

Cellular and organismal aging have been consistently associated with mitochondrial dysfunction and inflammation. Accumulating evidence indicates that aging-related inflammatory responses are mechanistically linked to compromised mitochondrial integrity coupled with mtDNA-driven CGAS activation, a process that is tonically inhibited by mitophagy.
Subject terms: Mitochondria, Ageing
Cellular and organismal aging have been consistently associated with mitochondrial dysfunction and inflammation. Accumulating evidence indicates that aging-related inflammatory responses are mechanistically linked to compromised mitochondrial integrity coupled with mtDNA-driven CGAS activation, a process that is tonically inhibited by mitophagy.
Both cellular senescence and organismal aging are accompanied by a multitude of pathophysiological alterations, encompassing (but not limited to) accumulating oxidative damage to macromolecules including DNA, impaired metabolism, and inflammation1. All these defects are intimately connected with yet another hallmark of aging: mitochondrial dysfunction1. Indeed, defective mitochondria are prone to overproduce reactive oxygen species (ROS), are unable to support physiological metabolism, and can elicit potent inflammatory reactions1,2. Importantly, aging biological systems also exhibit a decrease in the proficiency of homeostatic processes such as autophagy, a lysosome-dependent mechanism for the degradation of damaged or otherwise potentially harmful cytoplasmic entities1. In line with this notion, multiple experimental maneuvers that promote autophagy (such as caloric restriction) have been shown to extend lifespan in a variety of model organisms including mice1.
That said, whether aging-associated alterations emerge as a consequence of defects in cellular adaptation or instead perturbations of homeostasis ultimately overwhelm the cellular capacity for adaptation remains to be formally elucidated. Irrespective of this and other unknown, the acquisition of a senescent phenotype by aging cells has been shown to involve an autocrine/paracrine mechanism linked to type I interferon (IFN) signaling as elicited not only by DNA damage3, but also by mitochondrial dysfunction4. Thus, mitochondrial integrity stands out as a major gatekeeper for the control of genetic, metabolic, and inflammatory homeostasis1. Recent data from Jimenez-Loygorri et al. demonstrate that mitophagy (a specialized variant autophagy that degrades defective mitochondria)5 limits aging-associating neurological decline by suppressing inflammatory reactions driven by primary mitochondrial dysfunction coupled with mitochondrial DNA (mtDNA) release and consequent cyclic GMP-AMP synthase (CGAS) signaling6.
Jimenez-Loygorri et al. set out to examine the activation of the molecular machinery for mitophagy across different organs in old versus young mice expressing a pH-sensitive reporter that enables the discrimination of cytosolic (mCherry+GFP+) versus lysosomal (mCherry+GFP−) mitochondria (so-called mito-QC mice). Surprisingly, in some organs such as the retina, mitophagy was higher in old vs young mice. In line with this notion, the retina of aged mice exhibited increased markers of mitophagy (but not general autophagy) activation including the PTEN-induced kinase 1 (PINK1)-dependent phosphorylation of ubiquitin at S65. Such an increase in mitophagy was accompanied not only by ultrastructural markers of mitochondrial damage (e.g., swollen mitochondria, mitochondria exhibiting cristae disruption) but also by the cytosolic accumulation of mtDNA and consequent CGAS activation, culminating in (1) the stimulator of interferon response cGAMP interactor 1 (STING1)-dependent activating phosphorylation of the transcription factor interferon regulatory factor 3 (IRF3) at S396, and (2) the expression of multiple IRF3 targets, including several interferon-stimulated genes (ISGs) like interferon beta 1 (Ifnb1). Similar observations were obtained with primary normal human dermal fibroblasts (NHDFs) from aged individuals, globally suggesting that PINK1-mediated mitophagy is activated during aging in response to primary mitochondrial dysfunction6.
Next, Jimenez-Loygorri and colleagues investigated the impact of experimental interventions that alter mitophagic activity on the aging retina. Urolithin A (UA), which indirectly inhibits mechanistic target of rapamycin (MTOR) signaling, promoted mitophagy in the retina of both young and aged mice, a precess that in the latter setting was associated with significant improvements in recognition memory, night vision, synaptic integrity and limited aberrant integration of light stimuli compared to untreated old mice. Moreover, UA decreased the amount of total and CGAS-bound mtDNA in the cytosolic fraction of the retina from aged mice, which was accompanied by a reduction in genetic signatures of CGAS signaling and type I interferon (IFN) responses6. In line with this finding, cytosolic mtDNA accumulation as elicited by ABT-737 – which promotes mitochondrial outer membrane permeabilization (MOMP) by enabling BCL2 associated X, apoptosis regulator (BAX) and BCL2 antagonist/killer 1 (BAK1) oligomerization7 – and QVD – which blocks caspases to prevent the suppression of CGAS signaling downstream of MOMP8,9 – in immortalized retinal pigmented epithelial ARPE-19 cells was significantly inhibited by UA. Accordingly, UA not only abolished CGAS activation but also limited ROS production and restored oxidative phosphorylation in ARPE-19 cells exposed to ABT-737 and QVD. Of note, the CGAS inhibitor G140 also limited mitophagy as driven by MOMP in ARPE-19 cells, but failed to alter cytosolic mtDNA accumulation6. These findings suggest that mitophagic responses promoted by mitochondrial dysfunction in the aged retina may depend, at least in part, on CGAS signaling.
To obtain additional insights into their observations, Jimenez-Loygorri and collaborators co-silenced PINK1 and parkin RBR E3 ubiquitin-protein ligase (PRKN, which encodes another molecular component of the mitophagy apparatus)5 in ARPE-19 cells exposed to ABT-737 and QVD, finding that cytosolic mtDNA accumulation as elicited by MOMP in the context of caspase inhibition is aggravated by mitophagy defects, which also abolish the effects of UA. Conversely, inhibition of mitochondrial biogenesis with chloramphenicol reduced cytosolic mtDNA accumulation as driven by ABT-737 and QVD in ARPE-19 cells while preserving their sensitivity to UA6.
In conclusion, Jimenez-Loygorri and colleagues demonstrated that mitophagy decelerates aging by suppressing inflammatory responses downstream of primary mitochondrial dysfunction and consequent mtDNA-dependent CGAS signaling (Fig. 1). Together with recent data from us and others4,10–13, these findings point to a central role for the mitochondrial checkpoint in a multitude of pathophysiological settings, including aging, autoimmunity, adaptive immune responses as well as cancer sensitivity to (immuno)therapeutics. Thus, since mitophagy acts as a major gatekeeper of the mitochondrial checkpoint, pharmacological strategies to enforce it (mitophagy activators) or weaken it (mitophagy inhibitors) may have broad therapeutic applications. Importantly, the activation of apoptotic executioner caspases as elicited by widespread MOMP has been consistently shown to suppress CGAS signaling by a variety of mechanisms14,15. In line with this notion, aging cells appear to experience sublethal degrees of MOMP (also known as minority MOMP) that are compatible with cell survival but promote senescence and inflammatory responses2,4. These observations raise the intriguing possibility that strategies to elicit the sublethal activation of executioner caspases in the absence of accrued MOMP might suppress aging-associated inflammation without causing cell death, as would inhibitors of CGAS, STING1, or IRF3. Additional work is required to formally investigate these possibilities.
Fig. 1. Mitophagy-dependent enforcement of the mitochondrial checkpoint in stressed and aging cells.
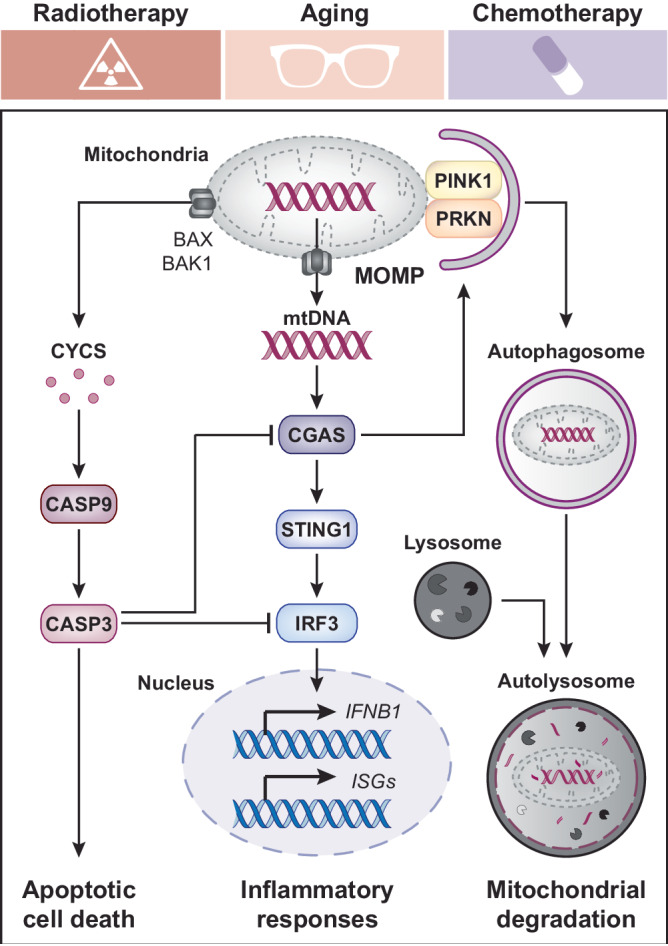
Mitochondrial dysfunction as spontaneously emerging in aging cells or as elicited by exogenous stressors such as radiation therapy and chemotherapy can be accompanied by the permeabilization of mitochondrial membranes, hence compromising the integrity of the mitochondrial checkpoint. In this context, mitochondrial DNA (mtDNA) released or bulging from permeabilized mitochondria operates as a potent activator of cyclic GMP-AMP synthase (CGAS), hence initiating a stimulator of interferon response cGAMP interactor 1 (STING1)-dependent signaling cascade that culminates with the transactivation of multiple interferon-stimulated genes (ISGs), including interferon beta 1 (IFNB1). The efficient removal of compromised mitochondria as ensured by PTEN induced kinase 1 (PINK1)- and parkin RBR E3 ubiquitin protein ligase (PRKN)-dependent mitophagy tonically suppresses such an aging- and stress-associated inflammatory phenotype. Of note, mitochondrial outer membrane permeabilization (MOMP) as mediated by BCL2 associated X, apoptosis regulator (BAX) and BCL2 antagonist/killer 1 (BAK1) is also associated with the release of cytochrome c, somatic (CYCS), culminating in at least some degree of caspase 9 (CASP9) and CASP3 activation, which precipitates apoptotic cell death as it suppresses CGAS signaling. Whether activating apoptotic caspases to sublethal degrees in the absence of accrued MOMP may decelerate aging by limiting mtDNA-driven CGAS activation remains to be elucidated. IRF3 interferon regulatory factor 3.
Acknowledgements
The LG lab is/has been supported (as a PI unless otherwise indicated) by one NIH R01 grant (#CA271915), by two Breakthrough Level 2 grants from the US DoD BCRP (#BC180476P1, #BC210945), by a grant from the STARR Cancer Consortium (#I16-0064), by a Transformative Breast Cancer Consortium Grant from the US DoD BCRP (#W81XWH2120034, PI: Formenti), by a U54 grant from NIH/NCI (#CA274291, PI: Deasy, Formenti, Weichselbaum), by the 2019 Laura Ziskin Prize in Translational Research (#ZP-6177, PI: Formenti) from the Stand Up to Cancer (SU2C), by a Mantle Cell Lymphoma Research Initiative (MCL-RI, PI: Chen-Kiang) grant from the Leukemia and Lymphoma Society (LLS), by a Rapid Response Grant from the Functional Genomics Initiative (New York, US), by a pre-SPORE grant (PI: Demaria, Formenti), a Collaborative Research Initiative Grant and a Clinical Trials Innovation Grant from the Sandra and Edward Meyer Cancer Center (New York, US), by startup funds from the Dept. of Radiation Oncology at Weill Cornell Medicine (New York, US), by industrial collaborations with Lytix Biopharma (Oslo, Norway), Promontory (New York, US) and Onxeo (Paris, France), as well as by donations from Promontory (New York, US), the Luke Heller TECPR2 Foundation (Boston, US), Sotio a.s. (Prague, Czech Republic), Lytix Biopharma (Oslo, Norway), Onxeo (Paris, France), Ricerchiamo (Brescia, Italy), and Noxopharm (Chatswood, Australia).
Author contributions
L.G. conceived the article. E.G. and L.G. wrote the first version of the manuscript with constructive input from K.A.S. E.G. generated display items under supervision from K.A.S. and L.G. All authors approved the submitted version of the article.
Competing interests
L.G. is/has been holding research contracts with Lytix Biopharma, Promontory, and Onxeo, has received consulting/advisory honoraria from Boehringer Ingelheim, AstraZeneca, OmniSEQ, Onxeo, The Longevity Labs, Inzen, Imvax, Sotio, Promontory, Noxopharm, EduCom, and the Luke Heller TECPR2 Foundation, and holds Promontory stock options. E.G. and K.A.S. have no conflicts of interest to declare.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lopez-Otin C, Pietrocola F, Roiz-Valle D, Galluzzi L, Kroemer G. Meta-hallmarks of aging and cancer. Cell Metab. 2023;35:12–35. doi: 10.1016/j.cmet.2022.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Marchi S, Guilbaud E, Tait SWG, Yamazaki T, Galluzzi L. Mitochondrial control of inflammation. Nat. Rev. Immunol. 2023;23:159–173. doi: 10.1038/s41577-022-00760-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dou Z, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550:402–406. doi: 10.1038/nature24050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Victorelli S, et al. Apoptotic stress causes mtDNA release during senescence and drives the SASP. Nature. 2023;622:627–636. doi: 10.1038/s41586-023-06621-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vargas JNS, Hamasaki M, Kawabata T, Youle RJ, Yoshimori T. The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol. 2023;24:167–185. doi: 10.1038/s41580-022-00542-2. [DOI] [PubMed] [Google Scholar]
- 6.Jimenez-Loygorri JI, et al. Mitophagy curtails cytosolic mtDNA-dependent activation of cGAS/STING inflammation during aging. Nat. Commun. 2024;15:830. doi: 10.1038/s41467-024-45044-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McArthur K, et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science. 2018;359:eaao6047. doi: 10.1126/science.aao6047. [DOI] [PubMed] [Google Scholar]
- 8.Rongvaux A, et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 2014;159:1563–1577. doi: 10.1016/j.cell.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.White MJ, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159:1549–1562. doi: 10.1016/j.cell.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao L, et al. BCL2 inhibition reveals a dendritic cell-specific immune checkpoint that controls tumor immunosurveillance. Cancer Discov. 2023;13:2448–2469. doi: 10.1158/2159-8290.CD-22-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamazaki T, et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat. Immunol. 2020;21:1160–1171. doi: 10.1038/s41590-020-0751-0. [DOI] [PubMed] [Google Scholar]
- 12.Kim J, et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science. 2019;366:1531–1536. doi: 10.1126/science.aav4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zecchini V, et al. Fumarate induces vesicular release of mtDNA to drive innate immunity. Nature. 2023;615:499–506. doi: 10.1038/s41586-023-05770-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez-Ruiz ME, et al. Apoptotic caspases inhibit abscopal responses to radiation and identify a new prognostic biomarker for breast cancer patients. Oncoimmunology. 2019;8:e1655964. doi: 10.1080/2162402X.2019.1655964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giampazolias E, et al. Mitochondrial permeabilization engages NF-kappaB-dependent anti-tumour activity under caspase deficiency. Nat. Cell Biol. 2017;19:1116–1129. doi: 10.1038/ncb3596. [DOI] [PMC free article] [PubMed] [Google Scholar]