12a, requires further investigation to draw mechanistic insights into how truncated crRNAs are recruited and bind to the dsDNA target.
Noncanonical crRNAs affect Cas12a thermal stability
Derived from mesophilic bacterial species, many of these Cas12a proteins have been repurposed for genome editing.4,7,13,15–20 LbCas12a, AsCas12a, and FnCas12a have been widely used in in vitro and in vivo applications, including gene editing in plants, insects, and mammalian cells.1–6 They exhibit various levels of editing efficiency under different temperatures ranging from 22°C to 42°C.1,28 For instance, Merker et al. showed that a mutant of LbCas12a (ttLbCas12a) displayed enhanced temperature-tolerant gene editing activity and worked robustly at 22°C in Arabidopsis thaliana.28 Fascinated by these observations, we sought to understand whether noncanonical crRNAs could affect the thermal stability of Cas12a. We therefore carried out differential scanning fluorimetry (DSF) of 22 Cas12a orthologs against 9 distinct groups of crRNAs (Figures 3A–3G and S6). We observed that the melting temperature (Tm) of the binary complex of Cas12a (crRNA:Cas12a) such as AsCas12a, CMaCas12a, PcCas12a, LbCas12a, BoCas12a, and BfCas12a did not differ from its apo form (Figure S6). On the other hand, TsCas12a, ArCas12a, and ErCas12a showed an increase in Tm of the binary complex compared to their apo form (Figures 3A–3F). Surprisingly, noncanonical crRNAs, such as crRNA6, had a tremendous effect on TsCas12a thermal stability, increasing its Tm by ~6°C from 44°C (apo) to 50°C (crRNA:Cas12a binary complex).
Figure 3. Noncanonical crRNAs increase the thermal stability of some Cas12a.
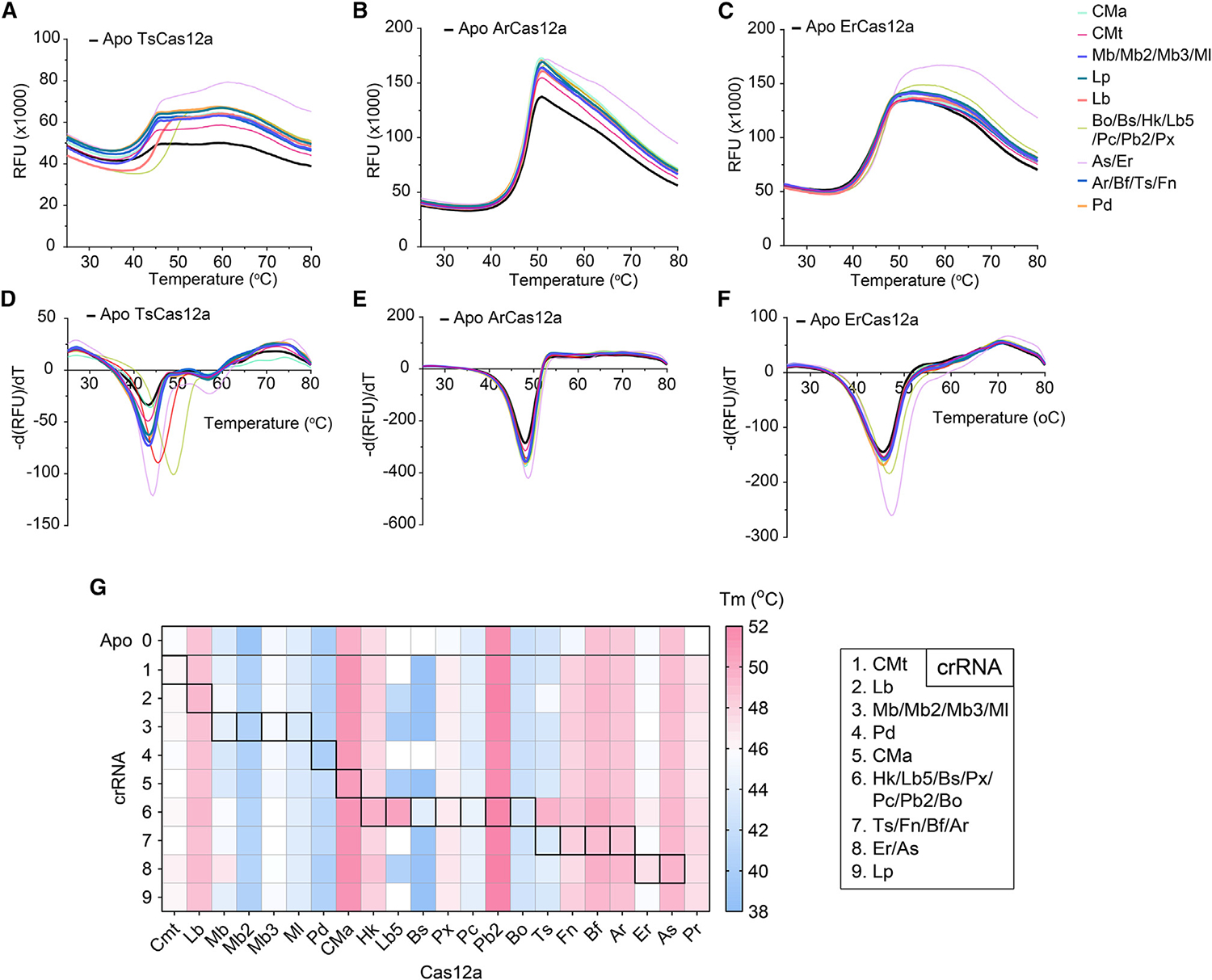
(A–F) Representation of DSF of 3 Cas12a (TsCas12a, ArCas12a, and ErCas12a), where (A), (C), and (E) represent melting curves, and (B), (D), and (F) represent the derivative of fluorescence from (A), (C), and (E), respectively (mean, n = 4, with n = 2 replicates per experiment over 2 independent experiments). For melting curves of the rest of Cas12a, refer to Figure S6. Except for the black curve, which represents apoCas, different color-coded curves refer to the crRNAs complexed with each respective protein.
(G) Summary of thermal stability of 22 Cas12a when complexing with 9 different crRNAs. Canonical crRNAs are highlighted in the boxes with borders. The fluorescence signal presented is the mean value with a total n = 4 (n = 2 replicates per experiment over 2 independent experiments).
Cas12a with noncanonical and extended crRNAs are active in mammalian cells
Intrigued by the above findings, we sought to investigate whether noncanonical and extended crRNAs complexed with Cas12a could induce insertions and deletions (indels) in mammalian cells. We performed plasmid cotransfection of 207 combinations of 9 distinct crRNAs and 23 Cas12a orthologs into HEK293T cells. Among the canonical Cas12a:crRNA pairings, ErCas12a, AsCas12a, FnCas12a, and LbCas12a showed the highest indel formation, corroborating previous studies (Figure 4A).1,3,29,30 Additionally, these Cas12a orthologs and others demonstrated comparable activity when paired with many of the noncanonical crRNAs (Figures 4A–4E).
Figure 4. Adaptability of noncanonical crRNAs in mammalian cells.

(A) Percentage of indels induced by cotransfection of various Cas12a orthologs and crRNAs targeting the CCR5 locus in HEK293T cells (n = 3 biological replicates). Canonical crRNAs are highlighted in the boxes with borders.
(B–E) A subset of the percentage of indels data was compared relative to the canonical crRNAs for AsCas12a, ErCas12a, BsCas12a, and FnCas12a, respectively (n = 4). Error bars represent ±1 SD.
(F) Pictorial representation of the WT and noncanonical MS2 crRNAs tested.
(G) Percentage of indels induced by RNP nucleofection of noncanonical MS2 crRNAs targeting the CCR5 locus in HEK293T cells (n = 3 biological replicates). Error bars represent ±1 SD. All indel percentages were generated with CRISPResso2.31
Given the high activity of ErCas12a with a variety of noncanonical crRNAs, we further examined the adaptability of crRNA by installing 5-nt, 10-nt, and full-length MS2 aptamer extensions to the stem-loop region of the canonical crRNA of ErCas12a (Figure 4F). RNP complexes were introduced to HEK293T cells via nucleofection. Interestingly, we observed that these crRNA modifications were tolerated and resulted in considerable indels, albeit lower than the WT crRNA (Figure 4G). These data indicate that the MS2crRNA:Cas12a complex is stable inside mammalian cells and is capable of localizing to its target genes, supporting the development of this modified CRISPR-ErCas12a system as a potential candidate for transcriptional activation/interference.
A combinatorial approach to clinical validation of SARS-CoV-2 detection
Through combinatorial screenings of trans-cleavage of Cas12a orthologs, we observed robust trans-cleavage activity when ErCas12a, TsCas12a, BsCas12a, and BoCas12a were paired with both canonical and noncanonical crRNAs (Figures 1D–1F and S3). Additionally, their less restrictive PAM requirements compared to LbCas12a and AsCas12a make them suitable candidates for nucleic acid detection. We hypothesized that these Cas12a effector proteins could distinguish a SARS-CoV-2 variant of concern, B.1.1.7 lineage (also known as the Alpha variant), which was first identified in the United Kingdom, from its WT original variant. The standard method for detecting the B.1.1.7 variant is the S-gene target failure test, wherein the qRT-PCR fails to detect the S-gene due to the 6-base deletions in codons H69–V70. As a proof of concept, we designed 2 gRNAs for ErCas12a targeting a region encoding the Spike protein of the virus. One gRNA is compatible with the WT S gene, and the other targets the same region of the B.1.1.7 S gene bearing a 6-base deletion (Figures S7 and S8). One significant difference is the PAM sequence, with the B.1.1.7 crRNA containing the preferred YTTN sequence for ErCas12a (Figure S7A). We performed a reverse-transcription-loop-mediated isothermal amplification (RT-LAMP) step with either WT SARS-CoV-2 or B.1.1.7 variant genomic RNA (obtained from BEI Resources).32–34 We proceeded to detect the amplicons with the crRNA (henceforth referred to as crS-WT and crS-B.1.1.7) to test each of the Cas12a specificities. Within a 30-min run from the trans-cleavage reporter assay, a 160-fold change in fluorescence signal compared to the nontemplate control control was observed for the crS-B.1.1.7 targeting positive B.1.1.7 samples for all Cas12a, indicating robust specificity (Figures S7B, S7D, S7F, S7H, S7J, and S7L). In the case of positive WT SARS-CoV-2, we observed some background activity from the crS-B.1.1.7 when complexed with AsCas12a, TsCas12a, BsCas12a, and BoCas12a, suggesting their various levels of tolerance toward sequence mismatches (Figures S7C, S7E, S7G, S7I, S7K, and S7M). On the other hand, LbCas12a and ErCas12a demonstrated minimal to no background fluorescence with crS-B.1.1.7 targeting WT SARS-CoV-2 gRNA (Figures S7F, S7G, S7J, and S7K). These results, along with PAM preference, enabled us to select ErCas12a for S gene detection in patient samples.
We proceeded to validate Cas12a detection capabilities in patient samples while comparing WT to noncanonical crRNAs. Primers were designed and optimized for the RT-LAMP reaction to amplify N and S segments of the genomic SARS-CoV-2 RNA.12,32–34 Once amplified, N gene segments were detected with a series of ErCas12a, TsCas12a, BsCas12a, and BoCas12a with both WT and noncanonical crRNAs. S gene segments were verified using ErCas12a with both WT and B.1.1.7 crRNA, and finally, a quality control check of RNase P with LbCas12a. In total, 50 nasopharyngeal swabs, 20 tracheal aspirates, and 45 saliva samples were validated with 96.5% accuracy (Figures 5A–5C; Tables S2 and S3). In some cases, noncanonical crRNAs outperformed WT detection, such as the case with ErCas12a combined with CMacrRNA (Figure 5D). Although the performance was not enhanced in the case of BoCas12a, BsCas12a, and TsCas12a with noncanonical crRNAs, we observed their flexibility in adapting to changes in the crRNA sequence (Figures 5E–5G). All of the samples were also confirmed with qRT-PCR, with a Ct standard of ≤40.0 considered a positive sample. In the failed detection instances (4 false negatives), all of the failed samples were nasopharyngeal swabs with a higher Ct value between 35 and 39 and it was expected that the RT-LAMP reaction failed, likely because these samples were obtained sometime in May 2020 and had undergone multiple freeze-thaw cycles. Interestingly, out of 50 nasopharyngeal swabs, we identified 3 swabs that tested positive for the B.1.1.7 lineage. Finally, we did not observe any false positives in our assay, indicating high specificity.
Figure 5. Validation of patient samples using new Cas12a variants with cCRISPR assay.

The 4 proteins, ErCas12a, TsCas12a, BsCas12a, and BoCas12a, with the best combination of crRNA candidates were selected and tested on patient samples. Notably, some noncanonical crRNAs exhibit enhanced trans-cleavage with higher fluorescence signals compared to the WT crRNA.
(A–C) CRISPR detection consisting of positive and negative SARS-CoV-2 determination and detection of B.1.1.7 variant via fluorescence reporter assay in nasopharyngeal swabs (A), saliva (B), and tracheal aspirate (C) samples, respectively. The fluorescence shown above was taken at t = 15 min, except for RNase P, taken at t = 30 min.
(D–G) Detection performance of ErCas12a (D), BoCas12a (E), BsCas12a (F), and TsCas12a (G) with canonical and noncanonical crRNA. N-gene+ clinical samples from nasopharyngeal swabs, saliva samples, and tracheal aspirates were pooled together to evaluate the combinatorial performance of these Cas12a nucleases. A single experiment was performed to simulate the real-world scenario.
DISCUSSION
CRISPR-Cas12a systems have been widely employed for gene editing and nucleic acid detection due to their robust cis- and trans-cleavage activities. Even though there exists a wide range of Cas12a orthologs, a limited number have been thoroughly investigated, with most applications relying on the common orthologs LbCas12a, AsCas12a, and FnCas12a and other groups characterizing novel Cas12a orthologs such as ObCas12a and ScCas12a.4,5,7,13,15–20,35 There is a need for in-depth exploration to develop highly efficient and broadly applicable Cas12a systems; hence, we carried out phylogenetic analyses of 23 Cas12a orthologs. Our analyses supported previous studies that showed that the stem-loop regions of the crRNA scaffold vary significantly, yet the stem left and stem right regions between orthologs are nearly identical.3,17,20 Using biochemical assays in a combinatorial fashion, we demonstrated that noncanonical crRNAs from different orthologs are tolerated by Cas12a, and in some cases enhance cis- and trans-cleavage activities. Furthermore, we determined that Cas12a can recruit truncated versions of crRNA that adequately facilitate protein function. Through these experiments, we confirmed that the variable loop region of the crRNA is not necessary, yet it can be engineered for enhanced activity and unique applications.
Teng et al. showed that optimization of the loop region of the crRNA can improve gene editing efficiency.17 Additionally, chemical modifications to nucleotides within the crRNA spacer sequence have been shown to increase Cas9 and Cas12a specificity.36 To further explore the adaptability of Cas12a for gene-editing purposes, we performed a screen of 207 different Cas12a:crRNA combinations for activity in mammalian cells. Many orthologs, including AsCas12a, ErCas12a, BsCas12a, and FnCas12a, produced indels when paired with noncanonical crRNA at levels equivalent to that of the WT. ErCas12a was observed to have the highest overall activity, even with noncanonical crRNA, making it a candidate for other genetic engineering systems. As a proof of concept, we installed an MS2 aptamer to the crRNA of ErCas12a. We demonstrated that the complex was stable and capable of indel formation at the region of interest inside of HEK293T cells. Future work is required to confirm the capability of this complex for targeted gene activation/interference.
In addition to gene editing applications, we explored the potential of combinatorial CRISPR-Cas12a systems for nucleic acid detection. CRISPR-based diagnostic platforms have been gaining attention and progressively advancing in the last 5 years. The development of CRISPR-Cas detection systems focuses on a next-generation diagnostic test that can outperform the gold standard qRT-PCR method in terms of cost, simplicity, and time.9,11,12,32,37,38 The STOPCovid (specific high-sensitivity enzymatic reporter unlocking testing in one pot) combines a straightforward magnetic-based extraction of SARS-CoV-2 and a thermostable Cas12b into an all-in-one detection reaction at a single temperature.9 This method exhibits a sensitivity of 93.1% and greatly reduces carryover contamination. Although these existing technologies rely on a handful of Cas12a, Cas12b, and Cas13a orthologs with robust trans-cleavage activity, a myriad of CRISPR-Cas systems remain unexplored.
To expand the diagnostic toolbox and to demonstrate the adaptability of Cas12a systems, we surveyed the detection capabilities of BoCas12a, BsCas12a, ErCas12a, and TsCas12a with both canonical and noncanonical crRNA in clinical samples. In tandem with the combinatorial detection of the SARS-CoV-2 N gene, we also used the high specificity of ErCas12a to develop an assay that has the potential to recognize SARS-CoV-2 B.1.1.7 lineage. We tested 45 saliva samples, 50 nasopharyngeal swabs, and 20 tracheal aspirates. Out of 115 patient samples, we detected no false positives and 4 false negatives from nasopharyngeal swabs, resulting in 96.5% accuracy.
Here, we demonstrated the adaptability of Cas12a systems and applied the use of noncanonical crRNA for next-generation CRISPR-based diagnostics and genetic engineering. Although we highlighted the enhanced activity and broad applicability of Cas12a with noncanonical crRNA, there is still much room for optimization. Particularly, studies on PAM preference, crRNA and protein sequence optimization, target specificity, and structural and mechanistic studies will advance sensitive and specific detection technologies and enable robust gene editing tools.
Limitations of the study
Although cCRISPR explores 23 distinct CRISPR-Cas12a effectors and 9 crRNAs, there remains a vast diversity of Cas12a systems that have yet to be characterized. Furthermore, we demonstrate that Cas12a tolerates many noncanonical crRNAs, but we have not confirmed the mechanism for this unique property. Our hypothesis, based on structural data, is that the solvent-accessible stem-loop makes minimal interactions with the Cas12a effector, which in turn allows crRNAs with modifications to this region to maintain activity. Additional structural and evolutionary analyses will provide mechanistic insights into the adaptability of Cas12a systems and guide the further engineering of Cas12a and crRNAs for diverse applications.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further inquiries and information regarding resources and reagents should be directed to and will be fulfilled by the lead contact, Piyush Jain (jainp@ufl.edu).
Materials availability
Distribution of Cas12a bacterial expression plasmids can be found on Addgene (plasmid # 174671–174687).
Data and code availability
Data are presented in the main text as well as in the supplementary information. Next generation raw sequencing data is deposited to Harvard Dataverse (https://doi.org/10.7910/DVN/IPMTKS)
This paper does not contain original code.
Any additional information required to re-analyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT PARTICIPANT DETAILS
Subject participant details
For collection and processing of de-identified patient samples, this study was approved as a non-human exempt study from the University of Florida Institutional Review Board (IRB202000781) and all ethical regulations were adhered to the guidelines. In the clinical validation phase, samples were sourced from the Clinical and Translational Science Institute Biorepository at the University of Florida (UF). These included 50 nasopharyngeal swabs (23 SARS-CoV-2 positive, 27 negative), 20 tracheal aspirates (19 SARS-CoV-2 positive, 1 negative), and 45 saliva samples (16 SARS-CoV-2 positive, 29 negative). Inclusion criteria for positive samples were males and females aged 2 years or older testing positive for SARS-CoV-2, while negative samples included those testing negative for SARS-CoV-2 by a CDC recommended method.
Cell lines
Cas12a orthologs were expressed in Rosetta 2(DE3)pLysS Singles Competent Cells (Millipore Sigma Catalog #70956) following the supplier’s protocol. To initiate transformation, the frozen cells were thawed from −80°C, incubated with plasmids for 5 min on ice, heat-shocked for 30 s at 42°C in a water bath, incubated on ice for 2 min, and then cultured at 37°C in SOC medium (New England Biolabs Catalog #B9020) at 250 rpm for 1 h before being spread on antibiotic-containing agar plates. Post spreading, the plates were incubated at 37°C for 12–48 h, and subsequently, the colonies were selected, expanded in 10 mL Luria Broth (Fisher Scientific Catalog #BP9723) containing the appropriate antibiotics, and subjected to Sanger sequencing. For additional details, please consult the protein expression and purification section.
HEK293T cells were sourced from ATCC (Cat# CRL-3216) and were cultured in D10 medium (Dulbecco’s Modified Eagle’s Medium [DMEM, Gibco] supplemented with 1% Penicillin/Streptomycin and 10% FBS) at 37°C with 5% CO2. Cells intended for downstream transfection and nucleofection procedures were maintained within passages 5 to 15. Any cells beyond the 15th passage were disposed of.
METHOD DETAILS
Phylogenetic analysis of Cas12a and mature crRNA orthologs
Protein sequences of 23 Cas12a and their corresponding mature crRNA orthologs were retrieved from previous studies and compiled into a collection in Geneious Prime. The collection was then subjected to multiple alignments with MUSCLE to generate phylogenetic clustering. The percent identities were extracted from Geneious Prime and plotted in Graphpad Prism 9.0. Alignment of crRNA orthologs was performed using Clustal Omega and visualized in Geneious.
Plasmid construction
Plasmids for mammalian cell expression of Cas12a were gifted from various labs (Zhang, Kleinstiver, Li, and Ekker).3,17,20,40,41 The plasmid encoding crRNAs under U6 promoter was obtained from Joung and Kleinstiver labs (Addgene #114087) and subsequently modified at the scaffold region to create the nine distinct crRNAs via site-directed mutagenesis.
A total of 23 proteins were expressed using the sample procedures described below. Nineteen plasmids containing human codon-optimized Cas12a genes were obtained from Addgene (a gift from Zhang lab, Ekker Lab, and Li Lab).3,17,20,40,41 and PCR amplified using Q5 hot start high-fidelity DNA polymerase (New England Biolabs, Catalog #M0493S), and subcloned into a bacterial expression vector (Addgene plasmid #29656, a gift from Scott Gradia). The product plasmids were then transformed into Rosetta(DE3)pLysS Competent Cells (Millipore Sigma, Catalog #70956) following the manufacturer’s protocols. For LbCas12a, AsCas12a, FnCas12a, and MbCas12a, protein expression plasmids were obtained from Addgene and used directly for protein expression.3,42,43
Protein expression and purification
For protein expression, individual colonies were picked and inoculated in 10 mL LB broth for 12 h. The culture was then transferred to a 1.5 L TB broth mix for scale-up and grown until an OD600 = 0.6–0.8. The culture was placed on ice for 45 min to 1 h prior to the addition of Isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM. The culture was then grown overnight at 16°C for 14–18 h.
Cell pellets were collected by centrifuging the overnight culture at 10,000×g for 5 min. The cells were then resuspended in lysis buffer (500 mM NaCl, 50 mM Tris-HCl, pH = 7.5, 20 mM Imidazole, 0.5 mM TCEP, 1 mM PMSF, 0.25 mg/mL Lysozyme, and DNase I). The cell mixture was subjected to sonication followed by centrifugation at 39800 ×g for 30 min. The cell lysate was filtered through a 0.22 μm syringe filter (Cytiva, Catalog #9913–2504) and then run through 5 mL Histrap FF (Cytiva, Catalog #17525501, Ni2+ was stripped off and recharged with Co2+) pre-equilibrated with Wash Buffer A (500 mM NaCl, 50 mM Tris-HCl, pH = 7.5, 20 mM Imidazole, 0.5 mM TCEP) connected to BioLogic DuoFlow FPLC system (Bio-rad). The column was eluted with Elution Buffer B (500 mM NaCl, 50 mM Tris-HCl, pH = 7.5, 250 mM Imidazole, 0.5 mM TCEP). The eluted fractions were pooled together and transferred to a 10–14 kDa MWCO dialysis bag. Homemade TEV protease (plasmid was obtained as a gift from David Waugh,39 Addgene #8827, and purified in-house) was added to the bag, submerged in Dialysis Buffer (500 mM NaCl, 50 mM HEPES, pH 7, 5 mM MgCl2, 2 mM DTT) and dialyzed at 4°C overnight.
The protein mixture was taken out of the dialysis bag and concentrated down to around 10 mL using a 30 kDa MWCO Vivaspin 20 concentrator. The concentrate was then equilibrated with 10 mL of Wash Buffer C (150 mM NaCl, 50 mM HEPES, pH = 7, 0.5 mM TCEP) prior to injecting into 1 mL Hitrap Heparin HP column pre-equilibrated with Wash Buffer C operated in the BioLogic DuoFlow FPLC system (Bio-rad). The protein was eluted from the column by running a gradient flowrate that exchanges Wash Buffer C and Elution Buffer D (2000 mM NaCl, 50 mM HEPES, pH = 7, 0.5 mM TCEP). Depending on how pure the protein samples were, additional size-exclusion chromatography may have been needed. In short, the eluted protein from the previous step was run through a HiLoad 16/600 Superdex (Cytiva, Catalog #28989335). The purest protein was picked from the eluted fractions, pooled together, concentrated using 30 kDa MWCO Vivaspin 20 concentrator, snap-frozen in liquid nitrogen, and stored at −80°C until use.
Nucleic acid preparation
The dsDNA activators used for screening and investigating the trans-cleavage activity of 22 Cas12a orthologs were purchased as 40-mer oligos from Integrated DNA Technologies. The crRNA sequences used for screening were purchased as ssDNA oligos with the T7 promoter sequence. The ssDNA was then annealed with the T7 complementary sequence followed by in vitro transcription to produce crRNAs. For detection of patient samples, synthetic crRNAs were purchased from Integrated DNA Technologies.
Trans-cleavage reporter assay
The experiment was carried out by pre-complexing 50 nM Cas12a and 62.5 nM crRNA in 1X NEBuffer 2.1 (New England Biolabs) at 37°C for 15 min. The mixture was then transferred to a medium-binding 384-well plate (FLUOTRAC 200, Greiner One) with 250 nM fluorophore-quencher reporter and nucleic acid activator to a final volume of 50 μL. The plate was immediately placed in BioTek Synergy 2 (Agilent) for fluorescence measurements. Fluorescence intensity for a FAM reporter was measured at wavelengths λex: 483/30 nm and λem: 530/30 nm every 2.5 min for 1 h. For trans-cleavage reactions involving truncated crRNAs, the crRNA:Cas12a was complexed at 37°C for 30 min.
In vitro cis-cleavage assay
Combinatorial investigation of crRNAs with each Cas12a was carried out by mixing crRNA, Cas12a, and dsDNA target to a final concentration of 100 nM:50 nM:6.15 nM in 1X NEBuffer 2.1. The mixing was performed in a PCR 96-well plate (Applied Biosystems) in the automated Opentrons OT-II robot and incubated at 37°C for 30 min in the robot thermal module. Proteinase K was added to the mixture to quench the reaction. The cleavage product was then analyzed in 1% agarose gel.
The above protocol was applied for all Cas12a except for PdCas12a due to its decreased reaction with the non-canonical PAM sequence. The RNP complex was assembled at a concentration 200 nM:200 nM:6.15 nM of PdCas12a:crRNA:dsDNA and incubated at 37°C for 1 h.
For cis-cleavage reactions involving truncated crRNA, the crRNA:Cas12a was complexed at 37°C for 30 min followed by the addition of dsDNA target and incubated at 37°C for 1 h. The reaction was then quenched with proteinase K prior to gel electrophoresis.
Differential scanning fluorimetry
The reaction was performed by combining Cas12a and crRNA to a final concentration of 1 μM: 2.5 μM in a combination of Protein Thermal Shift buffer (Thermo Fisher) and 1x cleavage buffer (100 nM NaCl, 50 nM Tris-HCl, pH = 7.5, 1 mM DTT, and 10 mM MgCl2). The complex was incubated at 37°C for 15 min. The product was then transferred to a PCR 96-well plate (Applied Biosystems) with pre-added Protein Thermal Shift dye (Thermo Fisher), and protein melting was carried out in the qPCR StepOne Plus system (Thermo Fisher) over a temperature range of 25–80°C with a ramp rate of 1%/second. The experiment was repeated twice with 2 replicates per experiment.
Patient sample collection and extraction
Nasopharyngeal swabs and saliva samples were obtained from the University of Florida (UF) Clinical and Translational Science Institute adhering to the guidelines approved by UF Institutional Review Board (protocol# IRB202000781). Nucleic acids extracted from the samples were carried out in an automated Maxwell RSC 16 system (Promega) using Maxwell RSC Viral Total Nucleic Acid Purification Kit (Promega) for both nasopharyngeal swabs and saliva, following separate protocols from the manufacturer. For tracheal aspirate, the extraction was carried out using the Maxwell RSC Blood DNA Kit (Promega).
RT-qPCR
Patient samples underwent RT-qPCR to determine the Ct values. The RT-qPCR experiments were carried out following the CDC 2019-Novel Coronavirus (2019-nCoV) Real-Time RT-PCR Diagnostic Panel. In short, a master mix of Luna Probe One-Step RT-qPCR 4X Mix with UDG, RNase-free water, and N1/N2/RNase P (RP) primer mix was assembled according to manufacturer’s instructions. The master mixes were transferred into a 96-well plate (Applied Biosystems). The reaction was initiated by adding 5 μL of the extracted patient into each well and transferring to the StepOnePlus Real-Time PCR system (Applied Biosystems) for fluorescence measurements.
SARS-CoV-2 detection assay
Extracted nucleic acid from patient samples above was input into the RT-LAMP reaction. 5 μL of the extracted sample was added to WarmStart Colorimetric LAMP 2X Master Mix with UDG to a final volume of 25 μL. The reaction was incubated at 62°C for 35 min. Next, 2 μL of the RT-LAMP product was added to the CRISPR reaction described in the trans-cleavage assay above, except the reaction was carried out in a high-throughput manner using the automated Opentrons OT-II robot. Fluorescence intensity measurements were recorded and analyzed in GraphPad Prism 9.0.
Mammalian cell culture conditions
HEK293T cells were obtained from ATCC (Cat# CRL-3216) and maintained in D10 medium (Dulbecco’s Modified Eagle’s Medium [DMEM, Gibco] supplemented with 1% Penicillin/Streptomycin and 10% FBS) at 37°C and 5% CO2. Cells used for downstream transfection and nucleofection were between passages 5 and 15. Cells were discarded after 15 passages.
Cell culture transfection and nucleofection
For plasmid transfection, cells were seeded in a 24-well plate at a density of 2.5 × 105 cells/mL (1.25 × 105 cells/well) 24 h prior to transfection. On the day of transfection, 500 ng each of Cas12a and crRNA plasmids were mixed with 2 μL of TransIT-X2 Dynamic Delivery System (Mirus Bio) in 50 μL of Opti-MEM I Reduced-Serum Medium (Thermo Fisher) and incubated for 15 min at room temperature. Solution was then added dropwise to wells. Cells were harvested after 48–72 h.
For nucleofection, 200 pmol of Cas12a was mixed with 720 pmol of crRNA in SF nucleofector solution followed by 20 min incubation at room temperature to create RNP complex. Next, 5 × 105 cells were added to the RNP to a final volume of 100 μL and transferred to the nucleofection cuvette (SF Cell Line 4D X Kit L, Lonza). The RNP/cells mixture was electroporated with the preset program DS-150 (4D-Nucleofector X Unit, Lonza) then mixed with 400 μL of DMEM and added to a 24-well plate. DMEM was replaced with D10 medium after 6 h. Cells were harvested after an additional 48–72 h.
Genomic DNA library preparation and next generation sequencing
Cells were harvested and lysed using QuickExtract DNA Extraction Solution (Biosearch Technologies) according to the manufacturer’s protocol. Genomic DNA from different transfected samples were amplified using homemade Pfu-sso7d polymerase and appended with Illumina adapters via two-step PCR. Next, samples were pooled together and loaded onto an Illumina Miseq. Sequencing data were demultiplexed then analyzed with CRISPResso2.31 Indel percentage was calculated as the number of reads containing insertions and/or deletions divided by the total reads.
QUANTIFICATION AND STATISTICAL ANALYSIS
Next generation sequencing data of gene editing outcomes were quantified using CRISPResso2 with default settings for Cpf1. For SARS-CoV-2 detection experiments, patient samples containing positive SARS-CoV-2 pathogens were randomized and blinded. Data were plotted using GraphPad Prism 9 (GraphPad Software, San Diego, CA) with mean ± SD.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Rosetta™ 2(DE3)pLysS Singles Competent Cells | Millipore Sigma | Cat# 71401 |
| NEB® 5-alpha Competent E. coli (High Efficiency) | New England Biolabs | Cat# C2987H |
|
| ||
| Biological samples | ||
|
| ||
| Nasopharyngeal Swab Samples | University of Florida Clinical and Translational Science Institute | N/A |
| 2019-nCoV positive Saliva Samples | University of Florida Clinical and Translational Science Institute | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Q5® Polymerase | New England BioLabs | Cat# M0493S |
| Thermo Scientific™ PMSF Protease Inhibitor | Fisher Scientific | Cat# 36978 |
| Lysozyme | MP Biomedicals, LLC | Cat# 02100831-CF |
| Deoxyribose Nuclease I (Bovine Pancreas) | Worthington Biomedical Corporation | Cat# LS002145 |
| TEV protease | Kapust et al.39 | Addgene #8827 |
| Proteinase K | New England BioLabs | Cat# P8107S |
| DMEM, high glucose | Thermo Fisher (Gibco) | Cat# 11965092 |
| TransIT-X2 Dynamic Delivery System | Mirus Bio | Cat# MIR 6004 |
| Opti-MEM™ I Reduced Serum Medium | Thermo Fisher (Gibco) | Cat# 31985062 |
| SF Cell Line 4D-Nucleofector™ X Kit L | Lonza | Cat# V4XC-2024 |
| QuickExtract DNA Extraction Solution | Biosearch Technologies | Cat# SS000035-D2 |
| See Table S4 (S4) for recombinant protein sequences | This paper | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| Q5® Site-Directed Mutagenesis Kit | New England BioLabs | Cat# E0554S |
| HiScribe® T7 High Yield RNA Synthesis Kit | New England BioLabs | Cat# E2040S |
| Protein Thermal Shift™ Dye Kit | Thermo Fisher Scientific | Cat# 4461146 |
| Maxwell® RSC Viral Total Nucleic Acid Purification Kit | Promega | Cat# ASB1330 |
| Maxwell® RSC Blood DNA Kit | Promega | Cat# AS1400 |
| Luna® Probe One-Step RT-qPCR 4X Mix with UDG | New England Biolabs | Cat# M3019 |
| WarmStart® Colorimetric LAMP 2X Master Mix with UDG | New England Biolabs | Cat# M1804 |
| MiSeq Reagent Nano Kit v2 (300-cycles) | Illumina | Cat# MS-103–1001 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK293T cell Line | ATCC | Cat# CRL-3216 |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S4 (S4) for sequences | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pET His6 MBP TEV LIC cloning vector (1M) | N/A | Addgene plasmid #29656 |
| pUC19-U6-FnCas12a crRNA-BsmBI cassette (BPK4446) | Kleinstiver et al.40 | Addgene plasmid #114087 |
| pY126 (pcDNA3.1-huBoCpf1) | Zetsche et al.3 | Addgene plasmid #92302 |
| pY125 (pcDNA3.1-huBfCpf1) | Zetsche et al.3 | Addgene plasmid #92301 |
| pY123 (pcDNA3.1-huCMaCpf1) | Zetsche et al.3 | Addgene plasmid #92299 |
| pY120 (pcDNA3.1-huFbCpf1) | Zetsche et al.3 | Addgene plasmid #92296 |
| pY119 (pcDNA3.1-huLb5Cpf1) | Zetsche et al.3 | Addgene plasmid #92295 |
| pY117 (pcDNA3.1-huMb3Cpf1) | Zetsche et al.3 | Addgene plasmid #92293 |
| pY116 (pcDNA3.1-huMb2Cpf1) | Zetsche et al.3 | Addgene plasmid #92292 |
| pY115 (pcDNA3.1-huMlCpf1) | Zetsche et al.3 | Addgene plasmid #92275 |
| pY113 (pcDNA3.1-huPb2Cpf1) | Zetsche et al.3 | Addgene plasmid #92272 |
| pY111 (pcDNA3.1-huTsCpf1) | Zetsche et al.3 | Addgene plasmid #92267 |
| pY016 (pcDNA3.1-hLbCpf1) | Zetsche et al.3 | Addgene plasmid #69988 |
| pY017 (pcDNA3.1-hPcCpf1) | Zetsche et al.3 | Addgene plasmid #69989 |
| pY018 (pcDNA3.1-hPdCpf1 ) | Zetsche et al.3 | Addgene plasmid #69990 |
| pY012 (pcDNA3.1-hCMtCpf1) | Zetsche et al.3 | Addgene plasmid #69984 |
| 6His-MBP-TEV-huLbCpf1 | Zetsche et al.3 | Addgene plasmid #90096 |
| 6His-MBP-TEV-huAsCpf1 | Zetsche et al.3 | Addgene plasmid #90095 |
| 6His-MBP-TEV-huFnCpf1 | Zetsche et al.3 | Addgene plasmid #90094 |
| pCAG-ArCas12a-2AeGFP | Teng et al.17 | Addgene plasmid #123632 |
| pCAG-BsCas12a-2AeGFP | Teng et al.17 | Addgene plasmid #123633 |
| pCAG-HkCas12a-2AeGFP | Teng et al.17 | Addgene plasmid #123634 |
| pCAG-LpCas12a-2AeGFP | Teng et al.17 | Addgene plasmid #123635 |
| pCAG-PrCas12a-2AeGFP | Teng et al.17 | Addgene plasmid #123636 |
| pCAG-PxCas12a-2AeGFP | Teng et al.17 | Addgene plasmid #123637 |
| pCMV-T7-AsCas12a-P2A-EGFP (RTW2861) | Kleinstiver et al.40 | Addgene plasmid #160140 |
| pCAG-hFnCas12a-NLS(nucleoplasmin)-3xHA (AAS1472) | Kleinstiver et al.40 | Addgene plasmid #114089 |
| pErCas12a EZ Clone | Wierson et al.41 | Addgene plasmid #132641 |
| pKEW336-MBP-TEV-MbCas12a-33362 | Watters et al.42 | Addgene plasmid #115670 |
| 6xHis-MBP-huArCas12a | This paper | Addgene plasmid #174670 |
| 6xHis-MBP-huBsCas12a | This paper | Addgene plasmid #174671 |
| 6xHis-MBP-huBfCas12a | This paper | Addgene plasmid #174672 |
| 6xHis-MBP-huBoCas12a | This paper | Addgene plasmid #174673 |
| 6xHis-MBP-huPrCas12a | This paper | Addgene plasmid #174674 |
| 6xHis-MBP-huPxCas12a | This paper | Addgene plasmid #174675 |
| 6xHis-MBP-huMb2Cas12a | This paper | Addgene plasmid #174676 |
|
| ||
| Software and algorithms | ||
|
| ||
| Geneious Prime | Dotmatics | https://www.geneious.com/ |
| MUSCLE | European Bioinformatics Institute | https://www.ebi.ac.uk/Tools/msa/muscle/ |
| Clustal Omega | European Bioinformatics Institute | https://www.ebi.ac.uk/Tools/msa/clustalo/ |
| Graphpad Prism 9.0. | Graphpad Software | https://www.graphpad.com/features |
| CRISPResso2 | Clement et al.31 | http://crispresso2.pinellolab.org/submission |
|
| ||
| Other | ||
|
| ||
| Terrific Broth | Cold Spring Harbor Protocols | https://cshprotocols.cshlp.org/content/2015/9/pdb.rec085894.full?rss=1 |
| Antifoam 204 | Millipore Sigma | Cat# A8311–50ML |
| Millex™-GP 0.22-mm Filter | Millipore Sigma | Cat# SLGPM33RS |
| Histrap 5 mL FF Column | Cytiva Life Sciences | Cat# 17525501 |
| 10–14 kDa MWCO Dialysis Bag | Ward’s Science | Cat# 470163–406 |
| 30 kDa MWCO Vivaspin® 20 concentrator | Cytiva Life Sciences | Cat# 28932361 |
| Hitrap 1 mL SP HP Column | Cytiva Life Sciences | Cat# 29051324 |
| HiLoad® 16/600 Superdex® | Cytiva Life Sciences | Cat# 28989335 |
| NEBuffer™ 2.1 | New England Biolabs | Cat# B9200S |
| Greiner Bio-One FLUOTRAC™ 200 96-well Microplate | Fisher Scientific | Cat# 07–000-722 |
| Applied Biosystems™ MicroAmp™ Optical 96-Well Reaction Plate | Fisher Scientific | Cat# N8010560 |
Highlights.
Engineered crRNAs with variable stem-loop designs demonstrate adaptability with Cas12a
cCRISPR broadens the gene editing toolbox by screening 207 combinations of Cas12a and crRNAs
cCRISPR detects SARS-CoV-2 in saliva, nasopharyngeal swabs, and tracheal aspirates
ACKNOWLEDGMENTS
We are thankful for valuable advice regarding experiment setup from members of the Jain lab at the University of Florida (UF). We are grateful to Dr. Timothy Hamerly from Dr. Rhoel Dinglasan’s lab for suggestions on optimizing the DSF experiments. We also thank Dr. Cuong Nguyen, Dr. David Ostrov, Dr. Rhoel Dinglasan, and Dr. Marco Salemi for their support in obtaining the patient samples. We also thank the UF ICBR NextGen DNA Sequencing facility staff, especially Benjamin Hoffman, for coordinating the next-generation sequencing experiments. Finally, we are grateful to UF, the UF Health Cancer Center, and the Clinical and Translational Science Institute Repository for their support. The following reagents were obtained through BEI Resources, the National Institute of Allergy and Infectious Diseases (NIAID), and the NIH: genomic RNA from SARS-Related Coronavirus 2, Isolate USA-WA1/2020, NR-52285. Genomic RNA from SARS-Related Coronavirus 2, Isolate USA/CA_CDC_5574/2020 (Lineage B.1.1.7), NR-55244, was contributed by the Centers for Disease Control and Prevention. This work was in part supported by funds from UF, the UF Herbert Wertheim College of Engineering, the Florida Breast Cancer Foundation (AGR00018466), the United States-India Science & Technology Endowment Fund (USISTEF/COVID-I/247/2020), the National Institute of General Medical Sciences (R35GM147788), and the NIAID (R21AI168795) of the NIH. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
DECLARATION OF INTERESTS
P.K.J. and L.T.N. are listed as inventors on patent applications related to the content of this work. Provisional patents (63/222,251 and 63/191,647) have been filed covering the methods and compositions of combinatorial CRISPR-Cas complexes. P.K.J. is a cofounder of Genable Biosciences LLC, Par Biosciences LLC, and CRISPR LLC.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.113777.
REFERENCES
- 1.Port F, Starostecka M, and Boutros M (2020). Multiplexed conditional genome editing with Cas12a in Drosophila. Proc. Natl. Acad. Sci. USA 117, 22890–22899. 10.1073/pnas.2004655117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dong Z, Qin Q, Hu Z, Zhang X, Miao J, Huang L, Chen P, Lu C, and Pan M (2020). CRISPR/Cas12a Mediated Genome Editing Enhances Bombyx mori Resistance to BmNPV. Front. Bioeng. Biotechnol. 8, 841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, et al. (2015). Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771. 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campa CC, Weisbach NR, Santinha AJ, Incarnato D, and Platt RJ (2019). Multiplexed genome engineering by Cas12a and CRISPR arrays encoded on single transcripts. Nat. Methods 16, 887–893. 10.1038/s41592-019-0508-6. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, Ren Q, Tang X, Liu S, Malzahn AA, Zhou J, Wang J, Yin D, Pan C, Yuan M, et al. (2021). Expanding the scope of plant genome engineering with Cas12a orthologs and highly multiplexable editing systems. Nat. Commun. 12, 1944. 10.1038/s41467-021-22330-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jia H, Orbovic V, and Wang N (2019). CRISPR-LbCas12a-mediated modification of citrus. Plant Biotechnol. J. 17, 1928–1937. 10.1111/pbi.13109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gootenberg JS, Abudayyeh OO, Kellner MJ, Joung J, Collins JJ, and Zhang F (2018). Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 360, 439–444. 10.1126/science.aaq0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, Verdine V, Donghia N, Daringer NM, Freije CA, et al. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442. 10.1126/science.aam9321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joung J, Ladha A, Saito M, Kim N-G, Woolley AE, Segel M, Barretto RPJ, Ranu A, Macrae RK, Faure G, et al. (2020). Detection of SARS-CoV-2 with SHERLOCK One-Pot Testing. N. Engl. J. Med. 383, 1492–1494. 10.1056/NEJMc2026172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kellner MJ, Koob JG, Gootenberg JS, Abudayyeh OO, and Zhang F (2019). SHERLOCK: nucleic acid detection with CRISPR nucleases. Nat. Protoc. 14, 2986–3012. 10.1038/s41596-019-0210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patchsung M, Jantarug K, Pattama A, Aphicho K, Suraritdechachai S, Meesawat P, Sappakhaw K, Leelahakorn N, Ruenkam T, Wong-satit T, et al. (2020). Clinical validation of a Cas13-based assay for the detection of SARS-CoV-2 RNA. Nat. Biomed. Eng. 4, 1140–1149. 10.1038/s41551-020-00603-x. [DOI] [PubMed] [Google Scholar]
- 12.Broughton JP, Deng X, Yu G, Fasching CL, Servellita V, Singh J, Miao X, Streithorst JA, Granados A, Sotomayor-Gonzalez A, et al. (2020). CRISPR–Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 38, 870–874. 10.1038/s41587-020-0513-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen JS, Ma E, Harrington LB, Da Costa M, Tian X, Palefsky JM, and Doudna JA (2018). CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436–439. 10.1126/science.aar6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nguyen LT, Rananaware SR, Pizzano BLM, Stone BT, and Jain PK (2022). Clinical validation of engineered CRISPR/Cas12a for rapid SARS-CoV-2 detection. Commun. Med. 2, 7. 10.1038/s43856-021-00066-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li S-Y, Cheng Q-X, Liu J-K, Nie X-Q, Zhao G-P, and Wang J (2018). CRISPR-Cas12a has both cis- and trans-cleavage activities on single-stranded DNA. Cell Res. 28, 491–493. 10.1038/s41422-018-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Z, Schiel JA, Maksimova E, Strezoska Z, Zhao G, Anderson EM, Wu Y, Warren J, Bartels A, van Brabant Smith A, et al. (2020). ErCas12a CRISPR-MAD7 for Model Generation in Human Cells, Mice, and Rats. CRISPR J. 3, 97–108. 10.1089/crispr.2019.0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teng F, Li J, Cui T, Xu K, Guo L, Gao Q, Feng G, Chen C, Han D, Zhou Q, and Li W (2019). Enhanced mammalian genome editing by new Cas12a orthologs with optimized crRNA scaffolds. Genome Biol. 20, 15. 10.1186/s13059-019-1620-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamano T, Zetsche B, Ishitani R, Zhang F, Nishimasu H, and Nureki O (2017). Structural Basis for the Canonical and Non-canonical PAM Recognition by CRISPR-Cpf1. Mol. Cell 67, 633–645.e3. 10.1016/j.molcel.2017.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yue H, Shu B, Tian T, Xiong E, Huang M, Zhu D, Sun J, Liu Q, Wang S, Li Y, and Zhou X (2021). Droplet Cas12a Assay Enables DNA Quantification from Unamplified Samples at the Single-Molecule Level. Nano Lett. 21, 4643–4653. 10.1021/acs.nanolett.1c00715. [DOI] [PubMed] [Google Scholar]
- 20.Zetsche B, Abudayyeh OO, Gootenberg JS, Scott DA, and Zhang F (2020). A Survey of Genome Editing Activity for 16 Cas12a Orthologs. Keio J. Med. 69, 59–65. 10.2302/kjm.2019-0009-OA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fonfara I, Richter H, Bratovič M, Le Rhun A, and Charpentier E (2016). The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532, 517–521. 10.1038/nature17945. [DOI] [PubMed] [Google Scholar]
- 22.Dong D, Ren K, Qiu X, Zheng J, Guo M, Guan X, Liu H, Li N, Zhang B, Yang D, et al. (2016). The crystal structure of Cpf1 in complex with CRISPR RNA. Nature 532, 522–526. 10.1038/nature17944. [DOI] [PubMed] [Google Scholar]
- 23.Swarts DC, van der Oost J, and Jinek M (2017). Structural Basis for Guide RNA Processing and Seed-Dependent DNA Targeting by CRISPR-Cas12a. Mol. Cell 66, 221–233.e4. 10.1016/j.molcel.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swarts DC, and Jinek M (2019). Mechanistic Insights into the cis- and trans-Acting DNase Activities of Cas12a. Mol. Cell 73, 589–600.e4. 10.1016/j.molcel.2018.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao P, Yang H, Rajashankar KR, Huang Z, and Patel DJ (2016). Type V CRISPR-Cas Cpf1 endonuclease employs a unique mechanism for crRNA-mediated target DNA recognition. Cell Res. 26, 901–913. 10.1038/cr.2016.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lorenz R, Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, and Hofacker IL (2011). ViennaRNA Package 2.0. Algorithm Mol. Biol. 6, 26. 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shebanova R, Nikitchina N, Shebanov N, Mekler V, Kuznedelov K, Ulashchik E, Vasilev R, Sharko O, Shmanai V, Tarassov I, et al. (2022). Efficient target cleavage by Type V Cas12a effectors programmed with split CRISPR RNA. Nucleic Acids Res. 50, 1162–1173. 10.1093/nar/gkab1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merker L, Schindele P, and Puchta H (2020). Using CRISPR/ttLbCas12a for in planta Gene Targeting in A. thaliana. Curr. Protoc. Plant Biol. 5, e20117. 10.1002/cppb.20117. [DOI] [PubMed] [Google Scholar]
- 29.Endo A, Masafumi M, Kaya H, and Toki S (2016). Efficient targeted mutagenesis of rice and tobacco genomes using Cpf1 from Francisella novicida. Sci. Rep. 6, 38169. 10.1038/srep38169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo LY, Bian J, Davis AE, Liu P, Kempton HR, Zhang X, Chemparathy A, Gu B, Lin X, Rane DA, et al. (2022). Multiplexed genome regulation in vivo with hyper-efficient Cas12a. Nat. Cell Biol. 24, 590–600. 10.1038/s41556-022-00870-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clement K, Rees H, Canver MC, Gehrke JM, Farouni R, Hsu JY, Cole MA, Liu DR, Joung JK, Bauer DE, and Pinello L (2019). CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226. 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ali Z, Aman R, Mahas A, Rao GS, Tehseen M, Marsic T, Salunke R, Subudhi AK, Hala SM, Hamdan SM, et al. (2020). iSCAN: An RT-LAMP-coupled CRISPR-Cas12 module for rapid, sensitive detection of SARS-CoV-2. Virus Res. 288, 198129. 10.1016/j.virusres.2020.198129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, and Hase T (2000). Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28, e63. 10.1093/nar/28.12.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fang X, Chen H, Yu S, Jiang X, and Kong J (2011). Predicting Viruses Accurately by a Multiplex Microfluidic Loop-Mediated Isothermal Amplification Chip. Anal. Chem. 83, 690–695. 10.1021/ac102858j. [DOI] [PubMed] [Google Scholar]
- 35.Liu X, Qiu X, Han L, Yue Y, Xu S, Li F, Yao J, Sun L, and Li Z (2023). Three novel Cas12a orthologs with robust DNA cleavage activity suitable for nucleic acid detection. Gene 852, 147055. 10.1016/j.gene.2022.147055. [DOI] [PubMed] [Google Scholar]
- 36.Krysler AR, Cromwell CR, Tu T, Jovel J, and Hubbard BP (2022). Guide RNAs containing universal bases enable Cas9/Cas12a recognition of polymorphic sequences. Nat. Commun. 13, 1617. 10.1038/s41467-022-29202-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ding X, Yin K, Li Z, Lalla RV, Ballesteros E, Sfeir MM, and Liu C (2020). Ultrasensitive and visual detection of SARS-CoV-2 using all-in-one dual CRISPR-Cas12a assay. Nat. Commun. 11, 4711. 10.1038/s41467-020-18575-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fozouni P, Son S, Díaz de León Derby M, Knott GJ, Gray CN, D’Ambrosio MV, Zhao C, Switz NA, Kumar GR, Stephens SI, et al. (2021). Amplification-free detection of SARS-CoV-2 with CRISPR-Cas13a and mobile phone microscopy. Cell 184, 323–333.e9. 10.1016/j.cell.2020.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kapust RB, Tözsér J, Fox JD, Anderson DE, Cherry S, Copeland TD, and Waugh DS (2001). Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 14, 993–1000. 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 40.Kleinstiver BP, Sousa AA, Walton RT, Tak YE, Hsu JY, Clement K, Welch MM, Horng JE, Malagon-Lopez J, Scarfò I, et al. (2019). Engineered CRISPR–Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat. Biotechnol. 37, 276–282. 10.1038/s41587-018-0011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wierson WA, Simone BW, WareJoncas Z, Mann C, Welker JM, Kar B, Emch MJ, Friedberg I, Gendron WAC, Barry MA, et al. (2019). Expanding the CRISPR Toolbox with ErCas12a in Zebrafish and Human Cells. CRISPR J. 2, 417–433. 10.1089/crispr.2019.0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watters KE, Fellmann C, Bai HB, Ren SM, and Doudna JA (2018). Systematic discovery of natural CRISPR-Cas12a inhibitors. Science 362, 236–239. 10.1126/science.aau5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, DeGennaro EM, Winblad N, Choudhury SR, Abudayyeh OO, Gootenberg JS, et al. (2017). Multiplex gene editing by CRISPR–Cpf1 using a single crRNA array. Nat. Biotechnol. 35, 31–34. 10.1038/nbt.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are presented in the main text as well as in the supplementary information. Next generation raw sequencing data is deposited to Harvard Dataverse (https://doi.org/10.7910/DVN/IPMTKS)
This paper does not contain original code.
Any additional information required to re-analyze the data reported in this work paper is available from the lead contact upon request.