

Abstract
High levels of lipoprotein(a) [Lp(a)] are causal for atherosclerotic cardiovascular disease (ASCVD). Lp(a) is the most prevalent inherited dyslipidemia and strongest genetic ASCVD risk factor. This risk persists in the presence of at target, guideline-recommended, LDL-C levels and adherence to lifestyle modifications. Epidemiological and genetic evidence supporting its causal role in ASCVD and calcific aortic stenosis continues to accumulate, although various facets regarding Lp(a) biology (genetics, pathophysiology, and expression across race/ethnic groups) are not yet fully understood. The evolving nature of clinical guidelines and consensus statements recommending universal measurements of Lp(a) and the scientific data supporting its role in multiple disease states reinforce the clinical merit to start population screening for Lp(a) now. There is a current gap in the implementation of recommendations for primary and secondary cardiovascular disease (CVD) prevention in those with high Lp(a), in part due to a lack of protocols for management strategies. Importantly, targeted apolipoprotein(a) [apo(a)]-lowering therapies that reduce Lp(a) levels in patients with high Lp(a) are in phase 3 clinical development. This review focuses on the identification and clinical management of patients with high Lp(a). Specifically, we highlight the clinical value of measuring Lp(a) and its use in determining Lp(a)-associated CVD risk by providing actionable guidance, based on scientific knowledge, that can be utilized now to mitigate risk caused by high Lp(a).
Keywords: Lipoprotein(a) [Lp(a)], Cardiovascular Disease (CVD), Lp(a) testing, Risk mitigation
1. Introduction
Despite being discovered several decades ago and having reported associations with cardiovascular disease (CVD) in the 1980s, it is only since the turn of the century that a resurgence of interest in lipoprotein(a) [Lp(a)] biology has occurred [1,2]. This resurgence has been instigated by high quality epidemiological studies, genome wide association studies (GWAS) and Mendelian randomization studies, revealing high Lp(a) as an inherited, independent, and causal CVD risk factor [3], [4], [5]. Furthermore, evidence suggests that high Lp(a) levels contribute to residual CVD risk in patients achieving target low-density lipoprotein cholesterol (LDL-C) goals from apolipoprotein B-100 (apoB)-lowering therapies and following healthy lifestyle changes [6], [7], [8], [9]. There continues to be a rising tide of studies investigating the mechanisms linking high Lp(a) levels to disease development, and the challenges associated with the measurement and assessment of risk. These have been the focus of a recently published scientific statement by the American Heart Association (AHA) [10] and consensus statement from the European Atherosclerosis Society (EAS) [5]. The primary purpose of this paper is to review the scientific evidence that supports management strategies for individuals with high Lp(a); including the importance of screening for Lp(a) levels and providing actionable guidance for mitigating CVD risk in individuals with high Lp(a).
1.1. Prevalence and supporting evidence for Lp(a) links to ASCVD
Using data from the Danish population [11] and UK Biobank [12], which include mostly White populations, it is estimated that approximately one in five individuals worldwide (∼1.5 billion individuals) have high Lp(a) levels (≥100–125 nmol/L [∼≥50 mg/dL]) (Fig. 1A) [13]. In comparison, heterozygous familial hypercholesterolemia (HeFH), a well-established atherosclerotic cardiovascular disease (ASCVD) risk factor, has an estimated worldwide prevalence of 1 in 311 individuals (∼25 million individuals) [14], while a common disease such as diabetes, affects ∼537 million adults worldwide [15]. Despite the high prevalence, the contribution of high Lp(a) to ASCVD risk remains underappreciated in clinical practice [5,16]. Importantly, the recent development of novel therapeutics (glucagon-like peptide [GLP]−1) that are changing the outcomes of individuals with obesity, insulin resistance, and type 2 diabetes mellitus (T2DM) will not address the risk of high Lp(a) as a driver for ASCVD [17]. High Lp(a) varies considerably by race, with sequential increments in population median Lp(a) levels reported in Chinese (16 nmol/L, [∼6.5 mg/dL]), White (19 nmol/L, [∼7.6 mg/dL]), South Asian (31 nmol/L, [∼12.4 mg/dL]) and Black (75 nmol/L, [∼30 mg/dL]) individuals [12]; however similar Lp(a) levels among White and Hispanic individuals have been reported [18]. Substantial differences in Lp(a) levels across racial subgroups exist, yet the relative risk of the incidence of ASCVD appears largely similar with increasing Lp(a) level [12,13,19]. Findings from the Lp(a)HERITAGE study suggest that approximately one in four patients with ASCVD have high Lp(a) (≥125 nmol/L, [∼≥50 mg/dL]); however, the prevalence of high Lp(a) is possibly overestimated due a selection bias of patients enrolled in this study [20]. When patients from Lp(a)HERITAGE were stratified by race in the United States, median Lp(a) levels were more than 2.5-fold higher in Black patients with ASCVD (132 nmol/L, [∼52.8 mg/dL]) compared to the overall group (52 nmol/L, [∼20.8 mg/dL]), with ∼one in two Black patients compared to one in three of the overall group having high Lp(a) [21]. The mechanisms and pathophysiology relating to these higher absolute levels of Lp(a) in Black individuals needs to be furthered studied. It must also be acknowledged that not all individuals with high Lp(a) levels will develop CVD, suggesting that the convergence of additional risk factors in the setting of high plasma Lp(a) is also important for CVD development [22].
Fig. 1.

Overview of the structure, regulation, measurement considerations and level interpretation of Lp(a) and CVD-associated risk. A) Listing of non-genetic and genetic factors that modolute lipoprotein(a) [Lp(a)] levels. Lp(a) levels are primarily determined by variability in the LPA gene which codes for the apolipoprotein(a) [apo(a)] component of the particle. [5]. The prevalence of High Lp(a) levels ranges from 10-30% and varies depending on ancestry [13]. B) The Lp(a) particle comprises a single apolipoprotein B-100 (apoB-100) containing lipoprotein covalently associated with apo(a). Oxidized phospholipids (OxPLs) are covalently associated with apo(a), apoB-100 and the lipid core. Apo(a) consists of 10 kringle IV (KIV) domains, a single KV domain and an inactive, protease-like domain. Different apo(a) isoforms have different KIV2 copy numbers [69]. Recent guidance on important considerations when choosing Lp(a) assays, along with the risk thresholds for determining risk from an Lp(a) measurement are described [5,10]. C) Adjusted hazard ratios for select cardiovascular disease (CVD) outcomes comparing participants in the top percentile of Lp(a) distribution (>95th percentile for calcific aortic valve stenosis [CAVS], ischemic stroke and CV mortality; >99th percentile for myocardial infarction [MI] and heart failure[HF]) versus those with lower Lp(a) levels (<22nd percentile for CAVS; <34th percentile for MI and HF; <50th percentile for ischemic stroke and CV mortality) [45]. Peripheral artery disease (PAD) risk is based on an adjusted odds ratio for Lp(a) >66th percentile compared to <33rd percentile [45]. Atrial fibrillation risk is based on a Mendelian randomization analysis of the odds ratio per 50 nmol/L increase in Lp(a) [55].
*Lp(a) levels are still reported in mg/dL and therefore knowledge of whether the Lp(a) measurement was in nmol/L or mg/dL is crucial; †conversion between nmol/L and mg/dL is a gross estimate. IFCC = International Federation of Clinical Chemistry.
As a lipoprotein, Lp(a) carries cholesterol, triglycerides (TG), phospholipids (of which some are oxidized), cholesteryl esters, and additional apoproteins, each of which can have their own disease-causing risk [23,24]. The Lp(a) particle comprises apolipoprotein(a) [apo(a)] covalently linked to an apoB-containing lipoprotein (Fig. 1B) [3]. ApoB-containing lipoproteins, exemplified by LDL, have a well-established mechanistic and clinical role in CVD [25]. The genetic inheritance of plasma Lp(a) levels is through its apo(a) component [24]. This apoprotein is coded by the LPA gene and shares high structural homology to plasminogen [26]. It is highly polymorphic in size and can potentiate atherothrombosis through additional mechanisms [3,27]. Investigators have described apo(a) as the major carrier of pro-inflammatory oxidized phospholipids (OxPL) with unique proteome characteristics when compared to other apoB containing lipoproteins. Of note, Lp(a) levels predict CVD risk even after apoB levels are accounted for in multivariate models [28], the latter highlighting independent apo(a) driven mechanisms towards development of CVD. Furthermore, despite a higher abundance of LDL particles, a recent analysis of UK Biobank data has determined that Lp(a) is approximately 6-fold more atherogenic than LDL on a per particle basis [29].
Key takeaway: Circulating Lp(a) particles are causal for ASCVD. The components of this particle, and in particular its specific apo(a) component, contribute to its role in disease development.
1.2. Genetics and clinical implications
The expression of the apo(a) component of Lp(a) is highly controlled by the LPA gene. Greater than 90 % of Lp(a) levels are genetically determined, and the high frequency of LPA gene variants makes Lp(a) the most common genetic dyslipidemia [30,31]. This compares to >160 genes that are required to explain between 50 and 70 % of the heritability of other lipoproteins [32]. It is worth noting that the LPA gene has been described as one of the strongest genetic risk factors for coronary artery disease (CAD) [33], with variants that are more potently associated with CAD than LDLR- and proprotein convertase subtilisin/kexin type 9 (PCSK9)-related variants [27]. While Lp(a) levels appear to be relatively constant over a lifetime, there are several conditions that influence Lp(a) levels [30]. Some of these have been linked to the LPA gene promoter region which contains transcription factor binding sites for growth hormones [34] and acute phase responders such as interleukin (IL)−6[35] and possibly other inflammatory cytokines [36], which increase plasma Lp(a) levels (Fig. 1A). Lp(a) levels increase during pregnancy and also increase after menopause suggesting an influence of endogenous estradiol levels on Lp(a) levels in women [37]. Furthermore, exogenous estradiol treatment in the form of menopausal hormone therapy is associated with lower Lp(a) levels [38], [39], [40], but hormone therapy is not recommended as a strategy for the primary purpose of CVD prevention or treatment of dyslipidemia. Androgen deprivation therapy results in an increase in LDL-C, TG, and Lp(a), and a decrease in high-density lipoprotein cholesterol (HDL-C) levels. The effect of testosterone replacement therapy on plasma lipids and lipoproteins is modest and variable but high-dose androgen therapy used by athletes can markedly decrease HDL-C and also reduce Lp(a) levels [41].
With increases in personalized medicine efforts and the observed racial/ethnic variations in Lp(a) levels, it is important to note numerous LPA variants that associate with high and low Lp(a) levels. Briefly, the LPA gene codes for various size polymorphisms within the apo(a) protein [32]. Notably, a highly variable region, the kringle IV type 2 (KIV2), can have between 3 and 40 repeats. With one allele inherited from each parent, unless a null allele is inherited, most individuals express two isoforms. Smaller apo(a) isoforms (containing up to 22 KIV2 repeats) are associated with higher plasma Lp(a) levels, while larger apo(a) isoforms (>22–40 KIV2 repeats) are associated with lower Lp(a) levels [32]. There is some evidence for the higher production of small apo(a) particles as the driver for high Lp(a) levels [42]. The number of KIV2 repeats and genetic variants (ie, single nucleotide polymorphisms [SNPs]) within the KIV2 region can explain 30–70 % of the variability in Lp(a) levels, depending on ancestry (Fig. 1A) [43]. However, substantial individual variations in Lp(a) levels exist within a given isoform size, which is often underappreciated [32]. Further, a variant in the pentanucleotide repeat of the LPA gene promoter region is reported to explain up to 14 % of Lp(a) variation in European individuals, but has no association in Black African individuals [30]. Considering this genetic regulation of Lp(a) levels, cascade screening of Lp(a) in individuals with a family history of premature CVD or high Lp(a) is warranted [5,22]. Moving beyond the LPA gene, variants in APOE and CETP loci are associated with lower Lp(a) levels, while an APOH loci has recently been associated with increased Lp(a) levels [44]. In Supplemental Table 1, we list specific examples of variants associated with LPA and non-LPA loci and their impact on Lp(a) levels in different racial groups.
Key takeaway: In the era of personalized medicine, efforts should be made to understand the role of LPA variants taking ancestry into account. Additionally, the links of variants to disease presentations and overall risk for CVD should be examined. The clinical significance of LPA gene associations with other genes is yet to be determined.
1.3. Lp(a) and clinical presentations
Key observations from a select number of studies support the role of high Lp(a) in CVD risk (summarized in Supplemental Table 2). Regarding ASCVD, high Lp(a) levels are strongly associated with CAD, ischemic stroke, and peripheral artery disease (PAD) (Fig. 1C) [45]. A recent analysis from the Copenhagen General Population Study involving >100,000 individuals has revealed that every 50 mg/dL (105 nmol/L) increment in genetically determined Lp(a) level was associated with a 39 % increased risk of PAD [46]. An observational analysis of the LipidCardio study, comprising a largely Caucasian population, found that patients with suspected or symptomatic CAD and high Lp(a) were associated with a more severe disease presentation and complex-to-treat form of CAD [47]. Beyond ASCVD, the strongest causal association between high Lp(a) and any cardiovascular (CV) endpoint has been observed with calcific aortic valve stenosis (CAVS), the most prevalent form of valvular heart disease (Fig. 1C) [45,[48], [49], [50]]. Indeed, the LPA gene remains the only identified monogenic risk factor for CAVS [10]. Reciprocally, it has been demonstrated that genetically-mediated lowering of Lp(a) is associated with a lower risk of CAVS (37 %), CAD (29 %), PAD (31 %) and stroke (13 %) [51]. This finding suggests the potential clinical benefit of lowering Lp(a) levels as a viable therapeutic strategy, although these observations represent lifetime lowering of Lp(a) and extrapolation to benefit in the secondary prevention setting should be done with caution [51]. Ongoing randomized controlled trials (RCTs) targeting apo(a) lowering and hence lowering Lp(a) levels will provide the first evidence on whether directed therapies provide improved outcomes.
An association between high Lp(a) levels and heart failure (HF) has also been reported. However, the association between Lp(a) and increased risk of HF (occurring in the >90th percentile) is lower compared to the increased risk between Lp(a) and CAD or CAVS (occurring in the >75th percentile) [5,52]. This observation likely reflects the role of CAD and CAVS as underlying causes, but an association with other pathophysiological pathways such as atherosclerosis-associated arterial stiffness and vascular non-compliance could not be excluded [45]. This association of high Lp(a) with HF has been shown in observational studies from the Atherosclerosis Risk in Communities (ARIC) study and the Multi-Ethnic Study of Atherosclerosis (MESA) [53,54].However, the association in the MESA study was only evident in White individuals suggesting further research is required to understand the association between Lp(a) and HF, including the possibility of race-based differences in Lp(a)-mediated risk. Furthermore, recent evidence has demonstrated high Lp(a) as a potential causal mediator of atrial fibrillation (AF); effects which were partly independent of its known effects on ASCVD [55].
Regarding venous thromboembolism (VTE), human genetic studies have not supported a causal role for high Lp(a) and VTE in the general population [56]. Interestingly, secondary analyses from the Further Cardiovascular Outcomes Research with PCSK9 inhibition in Subjects With Elevated Risk (FOURIER) [57] and ODYSSEY OUTCOMES [58] trials of patients with clinically evident CVD randomized to anti-PCSK9 mAbs that lower LDL-C levels, revealed that VTE incidence was associated with higher Lp(a) levels. The anti-PCSK9 mAbs from each trial, which also cause moderate Lp(a) level reductions, were associated with lower VTE incidence among patients with high Lp(a) levels [57,58]. This apparent discrepancy between findings from genetic studies and clinical studies is not clear, but could reflect patients enrolled in these clinical studies having a higher risk of VTE [9]. Definite physiological roles of Lp(a) within hemostasis and coagulation are yet to be defined [59].
Interestingly, studies over the last decade have also revealed associations between very low Lp(a) levels and incident type 2 diabetes mellitus (T2DM), yet the causality remains unknown [5,60]. From a clinical perspective, it will be interesting to monitor bleeding, thrombotic events, and T2DM incidence in RCTs with targeted Lp(a)-lowering therapies, which could shed light on Lp(a) physiology.
Key takeaway: Clinical research data supports high Lp(a) as a driver for the development of CVD, including CAD, MI, Stroke, PAD, and CAVS. The associations with AF and HF require additional validation. Individuals at risk for these events, or those who have been recently diagnosed, should have their Lp(a) level measured. Until targeted therapies are available and their benefit on outcomes is proven, high levels can be used to guide clinicians to enhance and optimize lowering of other CVD risk factors.
1.4. How does Lp(a) link to disease?: Atherogenesis, inflammation, and thrombosis/coagulation
The increased CVD risk mediated by high Lp(a) may be attributed to pro-atherogenic, pro-inflammatory and pro-thrombotic/anti-fibrinolytic properties [61]. Both apoB and apo(a) components, the main proteins on Lp(a), have been shown to be present in the arterial wall [25,62]. A strong, positive correlation between plasma Lp(a) level and its accumulation in atherosclerotic plaque was first reported in 1989 [63], with a subsequent study by van Dijk et al., detecting Lp(a) in all stages of the atherosclerotic process, with substantially greater abundance in advanced atherosclerotic lesions and ruptured plaques from coronary arteries [64]. More recent clinical investigations have demonstrated associations between high Lp(a) levels and vulnerable plaque characteristics, including thin-cap fibroatheroma and accelerated progression of low-attenuation plaque volume (necrotic core), providing a potential mechanistic explanation between high Lp(a) levels and clinical atherothrombotic events [65,66].
The most intriguing evidence for a pro-inflammatory role of Lp(a) in humans was reported by Van der Valk et al., [67]. Here, autologous, peripheral blood mononuclear cells that were reinfused, accumulated at the carotid and aortic arterial walls at significantly higher rates in individuals with high Lp(a) levels, suggesting that individuals with high Lp(a) have enhanced arterial wall inflammation, likely by facilitating monocyte entry [67]. Mechanistic evidence in support of this pro-inflammatory role is underpinned by findings where individuals with high Lp(a) have increased levels of endothelial and monocyte cell activation, and increased trans-endothelial migratory capacity, key initiating steps in atherosclerosis [67,68].
A prominent molecule that appears to drive the pro-inflammatory and subsequent pro-atherogenic properties of Lp(a) is OxPLs [67,69]. Furthermore, pro-calcific properties of Lp(a) have been attributed to its OxPL content, as this damage associated molecular pattern induces osteogenic gene expression and calcification in valvular interstitial cells and is associated with an increased incidence and progression of CAVS [5,70]. Both ASCVD and CAVS feature calcification, however, calcification of atherosclerotic plaque occurs relatively late in the disease process, whereas it features prominently in the earlier stages of CAVS and potentiates the progression of CAVS [69,71,72]. Clinical evidence has demonstrated that high Lp(a) is independently associated with the presence of early-stage aortic calcium, which precedes symptomatic CAVS by many years, and is believed to represent the most favorable timing for therapeutic interventions, such as Lp(a) lowering [73]. A phase 2 study “Lp(a)FRONTIERS CAVS” with the apo(a) antisense oligonucleotide (ASO), pelacarsen, will test this hypothesis (NCT05646381).
A pro-thrombotic role for Lp(a) has been hypothesized since the discovery of high homology (75–99 %) between the apo(a) component of Lp(a) and plasminogen, a key component of fibrinolysis that serves to limit excessive thrombus growth by facilitating clot lysis, via fibrin degradation [74]. Unlike plasminogen, apo(a) lacks an active protease domain, and experimental evidence with recombinant apo(a) supports an anti-fibrinolytic role through competition with plasminogen, directly or indirectly by fibrin binding for plasmin generation by tissue plasminogen activators [75]. However, ex vivo clot lysis times were unaltered following potent and specific apo(a) lowering with pelacarsen [74]. Evidence of a direct, pro-thrombotic role for Lp(a) via platelet activation has been demonstrated [76,77], while apo(a) also interacts with, and inhibits, the anti-coagulant protein, tissue factor pathway inhibitor, which could exacerbate thrombosis [78]. Further studies are required to fully understand the mechanistic (ie, pro-platelet, pro-coagulant, and/or anti-fibrinolytic) role(s) for Lp(a) in thrombosis.
Key takeaway: In patients with high Lp(a), clinicians should understand that the particle is not only pro-atherogenic, but also has pro-inflammatory and pro-thrombotic burden. In addition to optimizing therapies that decrease all CVD risk (ie, lipids, inflammation), imaging modalities such as coronary artery calcium (CAC; see the section on “imaging modalities”) could assist to assess Lp(a)-mediated risk [79].
2. The importance of Lp(a) screening now and how to manage individuals/patients with high Lp(a) levels
2.1. Why measure Lp(a) levels?
Measuring Lp(a) is recommended by numerous clinical guidelines including the 2018 AHA/American College of Cardiology (AHA/ACC) [80] and the 2019 European Society of Cardiology/EAS (ESC/EAS) [81] dyslipidemia guidelines, and by scientific or consensus statements from the National Lipid Association (NLA) [4] and the American Association of Clinical Endocrinologists and American College on Endocrinology (AACE/ACE) [31], respectively (Fig. 2). The AHA/ACC, NLA and AACE/ACE recommend measuring Lp(a) in those with personal and/or family history of premature ASCVD, while testing in moderate- to high-risk individuals is also recommended by the NLA and AACE/ACE [4,31,80]. However, the strongest stance is put forward by the ESC/EAS which recommends that an “Lp(a) measurement should be considered at least once in each adult person's lifetime” [81]. This stance of a universal measurement of Lp(a) at least once in a lifetime, is also now recommended by the Canadian Cardiovascular Society [82], a recent consensus statement on Lp(a) from the EAS [5] and a 2024 focused update to the scientific statement by the NLA [83]. The universal recommendation for an Lp(a) test is particularly important in the context of primary prevention and understanding the risk of CVD events in the absence of traditional risk factors [84]. In this regard, a recent observational analysis of participants from the MESA, followed for a median of 13.4 years, demonstrated that CVD risk in a primary prevention setting significantly increased with high Lp(a), even when LDL-C levels are optimal [85].
Fig. 2.

Clinical guidelines and consensus statements recommend screening for Lp(a): When, in whom, and why? Multiple global clinical guidelines and expert consensus statements recommend Lp(a) screening to manage dyslipidemia and mitigate Lp(a)-mediated CVD risk. Provided is a synopsis of key recommendations for measuring Lp(a) from the: American Heart Association/American College of Cardiology (AHA/ACC) [80], Canadian Cardiovascular Society [82], and European Society of Cardiology/European Atherosclerosis Society (ESC/EAS) clinical guidelines [81]; and the ACC, American Association Clinical Endocrinology/American College of Endocrinology (AACE/ACE) [31], EAS [5], and National Lipid Association (NLA) [4,83] consensus statements.
*Denotes clinical guideline; †Denotes scientific/consensus statement; ‡Premature atherosclerotic cardiovascular disease (ASCVD) is defined as occurring in men aged <55 years and women aged <65 years.
It is imperative for both clinicians and patients to understand that Lp(a) levels cannot be determined from a standard lipid profile, as high Lp(a) levels can occur alongside normal levels of LDL-C and TG [86]. Therefore, measuring Lp(a) levels is recommended for a comprehensive CVD risk assessment [50]. A seminal finding from the Bruneck prospective outcomes study determined that the addition of Lp(a) screening to a baseline CVD risk assessment (using the Framingham Risk Score and Reynolds Risk Score), significantly improved the discrimination and reclassification of CVD risk [87]. Lp(a) screening was particularly effective in reclassifying individuals at an intermediate risk, resulting in a net reclassification improvement of almost 40%. Consequently, this study laid the foundation for dyslipidemia management guidelines to classify Lp(a) as a CVD risk enhancer and recommend its measurement for risk mitigation, particularly in individuals with intermediate CVD risk [80,81]. Nurmohamed et al., reaffirmed the Bruneck study observation, by demonstrating that Lp(a) screening reclassified almost one-third and two-thirds of individuals with very high Lp(a) levels (>99th percentile) to a higher risk category for the SCORE (primary prevention) and SMART (secondary prevention) ASCVD risk algorithms, respectively [88]. Also of note, Afshar et al., revealed that a high Lp(a) level effectively reclassified individuals with moderate LDL-C levels (135–159 mg/dL) to a higher CVD risk category, who may otherwise not meet the criteria for lipid-lowering therapy [89]. Furthermore, the addition of Lp(a) levels to the pooled cohort equations improved ASCVD risk prediction in individuals with diabetes or prediabetes from the ARIC study [90]. Importantly, participants among these studies were of White/European descent [87], [88], [89], and therefore it remains to be determined if these observations extend to other racial or ethnic groups. A recent analysis of a large multi-ethnic pooled cohort of five prospective studies of CVD from a US population, shows consistent increases in ASCVD risk associated with higher Lp(a) levels by sex and self-reported race/ethnicity, with particularly stronger relationships noted in those with versus without diabetes [91].
Another recommendation to further enhance the comprehensive nature of a CVD risk assessment would be to measure apoB levels, which are known to be better predictors than LDL-C levels in estimating incident and residual CVD risk [25]. Accurate and inexpensive apoB assays are available [25]. Therefore, the contribution of measured Lp(a) levels to total apoB levels can be assessed by a simple conversion, as depicted in the recent AHA scientific statement on Lp(a) [10]. In the era of very potent lipid-lowering therapies that preferentially lower LDL-C, Lp(a) may persist as the predominant atherogenic lipoprotein in individuals who achieved ultra-low apoB levels, but high Lp(a).
Considering that Lp(a) is an independent ASCVD risk factor, knowledge of Lp(a) levels is crucial to identify patients who remain at risk, despite receiving apoB-lowering therapies [6]. Furthermore, an Lp(a) measurement can help explain cases of unexplained or premature ASCVD, which can enhance patient engagement and understanding of CVD risk [92]. From a risk mitigation perspective, Lp(a) screening can inform clinical practice, such that modifiable risk factors can be targeted more intensively (Fig. 2). Evidence in support of this was provided by the Copenhagen City Heart Study, where European males (>60 years of age) with high Lp(a) levels who did not smoke or have hypertension, had an ∼20 % reduction in the 10-year risk of ischemic heart disease compared to males who had similarly high Lp(a) levels, but smoked and were hypertensive [93]. Similarly, in patients with high Lp(a) levels, Perrot et al., demonstrated that those who had an ideal CV health score as determined by the seven AHA CV health metrics (ie, body mass index, diet, physical activity, blood pressure, smoking status, total cholesterol level and diabetes status), had a 67 % lower risk of CVD compared to those with unhealthy CV health scores, and therefore adoption of these health metrics should be encouraged in all individuals with high Lp(a) levels [94]. Interestingly, recent evidence from the MESA cohort, revealed that high Lp(a) levels enhanced the association of hypertension with incident CVD, and this association was greatest among Black participants [95]. Similarly, in the ARIC study, high Lp(a) levels were significantly associated with an increased incidence of ASCVD events among White individuals with diabetes or prediabetes at baseline, but not in those with normal fasting blood glucose [90]. Importantly, data from the ARIC and UK Biobank studies, that include diverse cohorts, support similar CVD risk ratios rates for all racial groups [12,19].
Although Lp(a) levels are highly genetically determined, various clinical conditions have been associated with high levels, as described in Fig. 1A [5,[96], [97], [98]]. Recent evidence also suggests that Lp(a) levels can increase by an average of 22% from childhood to adulthood [99]. Deshotels et al., demonstrated that a large proportion of adults with borderline high Lp(a) in middle life have high Lp(a) levels later in life; particularly women, Black individuals, and those with hypertension [100]. Therefore, the clinical presentation and timing of any previous Lp(a) measurement are important considerations in determining the need for a repeat Lp(a) measurement, while an Lp(a) value found within the “gray zone” (discussed in the following section) should merit consideration for another Lp(a) measurement [101].
Key takeaway: Lp(a) testing is recommended by multiple clinical guidelines and medical societies. CVD risk assessments that do not include an Lp(a) measurement, could underestimate an individual's risk and miss an opportunity to intensify global CVD risk factor management.
2.2. How can Lp(a) measurements be embedded into routine clinical practice?
In 2018, driven by efforts from patient advocacy groups and leaders in the lipid field, the Centers for Disease Control and Prevention approved two ICD-10 codes, E78.41 [Elevated Lp(a)] and Z83.430 [Family history of elevated Lp(a)], for diagnosing high Lp(a), emphasizing the importance of embedding this measurement into clinical practice now (Fig. 3) [4]. Utilization of these codes will further allow clinicians to document the prevalence of high Lp(a) in patient cohorts and generate large clinical research databases, which will also be particularly important for future policy decisions [2,61]. Furthermore, the CPT® code 83695 has been assigned to indicate an Lp(a) measurement allowing clinicians to electronically document this medical service (Fig. 3). However, despite the epidemiological and genetic evidence, guideline recommendations, and that approximately 60 million Americans (based on estimates from other general populations) are likely to have high Lp(a) levels, there remains a substantial gap in the implementation of Lp(a) measurements in clinical practice [50]. An analysis from the Family Heart Database™ in 2022 of over 112 million individuals determined that only 0.3 % (n = 333,726) had at least one Lp(a) measurement (Fig. 3) [102]. This low rate of Lp(a) screening (approximately 1 % or less) is supported by additional observational studies from Hu et al. [103], and Bhatia et al. [16], although the latter study observed that approximately 3–4 % of high risk patients with ASCVD or CAVS were screened.
Fig. 3.

Lp(a) screening rates in the United States are sub-optimal and are currently concentrated within a small number of health care providers (HCPs) [102]. Two dedicated ICD-10-CM codes, E78.41 and Z83.430, are available to enhance the diagnosis of high Lp(a) and allow familial risk to be assigned to patients with Lp(a)-mediated CVD risk. The available CPT® code (83695), reports an Lp(a) blood test, while direct-to-consumer Lp(a) assays that measure in nmol/L are available to order at a low cost.
To help embed Lp(a) measurements into routine clinical practice, barriers to implementing testing must be confronted and policy interventions are required. One commonly identified barrier is a lack of value perception among clinicians, mainly due to the perceived lack of targeted therapeutic options for an individual/patient with a high Lp(a) level [104]. Further, the lack of a universal Lp(a) threshold for CVD risk prediction among guidelines and of actionable recommendations on how to manage a patient with high Lp(a) fuels a misconception that Lp(a) testing is not necessary until a specific therapy targeting Lp(a) is available [104]. Identifying an appropriate threshold for high Lp(a) has been challenging, largely due to significant ethnic/racial differences in Lp(a) levels and population-level distribution [26].
There has been a recent increase in the use of digital health technologies to improve clinical care [105]. Various approaches have been studied, showing that using a text-message intervention may improve cardiovascular risk factors [106] and the use of electronic applications has showed improvements in physical activity and e-health literacy scores [107]. The application of these modalities to increase Lp(a) screening implementation practices needs further exploration.
2.3. A pragmatic approach to assess risk in those with high Lp(a)
2.3.1. What values are desirable and high?
Recent evidence from the UK Biobank, comprising a multiethnic population, challenges the use of a single number as an Lp(a) threshold associated with ASCVD risk, demonstrating that ASCVD risk increases linearly and consistently across all ethnic/racial subgroups with a rise in Lp(a) level, and is present at levels lower than reported thresholds [12,84]. In light of this, the EAS and NLA advised on a risk spectrum (Fig. 1B) where Lp(a)-associated CVD risk can be viewed as a continuum, with <75 nmol/L (∼30 mg/dL) generally considered desirable, ≥125 nmol/L (∼50 mg/dL) considered very high risk, and a gray zone of 75–<125 nmol/L (∼30–<50 mg/dL) [5,83]. The adoption of this is a pragmatic approach to determining Lp(a)-associated CVD risk and would allow clinicians to determine, based on overall risk, whether the Lp(a) level constitutes a clinically meaningful increase in risk [101]. A recent opinion piece by Wong ND highlights “The failure to screen and identify those with Lp(a)-associated risk represents a missed opportunity to address this risk, not only with our existing repertoire of treatments, but hopefully in the future with promising therapies in development targeting Lp(a).”[108]
2.3.2. Test cost and insurance coverage
In general, an Lp(a) test can cost between $25 to $100 dollars, which is comparable to a standard lipid profile [3]. Use of diagnostic codes may help with insurance coverage if there is a family history of high Lp(a) (Fig. 3). Some insurance plans may not cover the cost of measuring Lp(a) and some countries require patients to pay for their own Lp(a) measurement [104]. This cost of the test may be a barrier for some patients and it will be important to educate policy makers of the importance of this test to allow greater coverage by insurers. Individuals may also obtain direct-to-consumer Lp(a) assays for reasonable prices which may be an option to consider and may be useful for cascade screening of relatives.
2.3.3. Assay considerations
There are substantial challenges in assay standardization of Lp(a) particle molar and mass concentration, and these have been recently reviewed [101]. There is no unbiased conversion factor between the two measurement units (mg/dL and nmol/L) [10,109]. Efforts to further standardize and improve the accuracy of Lp(a) assays are ongoing, with more recent recommendations stating that Lp(a) should be measured in nmol/L and use an apo(a) isoform-insensitive assay traceable to the International Federation of Clinical Chemistry reference material (Fig. 1B) [5,10]. There are Lp(a) assays calibrated to international standards available, and these assays utilizing a calibration approach are suitable in determining Lp(a)-associated CVD risk in the general population [10,101,110].
Key takeaway: Once a suitable Lp(a) assay is adopted, it is recommended to continue using the same standardized assay for future measurements. If an optimized assay is not available at your site, clinicians are encouraged to use the available assays (despite limitations) and to continue using the same assay if repeated measures of Lp(a) levels are needed.
2.4. What is actionable following identification of a high Lp(a) level?
While CV outcome trials of targeted apo(a)-lowering therapies are pending, individuals with high Lp(a) may benefit from:
-
1.
Risk calculators: To facilitate actionable responses by clinicians following a high level of Lp(a), we have developed ASCVD risk mitigation flowcharts for primary (Central Illustration) and secondary (Fig. 4) prevention. The risk assessment component of the primary prevention flowchart incorporates an Lp(a) measurement into the ACC/AHA 10-year predicted ASCVD risk assessment to determine an individual's appropriate risk category and risk mitigation interventions (Central Illustration) [80,111]. Knowledge of an individual's Lp(a) level is incorporated as a risk enhancing factor and could adopt the observation by Patel et al., that the risk for ASCVD was 11 % higher for each 50 nmol/L increment of Lp(a) [hazard ratio 1.11 per 50 nmol/L Lp(a)] [10,12]. To incorporate Lp(a) into a 10-year risk estimate, the patient's modelled 10-year risk would be multiplied by a factor of 1.11^(patient's Lp(a) in nmol/L/50) (Central Illustration). As an example, the 10-year risk estimate for a patient with an Lp(a) of 200 nmol/L would have a 10-year risk that is 1.52-fold higher than modeled by the ACC/AHA pooled cohort equations calculator.
-
2.
Imaging modalities: There are risk modifiers not commonly included in ASCVD risk prediction models that also warrant consideration with an Lp(a) measurement, particularly CAC [112]. Mehta et al., demonstrated a joint association between high Lp(a) levels (upper quintile [Q5]) and CAC score (≥100) with ASCVD risk [112]. Asymptomatic individuals had an approximate five-fold increase in ASCVD risk compared to individuals within the lower Lp(a) quintiles (Q1–4) who had a CAC score of zero. Both risk factors were independently associated with ASCVD risk, and the authors acknowledged that individuals from the high-risk subgroup would likely benefit from intensive ASCVD risk reduction strategies [112]. Further supportive evidence showing a positive association between high Lp(a) levels and CAC progression over a mean follow-up of 7.3 years was demonstrated by Wong ND et al. [113]. It has also been recently proposed that CAC would be an appropriate imaging modality within certain clinical scenarios where risk-enhancing factors, including high Lp(a), are present [79].
-
3.
Assessment of inflammatory markers: Similar to Lp(a), hsCRP was added as an ASCVD risk enhancer in the 2018 AHA/ACC cholesterol guidelines [80]. Currently, there is conflicting evidence on whether hsCRP modifies Lp(a)-associated ASCVD risk. Using asymptomatic participants from the MESA population, Zhang et al., demonstrated that Lp(a)-associated ASCVD risk was only observed with concomitant elevation in hsCRP (≥2 mg/L) [114]. Similarly, a prespecified post hoc analysis of the multinational “A Study of Evacetrapib in High-Risk Vascular Disease (ACCELERATE)” trial revealed a stepwise relationship between high Lp(a) levels and CV death, myocardial infarction and stroke in patients with high CV risk only when hsCRP levels were >2 mg/L [115]. However, a more recent analysis of a large dataset of predominately White individuals from the Copenhagen General Population Study revealed that Lp(a) was the main driver of ASCVD (and CAVS) irrespective of hsCRP levels [116], suggesting contextual differences in population or ethnic/race in the interaction between systemic inflammation and Lp(a)-associated risk.
-
4.
Lifestyle modifications: A risk-based strategy for prevention is recommended for individuals with high Lp(a), tailoring interventions broadly effective at lowering ASCVD risk to the patient's comprehensive risk profile. Following the ASCVD risk assessment with Lp(a), it is advisable that all individuals, regardless of assigned risk category, adopt healthy lifestyle behaviors. This should be aligned with the AHA's enhanced approach to assessing CV health by “Life's Essential 8″, to target modifiable ASCVD risk factors through a healthy diet, physical activity, avoiding nicotine, healthy sleep, healthy weight, and healthy levels of blood lipids, blood glucose and blood pressure (Central Illustration) [117].
-
5.
CVD risk-modifying therapies/procedures: For individuals categorized as intermediate or high-risk, pharmacological therapies (eg, statins and other apoB-lowering therapies and anti-hypertensive therapies) are recommended to intensify modifiable risk factor management. For individuals in the high-risk category, more intensive targeting of risk factors is advised (Central Illustration); this could involve combined statin and anti-PCSK9 mAb or PCSK9 small-interfering RNA (siRNA) therapies, lipoprotein apheresis (FDA-approved only in those with high Lp(a) based on criteria in the setting of familial hypercholesterolemia [FH]), anti-platelet therapies (eg, aspirin) or additional CVD risk-modifying agents as discussed below. Importantly, where there is uncertainty regarding an appropriate risk mitigation approach, patient referral to a suitable specialist (eg, lipidologist or preventive cardiologist) should be considered in both primary (Central Illustration) and secondary (Fig. 4) prevention populations. Within the current repertoire of approved apoB-lowering therapies that are indicated for LDL-C lowering, several have reported effects on plasma Lp(a) levels (Table 1). Interestingly, statins tend to modestly increase Lp(a) levels by 9–20 % through mechanisms that remain to be determined [27]. Thus, while statins remains an essential therapeutic strategy in CVD risk mitigation, they do not produce clinically important changes in Lp(a) levels. Niacin can lower Lp(a) levels by ∼20 % [6], however no CV outcome benefit has been reported for niacin when added to statin therapy; considering the evidence of adverse effects associated with the use of niacin, it should be used with caution for Lp(a)-lowering [9]. Ezetimibe has modest Lp(a) lowering effects that are not clinically significant [9]. Bempedoic acid also does not lower Lp(a) levels [118]. Currently, the only available therapeutic approaches that lower Lp(a) levels and reduce CV risk are anti-PCSK9 mAbs and lipoprotein apheresis [9]. Anti-PCSK9 mAb (and PCSK9 siRNA) therapies are not approved for Lp(a)-lowering and to date there are no completed RCTs that have assessed CV risk reduction in patients with high Lp(a). The evidence in support of lipoprotein apheresis, which is FDA-approved for patients with high Lp(a) only in the setting of clinically diagnosed FH, is currently derived from observational data [61]. Lp(a) levels are reduced by ∼20–30 % with anti-PCSK9 mAbs [27], with similar reductions reported for inclisiran, a siRNA therapy that targets PCSK9 mRNA (Table 1) [119]. Secondary analyses of the CV outcome trials, FOURIER and ODYSSEY OUTCOMES, revealed that after lowering LDL-C with statins and anti-PCSK9 mAbs, high Lp(a) levels were associated with increased residual CVD risk [7,8]. The magnitude of clinical benefit with anti-PCSK9 mAbs appeared to be associated with the extent of Lp(a) reduction, suggesting that Lp(a) could be an important and modifiable risk factor in patients with nominally controlled LDL-C, but further investigation is required [9]. In the United States, lipoprotein apheresis performed every 2 weeks is the only FDA-approved therapeutic option for patients with high Lp(a) (>60 mg/dL [ ∼150 nmol/L]) in the setting of clinically diagnosed FH, and either coronary or peripheral artery disease, but uptake of this invasive therapeutic option is poor and not readily available to the vast majority of patients with high Lp(a) [10]. Lipoprotein apheresis lowers apoB-containing lipoproteins, LDL-C and Lp(a), by 70−75 %; however, due to the intermittent nature of the therapy, rebound occurs and the time-averaged reduction of Lp(a) with bi-weekly treatments is estimated to be between 30 and 35 % (Table 1) [120,121]. Retrospective and prospective lipoprotein apheresis trials have demonstrated CV event reduction by ∼80 % in patients with high Lp(a), but the benefit of apheresis requires prospective RCTs to make definitive conclusions [61].
-
6.
Management of other comorbidities: Other possible therapeutic options for targeting Lp(a)-associated risk include low-dose aspirin, as supported by two primary prevention trials: the Women's Healthy Study [122] and ASPirin in Reducing Events in the Elderly (ASPREE) [123]. In both trials, low-dose aspirin was beneficial in reducing CVD events in individuals with high Lp(a)-associated genotypes, with ASPREE suggesting CVD benefits outweighed bleeding risk. Therefore, aspirin − as a widely available, well tolerated, and cost-effective therapy − may be of benefit to individuals at moderate or high Lp(a)-associated CVD risk (Fig. 4). Further, in the absence of approved therapies for mitigating Lp(a)-associated risk, these data have been used to justify the prescription of aspirin by clinicians in patients with high Lp(a) [123]. Further evidence from RCTs and the analysis of directly measured Lp(a) levels are required to fully understand the role of aspirin in targeting Lp(a)-associated CVD risk.
Fig. 4.

Mitigation of Lp(a)-associated risk in patients with ASCVD. Patients with high Lp(a) and ASCVD should be categorized as being a high risk for future CVD event(s). As per the Central Illustration, adoption of “Life's Essential 8″ should be advised with initial risk mitigation discussions. Further understanding of the pro-atherogenic, pro-inflammatory and pro-thrombotic burden of Lp(a)-associated ASCVD risk should prompt consideration of the armamentarium of therapies that can mitigate risk (including lipid-lowering therapies and/or lipoprotein apheresis, and anti-inflammatory, anti-platelet and/or anti-coagulant therapies) with consideration of a referral to a specialist (eg, lipidologist or preventive cardiologist). Therapies in bold indicate those where current evidence suggests a clinical benefit could be derived in patients with high Lp(a).
*Anti-PCSK9 mAbs or PCSK9 siRNA therapies; †lipoprotein apheresis is currently FDA-approved for patients with high Lp(a) only in the setting of FH. FH = familial hypercholesterolemia; P2Y12 = purinergic receptor P2Y G-protein-coupled 12.
Table 1.
The effect of approved lipid-lowering procedures and therapies affecting Lp(a) levels.
| Therapeutic strategy | Effect on Lp(a) | Effect on LDL-C | Possible Lp(a)-lowering mechanism | Effect on CVD risk |
|---|---|---|---|---|
| Apheresis* | 30–35 % time-averaged reduction [27] | 70 % reduction [121] | Removal of circulating apoB-100 and/or apo(a) -containing lipoproteins [121] | An observational study showed a reduced MACE incidence of 58 % in a 2-year observation period vs 11 % in the 5-year treatment period [151] |
| Statins | 9–20 % increase [152] | 30–50 % reduction [153]† | Increased apo(a) synthesis and secretion [152] | A meta-analysis of 27 RCTs with statins showed a 21 % MACE reduction per 38.9 mg/dL reduction in LDL-C [154] |
| Ezetimibe | 0–7 % reduction [9] | 15–22 % reduction [81] | Unknown | An RCT with combined ezetimibe and statin vs statin monotherapy showed a ∼6–7 % MACE reduction [155] |
| Bempedoic acid | No significant change [118] | 17−28 % reduction [156] | – | An RCT showed a 15 % reduction in 3-point MACE [156] |
| Niacin | 21 % reduction [6] | 12 % reduction [6] | Inhibits LPA gene expression at the promotor level [152] | No incremental clinical benefit of adding niacin to statin therapy [157] |
| PCSK9 mAbs/siRNA | 19–27 % reduction [7,8,119] | 51–61 % reduction [158], [159], [160] | Enhanced clearance and reduced production of Lp(a) [8] | RCTs with different anti-PCSK9 mAbs have both demonstrated a 15 % MACE reduction in patients receiving statin therapy and LDL-C ≥ 70 mg/dL158,159 |
Apo(a) = apolipoprotein(a); apoB = apolipoprotein B-100; CVD = cardiovascular disease; FH, LDL-C = low-density lipoprotein cholesterol; Lp(a) = lipoprotein(a); MACE = major adverse cardiovascular event; PCSK9 = proprotein convertase subtilisin/kexin type 9; RCT = randomized control trial; siRNA = small interfering ribonucleic acid.
In the United States, apheresis is approved for patients with high Lp(a) only in the setting of FH.
Moderate intensity statins.
Importantly, there are intensive therapeutic interventions (as described) to mitigate the increased CVD risk caused by high Lp(a); however, this risk cannot be inferred from a standard lipid profile test [86] and can only be detected by an Lp(a) test following a simple, routine fasting or non-fasting venipuncture [50]. Notably, a cross-sectional and longitudinal analysis of patient records in Germany has uncovered that mortality rates of patients with prior ASCVD events and an Lp(a) test were significantly lower compared to a matched control group with ASCVD events and no Lp(a) test [124]. It has been suggested this observation is likely explained by intensified preventive treatment approaches along with specialized CV patient care as a result of Lp(a) testing [124].
Secondary prevention patients with ASCVD and high Lp(a) should be immediately categorized as high-risk. Patients with multiple risk factors in addition to high Lp(a) and those with recurrent ASCVD events may be considered very high risk [125]. High Lp(a) levels are common in those with FH and is associated with an ∼5 fold increased risk in an already high risk population [126,127]. Again, adoption of “Life's Essential 8″ components of CV health is recommended to complement pharmacologic risk reduction (Fig. 4) [117]. The best available evidence to date showing improved CV outcomes in patients with high Lp(a) comes from secondary analyses of RCTs with PCSK9 targeted-therapies and these therapies should be preferentially implemented when considering lipid-lowering therapies to achieve LDL-C/ApoB reduction goals [9]. Observational data suggest that lipoprotein apheresis, which acutely lowers Lp(a) and LDL-C by 70–80 % acutely and ∼35 % time averaged between bi-weekly treatment sessions, may also modify risk in patients with high Lp(a) [27]. Lipoprotein apheresis is an FDA-approved therapy for those with FH, high Lp(a) in the presence of FH, and ASCVD. Apheresis can be considered for this very high risk population especially if LDL-C/ApoB goals cannot be achieved with pharmacologic therapy alone or if recurrent events occur. Additional risk-based mitigation approaches should be considered based on Lp(a) pathophysiology, in specific the broad spectrum of evidence-based interventions that are effectively used in secondary prevention of ASCVD (Fig. 4).
Anti-platelet/anti-coagulant therapies, such as P2Y12 receptor antagonists [128] (as monotherapy or dual antiplatelet therapy with aspirin) and/or aspirin with a low-dose anticoagulant (rivaroxaban) [129] may potentially alleviate the additional pro-thrombotic burden posed by high Lp(a). The anti-inflammatory compound, colchicine, has emerged as an effective risk modifying therapy targeting the inflammatory axis of CVD; it is widely available and has a low cost (Fig. 4) [130]. The RCTs Low-Dose Colchicine (LoDoCo1, LoDoCo2) [131,132] and the Colchicine Cardiovascular Outcomes Trial (COLCOT) [133] have demonstrated that the risk of CV events is significantly lower with once daily colchicine compared to placebo in secondary prevention CVD populations. However, colchicine has not been tested in patients with high Lp(a). An inhibitor of IL-6, ziltivekimab, was shown to reduce Lp(a) among patients with chronic kidney disease [134], and this agent is currently under study in several CV outcome trials including patients with residual inflammatory risk at high CVD risk (NCT05021835, NCT05636176, NCT06118281). Autoimmune diseases, in particular rheumatoid arthritis, are associated with increased mortality due to CVD [135]; therefore, what effect (if any) can antirheumatic drugs, including ziltivekimab, have on Lp(a)-associated CVD risk? While evidence in support of these latter therapies mitigating Lp(a)-mediated CVD risk does not exist, it is anticipated that secondary analyses of ongoing trials will address this question and could prove beneficial while targeted Lp(a)-lowering therapies are pending.
Considering the heritable nature of Lp(a) levels, cascade screening of first-degree family members is another warranted and actionable response following the detection of a high Lp(a) level [136]. Indeed, the recent AHA scientific and EAS consensus statements have recommended cascade screening for high Lp(a), including those with a personal or family history (also noted by the ICD-10-CM code ‘Z83.430′) of ASCVD [5,10], while also incorporating this into services that already exist for cascade screening of FH, as ∼25 % of adults with clinical FH have high Lp(a) [5,137]. The clinical value of including systematic Lp(a) screening within cascade screening for FH was demonstrated by Ellis et al., where screening from index cases with both FH and high Lp(a) identified one new case of high Lp(a) for every 2.4 individuals screened; this was in comparison to opportunistic screening from index cases with FH, but without high Lp(a), identifying one individual for every 5.8 screened [138]. Notably, while FH and high Lp(a) alone were associated with increased ASCVD risk among family members, the greatest risk was observed in relatives with both FH and high Lp(a), with the authors also acknowledging the merit of Lp(a) screening outside of FH [138].
3. Emerging targeted Lp(a)-lowering therapies
Novel therapies that target apo(a) are at different clinical trial stages and substantially lower Lp(a) levels in patients with high Lp(a) [9]. This is critically important in the context of Lp(a) level population distribution, which is generally skewed, and has more than a 1000-fold range of concentrations between individuals [30]. Therefore, those with the highest levels of Lp(a) will likely require large, absolute reductions in Lp(a) levels to effectively manage their CVD risk [139,140]. There are currently five therapies in clinical development, four of which target the mRNA transcript of the LPA gene to inhibit Lp(a) translation [9] (NCT05565742, NCT05563246) (Table 2).
Table 2.
Emerging targeted Lp(a)-lowering therapies.
| Drug | Mechanism of action | Mean/median Lp(a) reduction (%) | Absolute Lp(a) reduction (nmol/L) | Current clinical trial stage/NCT identifier | Projected trial completion |
|---|---|---|---|---|---|
| Pelacarsen | GalNAc-conjugated ASO targeting apo(a) mRNA | Phase 2: 35–80 % [141] |
Phase 2: 96–188 |
Phase 3 [Lp(a)HORIZON]/ NCT04023552 |
2025 |
| Olpasiran | GalNAc-conjugated siRNA targeting apo(a) mRNA | Phase 2: 70–97 % [143] |
Phase 2: 250 |
Phase 3 [OCEAN(a)-Outcomes] NCT05581303 |
2026 |
| Zerlasiran | GalNAc-conjugated siRNA targeting apo(a) mRNA | Phase 1: 46–98 % [144] |
Phase 1: 183–259 |
Phase 2 NCT05537571 |
2024 |
| Lepodisiran | GalNAc-conjugated siRNA targeting apo(a) mRNA | Phase 1: 41–97 % [161] | Phase 1: 36–127 | Phase 2 NCT05565742 |
2024 |
| Muvalaplin | Small molecule inhibitor targeting Lp(a) | Phase 1: Up to 65 % [146] | Phase 1: N/A | Phase 2 [KRAKEN] NCT05563246 |
2024 |
The minimal Lp(a) level entry criteria for all trials described is ≥75 nmol/L (∼30 mg/dL). Lp(a)HORIZON and OCEAN(a)-Outcomes trials include patients with established ASCVD; KRAKEN and the zerlasiran phase 2 trials involve individuals at high-risk of CVD events. The phase 2 trial with lepodisiran involves healthy individuals with high Lp(a). Apo(a) = apolipoprotein(a); ASO = antisense oligonucleotide; ASCVD = atherosclerotic cardiovascular disease; GalNAc = N-acetyl galactosamine; Lp(a) = lipoprotein(a); mRNA = messenger ribonucleic acid; N/A, not available; siRNA = small interfering RNA.
Pelacarsen is a N-acetylgalactosamine (GalNAc)-conjugated ASO that specifically targets hepatic LPA mRNA [141]. In a phase 2, dose-ranging study in patients with ASCVD and Lp(a) levels ≥150 nmol/L (∼60 mg/dL), pelacarsen showed significant, dose-dependent reduction in Lp(a) levels of up to 80 %, while 98 % of treated patients achieved Lp(a) levels ≤125 nmol/L (∼50 mg/dL) at 6 months [141]. Adverse events were mostly mild, with injection-site reactions being the most common event, and there were no significant differences in terms of safety between any pelacarsen dose and placebo [141]. OxPLs have been proposed as drivers of Lp(a) pathogenicity and in this study both OxPL-apo(a) and OxPL-apoB were reduced by up to 70 % and 88 %, respectively [141]. Aligned with this finding, pelacarsen attenuated the pro-inflammatory gene expression signature of monocytes and their transendothelial migratory capacity, key initiating steps in arterial wall inflammation associated with ASCVD development [142]. A phase 3 CV outcomes trial with pelacarsen, Lp(a)HORIZON, is ongoing and has completed enrollment with over 8000 patients with ASCVD and high Lp(a) (≥70 mg/dL or ∼≥150 nmol/l) (NCT04023552).
Olpasiran is one of three candidate GalNAc-conjugated siRNA therapies that target hepatic LPA mRNA. Data from a phase 2 trial in patients with ASCVD and baseline Lp(a) ≥150 nmol/L revealed dose-dependent reductions in Lp(a) levels from baseline of 70–97 % at Week 36 following olpasiran administration every 12 weeks [143]. Furthermore, all patients who received olpasiran at doses ≥75 mg every 12 weeks achieved Lp(a) levels below 125 nmol/L [143]. From a safety perspective, olpasiran was well tolerated, as the incidence of serious adverse events was comparable among olpasiran- and placebo-treated patients [143]. The phase 3 CV outcomes study, OCEAN(a)-Outcomes, is ongoing in patients with high Lp(a) and a history of ASCVD, with the trial due to finish in late 2026 (NCT05581303).
Findings from a phase 1 trial of the siRNA zerlasiran have demonstrated a potent, Lp(a)-lowering effect of up to 98 % with a single dose in individuals with Lp(a) levels ≥150 nmol/L. Sustained suppression of Lp(a) levels up to 80 % below baseline levels was observed at 5 months following zerlasiran administration [144]. Zerlasiran was well tolerated, and any treatment emergent adverse events were generally mild [144]. A phase 2 trial investigating the efficacy, safety, and tolerability of zerlasiran in individuals with high Lp(a) levels at high risk of ASCVD is underway, with expected completion in 2024 (NCT05537571). The remaining GalNAc-conjugated siRNA that targets Lp(a) is lepodisiran (LY3819469), with recent findings from a phase 1 trial in healthy individuals with high Lp(a) (≥ 75 nmol/L, ∼30 mg/dL) reporting a maximal median percent reduction in Lp(a) levels of 97 % after a single dose of lepodisiran in the highest dose (608 mg) group, which was a sustained reduction out to 1 year [145]. A phase 2 trial with lepodisiran in individuals with high Lp(a) began in late 2022 (NCT05565742). Finally, a small oral molecule, muvalaplin (LY3473329), that disrupts the interaction between apo(a) and apoB, is under investigation. A phase 1 trial was completed in late 2021 and demonstrated a placebo-corrected reduction in Lp(a) levels by up to 65 % in doses ≥100 mg [146]. A phase 2 trial commenced in late 2022 (NCT05563246) (Table 2).
A next generation oral CETP inhibitor, obicetrapib, was also shown to reduce Lp(a) by 56 % in a phase 2 study of patients with dyslipidemia [147], and is moving on to further study in a phase 3 cardiovascular trial in patients with ASCVD and residual LDL-C elevation (NCT05202509).
Moving beyond RNA therapeutics, novel gene-editing approaches targeting dyslipidemia are on the horizon, offering alternative and exciting opportunities to target atherogenic lipoproteins as a ‘one-and-done’ therapy [148]. Most notably, CRISPR-based therapeutics have the capacity for genome editing directly at the DNA sequence level and have proven valuable in experimental research settings [149]. Gene-editing studies permanently targeting PCSK9 and ANGPTL3 are underway [148]. A study in primates has demonstrated that a single infusion of a CRISPR base-editing therapeutic, targeting liver PCSK9, nearly ablated gene expression and effectively lowered. LDL-C levels by 60 % at 8 months [150]. Following on from this landmark study, a phase 1 trial with this therapeutic, VERVE-101, is underway in patients with HeFH to evaluate long-term safety in humans, along with changes in LDL-C levels (NCT05398029). Undoubtably, the clinical adoption of such therapies will require resolution of ethical and safety issues [148].
4. Conclusion
High Lp(a) is a common CVD risk factor and conscious efforts, as directed by clinical guidelines and consensus statements, should be made to screen individuals/patients for high Lp(a) levels, to determine a more comprehensive CVD risk profile. The latter will facilitate appropriate risk mitigation strategies. Ongoing clinical trials with targeted apo(a)-lowering therapies that lower plasma Lp(a) levels provide hope to patients and clinicians for decreasing Lp(a)-mediated CVD risk and CVD events. Until targeted therapies are available, patients with high Lp(a) may be treated with a risk-based strategy using the armamentarium of available interventions effective at reducing CVD, preferentially those that have been tested and improve overall CVD risk.
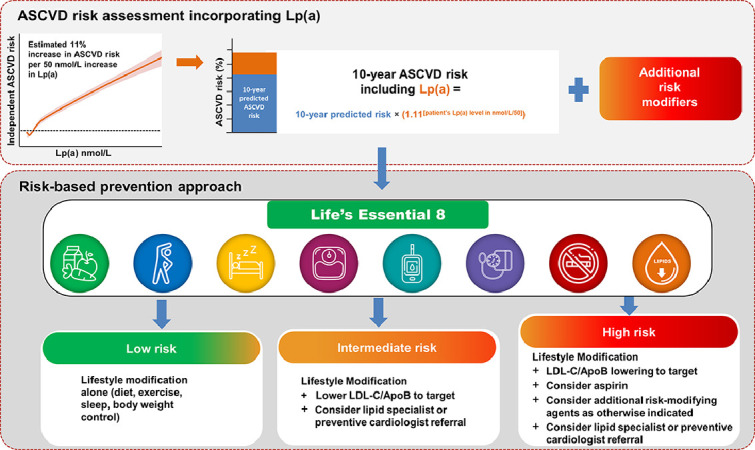
Central Illustration. Incorporation of an Lp(a) measurement into a primary prevention ASCVD risk assessment, with a risk-based prevention approach The upper panel describes how an Lp(a) measurement in nmol/L can be incorporated as a risk enhancer into a 10-year predicted ASCVD risk assessment. Lp(a) risk is derived from the standardized risk for ASCVD being 11 % higher per 50 nmol/L increment within a multiethnic population from the UK Biobank [12]. Additional risk modifiers should also be factored in. The bottom panel describes risk mitigation approaches depending on assigned risk category; all of these should initially adopt the AHA's holistic approach to CV health, “Life's Essential 8″, including healthy diet, physical activity, avoiding nicotine, healthy sleep, healthy weight, and healthy levels of blood lipids, blood glucose and blood pressure [117]. With increasing risk category (ie, moderate to high risk), pharmacotherapeutic intervention(s) should be considered to address overall ASCVD risk, with consideration for a specialist referral (eg, lipidologist or preventive cardiologist) to provide well-informed advice on Lp(a)-associated risk.
ApoB = apolipoprotein B-100; LDL-C = low-density lipoprotein cholesterol.
Disclosures
G.R.S is a consultant and receives research grants from Kaneka. Consulting Eli Lilly. These roles are not related to the current publication.
C.Y is a consultant for Kaneka.
E.D.M. has served as a consultant for Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Esperion, Edwards Lifescience, Medtronic, Merck, New Amsterdam, Novartis, Novo Nordisk, and Pfizer.
W.B reports being employed by Novartis at the time of this publication.
C.M.B has received grant/research support through his institution from Abbott Diagnostics, Ackea, Amgen, Arrowhead, Ionis, Eli Lilly, Merck, New Amsterdam, Novartis, Novo Nordisk, NIH, AHA and ADA; and consultant fees from Abbott Diagnostics, Amgen, Arrowhead, Astra Zeneca, Denka Seiken, Eli Lilly, Esperion, Illumina, Ionis, Merck, New Amsterdam, Novartis, Novo Nordisk, Roche Diagnostic, and TenSixteen Bio.
Funding
This manuscript was supported by Novartis Pharma AG, Basel, Switzerland.
CRediT authorship contribution statement
Gissette Reyes-Soffer: Writing – review & editing, Writing – original draft, Conceptualization. Calvin Yeang: Writing – review & editing, Writing – original draft, Conceptualization. Erin D Michos: Writing – review & editing, Writing – original draft, Conceptualization. Wess Boatwright: Writing – review & editing, Writing – original draft, Conceptualization. Christie M Ballantyne: Writing – review & editing, Writing – original draft, Conceptualization.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors thank Tony Walsh, PhD (Novartis Ireland Ltd.), and Shalini Verma, PhD (Novartis, Healthcare Pvt. Ltd. India) for providing medical writing support in accordance with Good Publication Practice 2022 guidelines (https://www.ismpp.org/gpp-2022).
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.ajpc.2024.100651.
Appendix. Supplementary materials
References
- 1.Berg K. A new serum type system in man–the Lp system. Acta Pathol Microbiol Scand. 1963;59:369–382. doi: 10.1111/j.1699-0463.1963.tb01808.x. [DOI] [PubMed] [Google Scholar]
- 2.Nestel P., Loh W.J., Ward N.C., Watts G.F. New horizons: revival of lipoprotein (a) as a risk factor for cardiovascular disease. J Clin Endocrinol Metab. 2022;107:e4281–e4294. doi: 10.1210/clinem/dgac541. [DOI] [PubMed] [Google Scholar]
- 3.Enas E.A., Varkey B., Dharmarajan T.S., Pare G., Bahl V.K. Lipoprotein(a): an independent, genetic, and causal factor for cardiovascular disease and acute myocardial infarction. Indian Heart J. 2019;71:99–112. doi: 10.1016/j.ihj.2019.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson D.P., Jacobson T.A., Jones P.H., et al. Use of lipoprotein(a) in clinical practice: a biomarker whose time has come. A scientific statement from the National Lipid Association. J Clin Lipidol. 2019;13:374–392. doi: 10.1016/j.jacl.2019.04.010. [DOI] [PubMed] [Google Scholar]
- 5.Kronenberg F., Mora S., Stroes E.S.G., et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: a European Atherosclerosis Society consensus statement. Eur Heart J. 2022;43:3925–3946. doi: 10.1093/eurheartj/ehac361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Albers J.J., Slee A., O'Brien K.D., et al. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes) J Am Coll Cardiol. 2013;62:1575–1579. doi: 10.1016/j.jacc.2013.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bittner V.A., Szarek M., Aylward P.E., et al. Effect of alirocumab on lipoprotein(a) and cardiovascular risk after acute coronary syndrome. J Am Coll Cardiol. 2020;75:133–144. doi: 10.1016/j.jacc.2019.10.057. [DOI] [PubMed] [Google Scholar]
- 8.O'Donoghue M.L., Fazio S., Giugliano R.P., et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation. 2019;139:1483–1492. doi: 10.1161/CIRCULATIONAHA.118.037184. [DOI] [PubMed] [Google Scholar]
- 9.Schwartz G.G., Ballantyne C.M. Existing and emerging strategies to lower lipoprotein(a) Atherosclerosis. 2022;349:110–122. doi: 10.1016/j.atherosclerosis.2022.04.020. [DOI] [PubMed] [Google Scholar]
- 10.Reyes-Soffer G., Ginsberg H.N., Berglund L., et al. Lipoprotein(a): a genetically determined, causal, and prevalent risk factor for atherosclerotic cardiovascular disease: a scientific statement From the American Heart Association. Arterioscler Thromb Vasc Biol. 2022;42:e48–e60. doi: 10.1161/ATV.0000000000000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nordestgaard B.G., Chapman M.J., Ray K., et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–2853. doi: 10.1093/eurheartj/ehq386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel A.P., Wang M., Pirruccello J.P., et al. Lp(a) (lipoprotein[a]) concentrations and incident atherosclerotic cardiovascular disease: new insights from a large National Biobank. Arterioscler Thromb Vasc Biol. 2021;41:465–474. doi: 10.1161/ATVBAHA.120.315291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsimikas S., Ancestry M.SM. Lipoprotein(a), and cardiovascular risk thresholds: JACC review topic of the week. J Am Coll Cardiol. 2022;80:934–946. doi: 10.1016/j.jacc.2022.06.019. [DOI] [PubMed] [Google Scholar]
- 14.Hu P., Dharmayat K.I., Stevens C.A.T., et al. Prevalence of familial hypercholesterolemia among the general population and patients with atherosclerotic cardiovascular disease: a systematic review and meta-analysis. Circulation. 2020;141:1742–1759. doi: 10.1161/CIRCULATIONAHA.119.044795. [DOI] [PubMed] [Google Scholar]
- 15.Sun H., Saeedi P., Karuranga S., et al. IDF Diabetes Atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. 2022;183 doi: 10.1016/j.diabres.2021.109119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhatia H.S., Hurst S., Desai P., Zhu W., Yeang C. Lipoprotein(a) testing trends in a large academic health system in the United States. J Am Heart Assoc. 2023;12 doi: 10.1161/JAHA.123.031255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Michos E.D., Lopez-Jimenez F., Gulati M. Role of glucagon-like peptide-1 receptor agonists in achieving weight loss and improving cardiovascular outcomes in people with overweight and obesity. J Am Heart Assoc. 2023;12 doi: 10.1161/JAHA.122.029282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guan W., Cao J., Steffen B.T., et al. Race is a key variable in assigning lipoprotein(a) cutoff values for coronary heart disease risk assessment: the Multi-Ethnic Study of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35:996–1001. doi: 10.1161/ATVBAHA.114.304785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Virani S.S., Brautbar A., Davis B.C., et al. Associations between lipoprotein(a) levels and cardiovascular outcomes in black and white subjects: the Atherosclerosis Risk in Communities (ARIC) study. Circulation. 2012;125:241–249. doi: 10.1161/CIRCULATIONAHA.111.045120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nissen S.E., Wolski K., Cho L., et al. Lipoprotein(a) levels in a global population with established atherosclerotic cardiovascular disease. Open Heart. 2022;9 doi: 10.1136/openhrt-2022-002060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shapiro M.D., Haddad T., Weintraub H.S., et al. Lipoprotein(a) levels in population with established atherosclerotic cardiovascular disease in the United States: a subanalysis from the Lp(a)heritage study - P1008-09. J Am Coll Cardiol. 2023;81:1633. [Google Scholar]
- 22.Reyes-Soffer G., Westerterp M. Beyond lipoprotein(a) plasma measurements: lipoprotein(a) and inflammation. Pharmacol Res. 2021;169 doi: 10.1016/j.phrs.2021.105689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koschinsky M.L., Boffa M.B. Oxidized phospholipid modification of lipoprotein(a): epidemiology, biochemistry and pathophysiology. Atherosclerosis. 2022;349:92–100. doi: 10.1016/j.atherosclerosis.2022.04.001. [DOI] [PubMed] [Google Scholar]
- 24.von Zychlinski A., Kleffmann T., Williams M.J., McCormick S.P. Proteomics of lipoprotein(a) identifies a protein complement associated with response to wounding. J Proteomics. 2011;74:2881–2891. doi: 10.1016/j.jprot.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 25.Sniderman A.D., Thanassoulis G., Glavinovic T., et al. Apolipoprotein B particles and cardiovascular disease: a narrative review. JAMA Cardiol. 2019;4:1287–1295. doi: 10.1001/jamacardio.2019.3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mehta A., Jain V., Saeed A., et al. Lipoprotein(a) and ethnicities. Atherosclerosis. 2022;349:42–52. doi: 10.1016/j.atherosclerosis.2022.04.005. [DOI] [PubMed] [Google Scholar]
- 27.Tsimikas S. A test in context: lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. 2017;69:692–711. doi: 10.1016/j.jacc.2016.11.042. [DOI] [PubMed] [Google Scholar]
- 28.Trinder M., Zekavat S.M., Uddin M.M., Pampana A., Natarajan P. Apolipoprotein B is an insufficient explanation for the risk of coronary disease associated with lipoprotein(a) Cardiovasc Res. 2021;117:1245–1247. doi: 10.1093/cvr/cvab060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bjornson E., Adiels M., Taskinen M.R., et al. Lipoprotein(a) is markedly more atherogenic than LDL: an Apolipoprotein B-based genetic analysis. J Am Coll Cardiol. 2024;83:385–395. doi: 10.1016/j.jacc.2023.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kronenberg F. Human genetics and the causal role of lipoprotein(a) for various diseases. Cardiovasc Drugs Ther. 2016;30:87–100. doi: 10.1007/s10557-016-6648-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Handelsman Y., Jellinger P.S., Guerin C.K., et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the management of dyslipidemia and prevention of cardiovascular disease algorithm - 2020 executive summary. Endocr Pract. 2020;26:1196–1224. doi: 10.4158/CS-2020-0490. [DOI] [PubMed] [Google Scholar]
- 32.Coassin S., Kronenberg F. Lipoprotein(a) beyond the kringle IV repeat polymorphism: the complexity of genetic variation in the LPA gene. Atherosclerosis. 2022;349:17–35. doi: 10.1016/j.atherosclerosis.2022.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Consortium C.A.D., Deloukas P., Kanoni S., et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eden S., Wiklund O., Oscarsson J., Rosen T., Bengtsson B.A. Growth hormone treatment of growth hormone-deficient adults results in a marked increase in Lp(a) and HDL cholesterol concentrations. Arterioscler Thromb. 1993;13:296–301. doi: 10.1161/01.atv.13.2.296. [DOI] [PubMed] [Google Scholar]
- 35.Muller N., Schulte D.M., Turk K., et al. IL-6 blockade by monoclonal antibodies inhibits apolipoprotein (a) expression and lipoprotein (a)synthesis in humans. J Lipid Res. 2015;56:1034–1042. doi: 10.1194/jlr.P052209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tzanatos H.A., Agroyannis B., Chondros C., et al. Cytokine release and serum lipoprotein (a) alterations during hemodialysis. Artif Organs. 2000;24:329–333. doi: 10.1046/j.1525-1594.2000.06483.x. [DOI] [PubMed] [Google Scholar]
- 37.Roeters van Lennep J.E., Tokgozoglu L.S., Badimon L., et al. Women, lipids, and atherosclerotic cardiovascular disease: a call to action from the European Atherosclerosis Society. Eur Heart J. 2023;44:4157–4173. doi: 10.1093/eurheartj/ehad472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim C.J., Jang H.C., Cho D.H., Min Y.K. Effects of hormone replacement therapy on lipoprotein(a) and lipids in postmenopausal women. Arterioscler Thromb. 1994;14:275–281. doi: 10.1161/01.atv.14.2.275. [DOI] [PubMed] [Google Scholar]
- 39.Honigberg M.C., Trinder M., Natarajan P. Lipoprotein(a), menopausal hormone therapy, and risk of coronary heart disease in postmenopausal individuals. JAMA Cardiol. 2022;7:565–568. doi: 10.1001/jamacardio.2022.0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suk Danik J., Rifai N., Buring J.E., Ridker P.M. Lipoprotein(a), hormone replacement therapy, and risk of future cardiovascular events. J Am Coll Cardiol. 2008;52:124–131. doi: 10.1016/j.jacc.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feingold K.R. In: Endotext. Feingold K.R., Anawalt B., Blackman M.R., Boyce A., Chrousos G., Corpas E., de Herder W.W., Dhatariya K., Dungan K., Hofland J., Kalra S., Kaltsas G., Kapoor N., Koch C., Kopp P., Korbonits M., Kovacs C.S., Kuohung W., Laferrere B., Levy M., McGee E.A., McLachlan R., New M., Purnell J., Sahay R., Shah A.S., Singer F., Sperling M.A., Stratakis C.A., Trence D.L., Wilson D.P., editors. 2000. The effect of endocrine disorders on lipids and lipoproteins. South Dartmouth (MA) [PubMed] [Google Scholar]
- 42.Matveyenko A., Matienzo N., Ginsberg H., et al. Relationship of apolipoprotein(a) isoform size with clearance and production of lipoprotein(a) in a diverse cohort. J Lipid Res. 2023;64 doi: 10.1016/j.jlr.2023.100336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kronenberg F., Utermann G. Lipoprotein(a): resurrected by genetics. J Intern Med. 2013;273:6–30. doi: 10.1111/j.1365-2796.2012.02592.x. [DOI] [PubMed] [Google Scholar]
- 44.Hoekstra M., Chen H.Y., Rong J., et al. Genome-wide association study highlights APOH as a novel locus for lipoprotein(a) levels-brief report. Arterioscler Thromb Vasc Biol. 2021;41:458–464. doi: 10.1161/ATVBAHA.120.314965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arsenault B.J., Kamstrup P.R. Lipoprotein(a) and cardiovascular and valvular diseases: a genetic epidemiological perspective. Atherosclerosis. 2022;349:7–16. doi: 10.1016/j.atherosclerosis.2022.04.015. [DOI] [PubMed] [Google Scholar]
- 46.Thomas P.E., Vedel-Krogh S., Nielsen S.F., Nordestgaard B.G., Kamstrup P.R. Lipoprotein(a) and risks of peripheral artery disease, abdominal aortic aneurysm, and major adverse limb events. J Am Coll Cardiol. 2023;82:2265–2276. doi: 10.1016/j.jacc.2023.10.009. [DOI] [PubMed] [Google Scholar]
- 47.Leistner D.M., Laguna-Fernandez A., Haghikia A., et al. Impact of elevated lipoprotein(a) on coronary artery disease phenotype and severity. Eur J Prev Cardiol. 2024 doi: 10.1093/eurjpc/zwae007. [DOI] [PubMed] [Google Scholar]
- 48.Arsenault B.J., Boekholdt S.M., Dube M.P., et al. Lipoprotein(a) levels, genotype, and incident aortic valve stenosis: a prospective Mendelian randomization study and replication in a case-control cohort. Circ Cardiovasc Genet. 2014;7:304–310. doi: 10.1161/CIRCGENETICS.113.000400. [DOI] [PubMed] [Google Scholar]
- 49.Kamstrup P.R., Tybjaerg-Hansen A., Nordestgaard B.G. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63:470–477. doi: 10.1016/j.jacc.2013.09.038. [DOI] [PubMed] [Google Scholar]
- 50.Thanassoulis G. Screening for high lipoprotein(a) Circulation. 2019;139:1493–1496. doi: 10.1161/CIRCULATIONAHA.119.038989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emdin C.A., Khera A.V., Natarajan P., et al. Phenotypic characterization of genetically lowered human lipoprotein(a) levels. J Am Coll Cardiol. 2016;68:2761–2772. doi: 10.1016/j.jacc.2016.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kamstrup P.R., Nordestgaard B.G. Elevated Lipoprotein(a) Levels, LPA risk genotypes, and increased risk of heart failure in the general population. JACC Heart failure. 2016;4:78–87. doi: 10.1016/j.jchf.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 53.Agarwala A., Pokharel Y., Saeed A., et al. The association of lipoprotein(a) with incident heart failure hospitalization: atherosclerosis risk in communities study. Atherosclerosis. 2017;262:131–137. doi: 10.1016/j.atherosclerosis.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Steffen B.T., Duprez D., Bertoni A.G., Guan W., Tsai M.Y. Lp(a) [lipoprotein(a)]-related risk of heart failure is evident in whites but not in other racial/ethnic groups. Arterioscler Thromb Vasc Biol. 2018;38:2498–2504. doi: 10.1161/ATVBAHA.118.311220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mohammadi-Shemirani P., Chong M., Narula S., et al. Elevated lipoprotein(a) and risk of atrial fibrillation: an observational and mendelian randomization study. J Am Coll Cardiol. 2022;79:1579–1590. doi: 10.1016/S0735-1097(22)02570-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boffa M.B. Beyond fibrinolysis: the confounding role of Lp(a) in thrombosis. Atherosclerosis. 2022;349:72–81. doi: 10.1016/j.atherosclerosis.2022.04.009. [DOI] [PubMed] [Google Scholar]
- 57.Marston N.A., Gurmu Y., Melloni G.E.M., et al. The effect of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) inhibition on the risk of venous thromboembolism. Circulation. 2020;141:1600–1607. doi: 10.1161/CIRCULATIONAHA.120.046397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schwartz G.G., Steg P.G., Szarek M., et al. Peripheral artery disease and venous thromboembolic events after acute coronary syndrome: role of lipoprotein(a) and modification by alirocumab: prespecified analysis of the ODYSSEY OUTCOMES randomized clinical trial. Circulation. 2020;141:1608–1617. doi: 10.1161/CIRCULATIONAHA.120.046524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Langsted A., Kamstrup P.R., Nordestgaard B.G. High Lipoprotein(a) and low risk of major bleeding in brain and airways in the general population: a Mendelian randomization study. Clin Chem. 2017;63:1714–1723. doi: 10.1373/clinchem.2017.276931. [DOI] [PubMed] [Google Scholar]
- 60.Mora S., Kamstrup P.R., Rifai N., et al. Lipoprotein(a) and risk of type 2 diabetes. Clin Chem. 2010;56:1252–1260. doi: 10.1373/clinchem.2010.146779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsimikas S., Fazio S., Ferdinand K.C., et al. NHLBI working group recommendations to reduce lipoprotein(a)-mediated risk of cardiovascular disease and aortic stenosis. J Am Coll Cardiol. 2018;71:177–192. doi: 10.1016/j.jacc.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boonmark N.W., Lou X.J., Yang Z.J., et al. Modification of apolipoprotein(a) lysine binding site reduces atherosclerosis in transgenic mice. J Clin Invest. 1997;100:558–564. doi: 10.1172/JCI119565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rath M., Niendorf A., Reblin T., et al. Detection and quantification of lipoprotein(a) in the arterial wall of 107 coronary bypass patients. Arteriosclerosis. 1989;9:579–592. doi: 10.1161/01.atv.9.5.579. [DOI] [PubMed] [Google Scholar]
- 64.van Dijk R.A., Kolodgie F., Ravandi A., et al. Differential expression of oxidation-specific epitopes and apolipoprotein(a) in progressing and ruptured human coronary and carotid atherosclerotic lesions. J Lipid Res. 2012;53:2773–2790. doi: 10.1194/jlr.P030890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Muramatsu Y., Minami Y., Kato A., et al. Lipoprotein level is associated with plaque vulnerability in patients with coronary artery disease: an optical coherence tomography study. Int J Cardiol Heart Vasc. 2019;24 doi: 10.1016/j.ijcha.2019.100382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kaiser Y., Daghem M., Tzolos E., et al. Association of Lipoprotein(a) with atherosclerotic plaque progression. J Am Coll Cardiol. 2022;79:223–233. doi: 10.1016/j.jacc.2021.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van der Valk F.M., Bekkering S., Kroon J., et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. 2016;134:611–624. doi: 10.1161/CIRCULATIONAHA.116.020838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schnitzler J.G., Hoogeveen R.M., Ali L., et al. Atherogenic lipoprotein(a) increases vascular glycolysis, thereby facilitating inflammation and leukocyte extravasation. Circ Res. 2020;126:1346–1359. doi: 10.1161/CIRCRESAHA.119.316206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boffa M.B., Koschinsky M.L. Oxidized phospholipids as a unifying theory for lipoprotein(a) and cardiovascular disease. Nat Rev Cardiol. 2019;16:305–318. doi: 10.1038/s41569-018-0153-2. [DOI] [PubMed] [Google Scholar]
- 70.Zheng K.H., Tsimikas S., Pawade T., et al. Lipoprotein(a) and oxidized phospholipids promote valve calcification in patients with aortic stenosis. J Am Coll Cardiol. 2019;73:2150–2162. doi: 10.1016/j.jacc.2019.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Blaser M.C., Buffolo F., Halu A., et al. Multiomics of tissue extracellular vesicles identifies unique modulators of atherosclerosis and calcific aortic valve stenosis. Circulation. 2023;148:661–678. doi: 10.1161/CIRCULATIONAHA.122.063402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rogers M.A., Aikawa E. A not-so-little role for lipoprotein(a) in the development of calcific aortic valve disease. Circulation. 2015;132:621–623. doi: 10.1161/CIRCULATIONAHA.115.018139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kaiser Y., Singh S.S., Zheng K.H., et al. Lipoprotein(a) is robustly associated with aortic valve calcium. Heart. 2021;107:1422–1428. doi: 10.1136/heartjnl-2021-319044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boffa M.B., Marar T.T., Yeang C., et al. Potent reduction of plasma lipoprotein (a) with an antisense oligonucleotide in human subjects does not affect ex vivo fibrinolysis. J Lipid Res. 2019;60:2082–2089. doi: 10.1194/jlr.P094763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moriarty P.M., Gorby L.K., Stroes E.S., et al. Lipoprotein(a) and its potential association with thrombosis and inflammation in COVID-19: a testable hypothesis. Curr Atheroscler Rep. 2020;22:48. doi: 10.1007/s11883-020-00867-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martinez C., Rivera J., Loyau S., et al. Binding of recombinant apolipoprotein(a) to human platelets and effect on platelet aggregation. Thromb Haemost. 2001;85:686–693. [PubMed] [Google Scholar]
- 77.Liu H., Fu D., Luo Y., Peng D. Independent association of Lp(a) with platelet reactivity in subjects without statins or antiplatelet agents. Sci Rep. 2022;12:16609. doi: 10.1038/s41598-022-21121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Caplice N.M., Panetta C., Peterson T.E., et al. Lipoprotein (a) binds and inactivates tissue factor pathway inhibitor: a novel link between lipoproteins and thrombosis. Blood. 2001;98:2980–2987. doi: 10.1182/blood.v98.10.2980. [DOI] [PubMed] [Google Scholar]
- 79.Multimodality Writing Group for Chronic Coronary D. Winchester D.E., Maron D.J., et al. ACC/AHA/ASE/ASNC/ASPC/HFSA/HRS/SCAI/SCCT/SCMR/STS 2023 multimodality appropriate use criteria for the detection and risk assessment of chronic coronary disease. J Am Coll Cardiol. 2023;81:2445–2467. doi: 10.1016/j.jacc.2023.03.410. [DOI] [PubMed] [Google Scholar]
- 80.Grundy S.M., Stone N.J., Bailey A.L., et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Circulation. 2019;139:e1082–e1143. doi: 10.1161/CIR.0000000000000625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mach F., Baigent C., Catapano A.L., et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41:111–188. doi: 10.1093/eurheartj/ehz455. [DOI] [PubMed] [Google Scholar]
- 82.Pearson G.J., Thanassoulis G., Anderson T.J., et al. 2021 Canadian cardiovascular society guidelines for the management of dyslipidemia for the prevention of cardiovascular disease in adults. Can J Cardiol. 2021;37:1129–1150. doi: 10.1016/j.cjca.2021.03.016. [DOI] [PubMed] [Google Scholar]
- 83.Koschinsky M., Bajaj A., Boffa M., et al. A focused update to the 2019 NLA scientific statement on use of lipoprotein(a) in clinical practice. J Clin Lipidol. 2024 doi: 10.1016/j.jacl.2024.03.001. (In Press) [DOI] [PubMed] [Google Scholar]
- 84.Kronenberg F. Measuring lipoprotein(a): do it without ifs and buts. Eur J Prev Cardiol. 2022;29:766–768. doi: 10.1093/eurjpc/zwab180. [DOI] [PubMed] [Google Scholar]
- 85.Rikhi R., Hammoud A., Ashburn N., et al. Relationship of low-density lipoprotein-cholesterol and lipoprotein(a) to cardiovascular risk: the multi-ethnic study of atherosclerosis (MESA) Atherosclerosis. 2022;363:102–108. doi: 10.1016/j.atherosclerosis.2022.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fonseca A.F., Lahoz R., Ferber P., et al. A real-world assessment of the distribution of lipid levels across increasing lipoprotein(a) levels in patients with established cardiovascular disease in the United States. Eur Heart J. 2020;41:2989. [Google Scholar]
- 87.Willeit P., Kiechl S., Kronenberg F., et al. Discrimination and net reclassification of cardiovascular risk with lipoprotein(a): prospective 15-year outcomes in the Bruneck Study. J Am Coll Cardiol. 2014;64:851–860. doi: 10.1016/j.jacc.2014.03.061. [DOI] [PubMed] [Google Scholar]
- 88.Nurmohamed N.S., Kaiser Y., Schuitema P.C.E., et al. Finding very high lipoprotein(a): the need for routine assessment. Eur J Prev Cardiol. 2022;29:769–776. doi: 10.1093/eurjpc/zwab167. [DOI] [PubMed] [Google Scholar]
- 89.Afshar M., Rong J., Zhan Y., et al. Risks of incident cardiovascular disease associated with concomitant elevations in lipoprotein(a) and low-density lipoprotein cholesterol-the framingham heart study. J Am Heart Assoc. 2020;9 doi: 10.1161/JAHA.119.014711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Saeed A., Sun W., Agarwala A., et al. Lipoprotein(a) levels and risk of cardiovascular disease events in individuals with diabetes mellitus or prediabetes: the Atherosclerosis Risk in Communities study. Atherosclerosis. 2019;282:52–56. doi: 10.1016/j.atherosclerosis.2018.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wong N., Fan W., Hu X., et al. Lipoprotein(a) and long-term cardiovascular risk in a multi-ethnic pooled prospective cohort. J Am Coll Cardiol. 2024 doi: 10.1016/j.jacc.2024.02.031. (In Press) [DOI] [PubMed] [Google Scholar]
- 92.Beheshtian A., Shitole S.G., Segal A.Z., et al. Lipoprotein (a) level, apolipoprotein (a) size, and risk of unexplained ischemic stroke in young and middle-aged adults. Atherosclerosis. 2016;253:47–53. doi: 10.1016/j.atherosclerosis.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kamstrup P.R., Benn M., Tybjaerg-Hansen A., Nordestgaard B.G. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen City heart study. Circulation. 2008;117:176–184. doi: 10.1161/CIRCULATIONAHA.107.715698. [DOI] [PubMed] [Google Scholar]
- 94.Perrot N., Verbeek R., Sandhu M., et al. Ideal cardiovascular health influences cardiovascular disease risk associated with high lipoprotein(a) levels and genotype: the EPIC-Norfolk prospective population study. Atherosclerosis. 2017;256:47–52. doi: 10.1016/j.atherosclerosis.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rikhi R., Bhatia H.S., Schaich C.L., et al. Association of Lp(a) (lipoprotein[a]) and hypertension in primary prevention of cardiovascular disease: the MESA. Hypertension. 2022;80:352–360. doi: 10.1161/HYPERTENSIONAHA.122.20189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Derby C.A., Crawford S.L., Pasternak R.C., et al. Lipid changes during the menopause transition in relation to age and weight: the Study of Women's Health Across the Nation. Am J Epidemiol. 2009;169:1352–1361. doi: 10.1093/aje/kwp043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dzobo K.E., Kraaijenhof J.M., Stroes E.S.G., Nurmohamed N.S., Kroon J. Lipoprotein(a): an underestimated inflammatory mastermind. Atherosclerosis. 2022;349:101–109. doi: 10.1016/j.atherosclerosis.2022.04.004. [DOI] [PubMed] [Google Scholar]
- 98.Enkhmaa B., Berglund L. Non-genetic influences on lipoprotein(a) concentrations. Atherosclerosis. 2022;349:53–62. doi: 10.1016/j.atherosclerosis.2022.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.de Boer L.M., Hof M.H., Wiegman A., et al. Lipoprotein(a) levels from childhood to adulthood: data in nearly 3,000 children who visited a pediatric lipid clinic. Atherosclerosis. 2022;349:227–232. doi: 10.1016/j.atherosclerosis.2022.03.004. [DOI] [PubMed] [Google Scholar]
- 100.Deshotels M.R., Sun C., Nambi V., et al. Temporal trends in lipoprotein(a) concentrations: the atherosclerosis risk in communities study. J Am Heart Assoc. 2022;11 doi: 10.1161/JAHA.122.026762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kronenberg F. Lipoprotein(a) measurement issues: are we making a mountain out of a molehill? Atherosclerosis. 2022;349:123–135. doi: 10.1016/j.atherosclerosis.2022.04.008. [DOI] [PubMed] [Google Scholar]
- 102.MacDougall D.E., McGowan M.P., Wilemon K.A., Ahmed C.D., Myers K.D. Characterization of Lp(a) measurement in a large U.S. Health Care Dataset. J Clin Lipidol. 2022;16:e36–e37. [Google Scholar]
- 103.Hu X., Cristino J., Gautam R., et al. Characteristics and lipid lowering treatment patterns in patients tested for lipoprotein(a): a real-world US study. Am J Prev Cardiol. 2023;14 doi: 10.1016/j.ajpc.2023.100476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Catapano A.L., Daccord M., Damato E., et al. How should public health recommendations address Lp(a) measurement, a causative risk factor for cardiovascular disease (CVD)? Atherosclerosis. 2022;349:136–143. doi: 10.1016/j.atherosclerosis.2022.02.013. [DOI] [PubMed] [Google Scholar]
- 105.Zwack C.C., Haghani M., Hollings M., et al. The evolution of digital health technologies in cardiovascular disease research. NPJ Digit Med. 2023;6:1. doi: 10.1038/s41746-022-00734-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chow C.K., Redfern J., Hillis G.S., et al. Effect of lifestyle-focused text messaging on risk factor modification in patients with coronary heart disease: a randomized clinical trial. JAMA. 2015;314:1255–1263. doi: 10.1001/jama.2015.10945. [DOI] [PubMed] [Google Scholar]
- 107.Redfern J., Coorey G., Mulley J., et al. A digital health intervention for cardiovascular disease management in primary care (CONNECT) randomized controlled trial. NPJ Digit Med. 2020;3:117. doi: 10.1038/s41746-020-00325-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wong ND. Lipoprotein(a): ready for prime time. J am Coll Cardiol. 2024 doi: 10.1016/j.jacc.2024.01.004. (In Press) [DOI] [PubMed] [Google Scholar]
- 109.Morris P.B., Narula J., Tsimikas S. Lipoprotein(a) and LDL-C: the relevance of equivalence. J Am Coll Cardiol. 2022;80:2011–2013. doi: 10.1016/j.jacc.2022.09.026. [DOI] [PubMed] [Google Scholar]
- 110.Marcovina S.M., Shapiro M.D. Measurement of lipoprotein(a): a once in a lifetime opportunity. J Am Coll Cardiol. 2022;79:629–631. doi: 10.1016/j.jacc.2021.11.053. [DOI] [PubMed] [Google Scholar]
- 111.Lloyd-Jones D.M., Braun L.T., Ndumele C.E., et al. Use of risk assessment tools to guide decision-making in the primary prevention of atherosclerotic cardiovascular disease: a special report from the American Heart Association and American College of Cardiology. Circulation. 2019;139:e1162–e1177. doi: 10.1161/CIR.0000000000000638. [DOI] [PubMed] [Google Scholar]
- 112.Mehta A., Vasquez N., Ayers C.R., et al. Independent association of lipoprotein(a) and coronary artery calcification with atherosclerotic cardiovascular risk. J Am Coll Cardiol. 2022;79:757–768. doi: 10.1016/j.jacc.2021.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wong N.D., Fan W., Hu X., et al. Relation of lipoprotein(a) to progression of coronary artery calcium: results from a pooled cohort of mesa, cardia, and framingham offspring studies. Circulation. 2022;146:A10296. [Google Scholar]
- 114.Zhang W., Speiser J.L., Ye F., et al. High-sensitivity C-reactive protein modifies the cardiovascular risk of lipoprotein(a): multi-ethnic study of atherosclerosis. J Am Coll Cardiol. 2021;78:1083–1094. doi: 10.1016/j.jacc.2021.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Puri R., Nissen S.E., Arsenault B.J., et al. Effect of C-reactive protein on lipoprotein(a)-associated cardiovascular risk in optimally treated patients with high-risk vascular disease: a prespecified secondary analysis of the ACCELERATE trial. JAMA Cardiol. 2020;5:1136–1143. doi: 10.1001/jamacardio.2020.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Thomas P.E., Vedel-Krogh S., Kamstrup P.R., Nordestgaard B.G. Lipoprotein(a) is linked to atherothrombosis and aortic valve stenosis independent of C-reactive protein. Eur Heart J. 2023;44:1449–1460. doi: 10.1093/eurheartj/ehad055. [DOI] [PubMed] [Google Scholar]
- 117.Lloyd-Jones D.M., Allen N.B., Anderson C.A.M., et al. Life's Essential 8: updating and enhancing the American Heart Association's Construct of Cardiovascular Health: a presidential advisory from the American Heart Association. Circulation. 2022;146:e18–e43. doi: 10.1161/CIR.0000000000001078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ridker P.M., Lei L., Ray K.K., et al. Effects of bempedoic acid on CRP, IL-6, fibrinogen and lipoprotein(a) in patients with residual inflammatory risk: a secondary analysis of the CLEAR harmony trial. J Clin Lipidol. 2023;17:297–302. doi: 10.1016/j.jacl.2023.02.002. [DOI] [PubMed] [Google Scholar]
- 119.Cupido A.J., Kastelein J.J.P. Inclisiran for the treatment of hypercholesterolaemia: implications and unanswered questions from the ORION trials. Cardiovasc Res. 2020;116:e136–e139. doi: 10.1093/cvr/cvaa212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tsimikas S., Moriarty P.M., Stroes E.S. Emerging RNA therapeutics to lower blood levels of Lp(a): JACC focus seminar 2/4. J Am Coll Cardiol. 2021;77:1576–1589. doi: 10.1016/j.jacc.2021.01.051. [DOI] [PubMed] [Google Scholar]
- 121.van Capelleveen J.C., van der Valk F.M., Stroes E.S. Current therapies for lowering lipoprotein(a) J Lipid Res. 2016;57:1612–1618. doi: 10.1194/jlr.R053066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chasman D.I., Shiffman D., Zee R.Y., et al. Polymorphism in the Apolipoprotein(a) gene, plasma lipoprotein(a), cardiovascular disease, and low-dose aspirin therapy. Atherosclerosis. 2009;203:371–376. doi: 10.1016/j.atherosclerosis.2008.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lacaze P., Bakshi A., Riaz M., et al. Aspirin for primary prevention of cardiovascular events in relation to lipoprotein(a) genotypes. J Am Coll Cardiol. 2022;80:1287–1298. doi: 10.1016/j.jacc.2022.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sturzebecher P.E., Schorr J.J., Klebs S.H.G., Laufs U. Trends and consequences of lipoprotein(a) testing: cross-sectional and longitudinal health insurance claims database analyses. Atherosclerosis. 2023;367:24–33. doi: 10.1016/j.atherosclerosis.2023.01.014. [DOI] [PubMed] [Google Scholar]
- 125.Writing C., Lloyd-Jones D.M., Morris P.B., et al. 2022 ACC expert consensus decision pathway on the role of nonstatin therapies for LDL-cholesterol lowering in the management of atherosclerotic cardiovascular disease risk: a report of the American College of Cardiology Solution Set Oversight Committee. J Am Coll Cardiol. 2022;80:1366–1418. doi: 10.1016/j.jacc.2022.07.006. [DOI] [PubMed] [Google Scholar]
- 126.Alonso R., Andres E., Mata N., et al. Lipoprotein(a) levels in familial hypercholesterolemia: an important predictor of cardiovascular disease independent of the type of LDL receptor mutation. J Am Coll Cardiol. 2014;63:1982–1989. doi: 10.1016/j.jacc.2014.01.063. [DOI] [PubMed] [Google Scholar]
- 127.Langsted A., Kamstrup P.R., Benn M., Tybjaerg-Hansen A., Nordestgaard B.G. High lipoprotein(a) as a possible cause of clinical familial hypercholesterolaemia: a prospective cohort study. Lancet Diabetes Endocrinol. 2016;4:577–587. doi: 10.1016/S2213-8587(16)30042-0. [DOI] [PubMed] [Google Scholar]
- 128.Wallentin L., Becker R.C., Budaj A., et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361:1045–1057. doi: 10.1056/NEJMoa0904327. [DOI] [PubMed] [Google Scholar]
- 129.Eikelboom J.W., Connolly S.J., Bosch J., et al. Rivaroxaban with or without aspirin in stable cardiovascular disease. N Engl J Med. 2017;377:1319–1330. doi: 10.1056/NEJMoa1709118. [DOI] [PubMed] [Google Scholar]
- 130.Deftereos S.G., Beerkens F.J., Shah B., et al. Colchicine in cardiovascular disease: in-depth review. Circulation. 2022;145:61–78. doi: 10.1161/CIRCULATIONAHA.121.056171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nidorf S.M., Eikelboom J.W., Budgeon C.A., Thompson P.L. Low-dose colchicine for secondary prevention of cardiovascular disease. J Am Coll Cardiol. 2013;61:404–410. doi: 10.1016/j.jacc.2012.10.027. [DOI] [PubMed] [Google Scholar]
- 132.Nidorf S.M., Fiolet A.T.L., Mosterd A., et al. Colchicine in patients with chronic coronary disease. N Engl J Med. 2020;383:1838–1847. doi: 10.1056/NEJMoa2021372. [DOI] [PubMed] [Google Scholar]
- 133.Tardif J.C., Kouz S., Waters D.D., et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. 2019;381:2497–2505. doi: 10.1056/NEJMoa1912388. [DOI] [PubMed] [Google Scholar]
- 134.Ridker P.M., Devalaraja M., Baeres F.M.M., et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. 2021;397:2060–2069. doi: 10.1016/S0140-6736(21)00520-1. [DOI] [PubMed] [Google Scholar]
- 135.Agca R., Smulders Y., Nurmohamed M. Cardiovascular disease risk in immune-mediated inflammatory diseases: recommendations for clinical practice. Heart. 2022;108:73–79. doi: 10.1136/heartjnl-2019-316378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Agarwala A., Ballantyne C., Stone N.J. Primary prevention management of elevated lipoprotein(a) JAMA Cardiol. 2023;8:96–97. doi: 10.1001/jamacardio.2022.4063. [DOI] [PubMed] [Google Scholar]
- 137.Averna M.R., Cefalu A.B. Lp(a): a genetic cause of clinical FH in children. Eur Heart J. 2022 doi: 10.1093/eurheartj/ehac789. [DOI] [PubMed] [Google Scholar]
- 138.Ellis K.L., Perez de Isla L., Alonso R., et al. Value of measuring lipoprotein(a) during cascade testing for familial hypercholesterolemia. J Am Coll Cardiol. 2019;73:1029–1039. doi: 10.1016/j.jacc.2018.12.037. [DOI] [PubMed] [Google Scholar]
- 139.Nurmohamed N.S., Kraaijenhof J.M., Stroes E.S.G. Lp(a): a new pathway to target? Curr Atheroscler Rep. 2022;24:831–838. doi: 10.1007/s11883-022-01060-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kamstrup P.R., Tybjaerg-Hansen A., Steffensen R., Nordestgaard B.G. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–2339. doi: 10.1001/jama.2009.801. [DOI] [PubMed] [Google Scholar]
- 141.Tsimikas S., Karwatowska-Prokopczuk E., Gouni-Berthold I., et al. Lipoprotein(a) reduction in persons with cardiovascular disease. N Engl J Med. 2020;382:244–255. doi: 10.1056/NEJMoa1905239. [DOI] [PubMed] [Google Scholar]
- 142.Stiekema L.C.A., Prange K.H.M., Hoogeveen R.M., et al. Potent lipoprotein(a) lowering following apolipoprotein(a) antisense treatment reduces the pro-inflammatory activation of circulating monocytes in patients with elevated lipoprotein(a) Eur Heart J. 2020;41:2262–2271. doi: 10.1093/eurheartj/ehaa171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.O’Donoghue M.L., Rosenson R.S., Gencer B., et al. Small interfering RNA to reduce lipoprotein(a) in cardiovascular disease. N Engl J Med. 2022;387:1855–1864. doi: 10.1056/NEJMoa2211023. [DOI] [PubMed] [Google Scholar]
- 144.Nissen S.E., Wolski K., Balog C., et al. Single ascending dose study of a short interfering RNA targeting lipoprotein(a) production in individuals with elevated plasma lipoprotein(a) levels. JAMA. 2022;327:1679–1687. doi: 10.1001/jama.2022.5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nissen S.E., Linnebjerg H., Shen X., et al. Lepodisiran, an extended-duration short interfering RNA targeting lipoprotein(a): a randomized dose-ascending clinical trial. JAMA. 2023;330:2075–2083. doi: 10.1001/jama.2023.21835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nicholls S.J., Nissen S.E., Fleming C., et al. Muvalaplin, an oral small molecule inhibitor of lipoprotein(a) formation: a randomized clinical trial. JAMA. 2023;330:1042–1053. doi: 10.1001/jama.2023.16503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nicholls S.J., Ditmarsch M., Kastelein J.J., et al. Lipid lowering effects of the CETP inhibitor obicetrapib in combination with high-intensity statins: a randomized phase 2 trial. Nat Med. 2022;28:1672–1678. doi: 10.1038/s41591-022-01936-7. [DOI] [PubMed] [Google Scholar]
- 148.Tokgozoglu L., Libby P. The dawn of a new era of targeted lipid-lowering therapies. Eur Heart J. 2022;43:3198–3208. doi: 10.1093/eurheartj/ehab841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Landmesser U., Poller W., Tsimikas S., et al. From traditional pharmacological towards nucleic acid-based therapies for cardiovascular diseases. Eur Heart J. 2020;41:3884–3899. doi: 10.1093/eurheartj/ehaa229. [DOI] [PubMed] [Google Scholar]
- 150.Musunuru K., Chadwick A.C., Mizoguchi T., et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature. 2021;593:429–434. doi: 10.1038/s41586-021-03534-y. [DOI] [PubMed] [Google Scholar]
- 151.Roeseler E., Julius U., Heigl F., et al. Lipoprotein apheresis for lipoprotein(a)-associated cardiovascular disease: prospective 5 years of follow-up and Apolipoprotein(a) characterization. Arterioscler Thromb Vasc Biol. 2016;36:2019–2027. doi: 10.1161/ATVBAHA.116.307983. [DOI] [PubMed] [Google Scholar]
- 152.Tsimikas S., Gordts P., Nora C., Yeang C., Witztum J.L. Statin therapy increases lipoprotein(a) levels. Eur Heart J. 2020;41:2275–2284. doi: 10.1093/eurheartj/ehz310. [DOI] [PubMed] [Google Scholar]
- 153.Grundy S.M., Stone N.J., Bailey A.L., et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines. J Am Coll Cardiol. 2019;73:3168–3209. doi: 10.1016/j.jacc.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 154.Cholesterol Treatment Trialists C., Mihaylova B., Emberson J., et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet. 2012;380:581–590. doi: 10.1016/S0140-6736(12)60367-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cannon C.P., Blazing M.A., Giugliano R.P., et al. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387–2397. doi: 10.1056/NEJMoa1410489. [DOI] [PubMed] [Google Scholar]
- 156.Nissen S.E., Lincoff A.M., Brennan D., et al. Bempedoic acid and cardiovascular outcomes in statin-intolerant patients. N Engl J Med. 2023;388:1353–1364. doi: 10.1056/NEJMoa2215024. [DOI] [PubMed] [Google Scholar]
- 157.Investigators A.-H., Boden W.E., Probstfield J.L., et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 158.Sabatine M.S., Giugliano R.P., Keech A.C., et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–1722. doi: 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- 159.Schwartz G.G., Steg P.G., Szarek M., et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379:2097–2107. doi: 10.1056/NEJMoa1801174. [DOI] [PubMed] [Google Scholar]
- 160.Wright R.S., Ray K.K., Raal F.J., et al. Pooled patient-level analysis of inclisiran trials in patients with familial hypercholesterolemia or atherosclerosis. J Am Coll Cardiol. 2021;77:1182–1193. doi: 10.1016/j.jacc.2020.12.058. [DOI] [PubMed] [Google Scholar]
- 161.Nissen S.E., Linnebjerg H. Shen X.,et al. Lepodisiran,an extended-duration short interfering RNA targeting lipoprotein(a): a randomized dose-ascending clinical trial. JAMA. 2023;330:2075–2083. doi: 10.1001/jama.2023.21835. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.