

Significance
Cancer relies on genomic changes to drive evolution. One type of change, chromosomal instability (CIN), promotes plasticity and heterogeneity of chromosome sets via ongoing errors in mitosis. The rate of these errors informs patient prognosis, drug response, and risk of metastasis. However, measuring CIN in patient tissues is challenging, hindering the emergence of CIN rate as a prognostic and predictive clinical biomarker. To advance clinical measures of CIN, we quantitatively tested the relative performance of several CIN measures in tandem using four well-defined, inducible CIN models. This survey revealed poor sensitivity in several common CIN assays and highlights the primacy of single-cell approaches. Further, we propose a standard, normalized unit of CIN, permitting comparison across methods and studies.
Keywords: chromosomal instability, aneuploidy, mitosis, clinical biomarkers
Abstract
Chromosomal instability (CIN) is the persistent reshuffling of cancer karyotypes via chromosome mis-segregation during cell division. In cancer, CIN exists at varying levels that have differential effects on tumor progression. However, mis-segregation rates remain challenging to assess in human cancer despite an array of available measures. We evaluated measures of CIN by comparing quantitative methods using specific, inducible phenotypic CIN models of chromosome bridges, pseudobipolar spindles, multipolar spindles, and polar chromosomes. For each, we measured CIN fixed and timelapse fluorescence microscopy, chromosome spreads, six-centromere FISH, bulk transcriptomics, and single-cell DNA sequencing (scDNAseq). As expected, microscopy of tumor cells in live and fixed samples significantly correlated (R = 0.72; P < 0.001) and sensitively detect CIN. Cytogenetics approaches include chromosome spreads and 6-centromere FISH, which also significantly correlate (R = 0.76; P < 0.001) but had limited sensitivity for lower rates of CIN. Bulk genomic DNA signatures and bulk transcriptomic scores, CIN70 and HET70, did not detect CIN. By contrast, scDNAseq detects CIN with high sensitivity, and significantly correlates with imaging methods (R = 0.82; P < 0.001). In summary, single-cell methods such as imaging, cytogenetics, and scDNAseq can measure CIN, with the latter being the most comprehensive method accessible to clinical samples. To facilitate the comparison of CIN rates between phenotypes and methods, we propose a standardized unit of CIN: Mis-segregations per Diploid Division. This systematic analysis of common CIN measures highlights the superiority of single-cell methods and provides guidance for measuring CIN in the clinical setting.
David von Hansemann and Theodor Boveri described chromosomal instability (CIN) and proposed its role in human cancer over 100 y ago (1, 2). Indeed, CIN accelerates tumor evolution, and portend increased metastasis (3, 4), therapeutic resistance (5–7), and worse prognosis (8–10). Importantly, tumors’ CIN levels vary considerably. While increasing CIN levels correlate with features of advanced cancer, the relationship is not linear or simple. Very high CIN levels inhibit tumors in mouse models (11–13). In human cancer, CIN level categories, such as “low” and “high” CIN, have been associated with survival in both directions—improved (14–18) and impaired (8, 10, 19, 20). This discrepancy may be due, in part, to context dependent effects of CIN in different tissues, tumors, and model systems (12). However, suboptimal CIN measurements and poorly defined categories likely contribute, highlighting the need for accurate, quantitative measures of CIN.
The term CIN has been used to describe elevated mis-segregation of whole chromosomes (numerical CIN) and structural and copy number alterations that result from chromosome mis-segregation (structural CIN, e.g., chromothripsis). It has also frequently been erroneous equated to aneuploidy, which is a result of, but not the same as CIN. How CIN is defined impacts the interpretation of its measure and generalization of results. For this study, we define CIN as transient or persistent mis-segregation of chromosomes or chromosome fragments via abnormal mitosis which results in copy number gains or losses of any fraction of a chromosome.
Current measures of CIN vary in accuracy as well as how comprehensively and directly they assess CIN, leading to disagreement among measures (21, 22). Given these discrepancies, it is important to compare and judge the analytic validity of these approaches.
Among cell biologists, direct observation of mitotic abnormalities is a common measure of CIN. Fixed and time-lapse microscopy rely on the visual identification of abnormal mitotic phenotypes such as lagging, polar, and bridging chromosomes as well as spindle multipolarity. This direct observation of mitotic defects is considered the “gold standard.” However, the identity of the mis-segregated chromosome(s) remains unclear and, in some cases, even direct observation requires assumptions to infer chromosome mis-segregation, such as whether a lagging chromosome segregates to the correct or incorrect daughter cell (5–7, 23). Therefore, imaging detects CIN by association with mitotic defects. Further, these methods are not readily applicable to human tumors.
Cytogenetic methods are used to infer CIN by association with its resulting cell-to-cell variation in chromosome numbers or karyotypes. Cycling cells are captured in mitosis for chromosome counts and karyotypes. However, the reliance on mitotic cells may bias the sample, particularly as de novo aneuploidy may delay progression through interphase (24–26). By contrast, centromeric fluorescence in situ hybridization (cenFISH) identifies abnormal chromosome sets in fixed interphase cells without bias for cycling cells. However, cenFISH is limited to a small number of chromosomes and may suffer from other limitations such as sectioning artifacts. Further, any analysis of living cells in a tumor is biased by cellular selection, as dead cells are cleared (7, 27–29).
High-throughput sequencing enables several methods to study intratumoral heterogeneity and CIN. Bulk transcriptional data (bRNAseq) assigns CIN scores to tumor samples based on expression of a selected set of genes. The CIN70 expression score (30), meant to reflect the level of CIN, is derived from an inferred level of aneuploidy and correlates with proliferation and structural aneuploidy. Similarly, the HET70 score correlates with high karyotype heterogeneity in the NCI60 cell line panel, and, unlike CIN70, is independent of proliferation (31). Despite these advances, it is unclear whether bulk transcriptional scores can discriminate ongoing CIN in the tumor from historical CIN—chromosome aberrations that previously arose but do not continue. Similarly, bulk DNA sequencing (bDNAseq) CIN signatures are proposed to quantify and identify the initial cause of CIN, by inferring mechanisms that could produce an observed cell-averaged DNA copy number profile. Whether bulk sequencing measures ongoing CIN has not been tested.
Single-cell genomic measures of cell-to-cell variation in copy number analysis are less commonly used in clinical samples but have an advantage over bulk methods as they are not averaged across cells, allowing for direct evaluation of cell-to-cell variation (27). Low-coverage single-cell DNA sequencing (scDNAseq) can determine numerical copy numbers of all chromosomes in single cells in experimental models and patient tumors (32, 33). Like FISH, this information can be used to measure CIN by quantifying cell-to-cell heterogeneity in genomics-inferred karyotypes. scDNAseq can be further improved by accounting for cellular selection against highly aneuploid cells using computational modeling and approximate Bayesian computation (ABC) (27).
We sought to understand which of these measures, under identical well-controlled circumstances, most readily detect ongoing CIN. Toward this, we directly compared these measures of CIN within biological replicates of well-defined inducible cell-based models. The results reveal the pre-eminence of single-cell measures (microscopic analysis of mitosis, cytogenetic methods, scDNAseq) in measuring ongoing CIN and the inability of bulk molecular sequencing (transcriptional profiles and bulk DNA sequencing) to detect CIN. Among single-cell measures, those that survey all chromosomes across hundreds of cells are most sensitive. The data collected here are made available to investigators who seek to accurately quantify CIN and are an important touchstone to establish the directness of specific measures of ongoing CIN. We anticipate that this comprehensive comparison will serve as a foundation to identify accurate CIN measures for use in clinical samples. While additional work in tumors is required to validate clinical measures of CIN, this work reveals which measures will advance mechanistic insight, reveal clinical significance, and allow use of CIN as a predictive biomarker, such as for microtubule-targeted therapies (17).
Results
Design and Validation of CIN Models with Distinct Mechanisms.
We developed four phenotypic models of CIN induced by distinct mechanisms (Fig. 1A): bridging chromosomes (Br), pseudobipolar spindles (Pb), multipolar spindles (Mp), and polar chromosomes (Po). Bridging chromosomes are modeled in CAL51 cells via tetracycline-inducible expression of a dominant negative mutant of telomeric repeat binding factor (TERF2-DN-tetOn) lacking both its basic domain and Myb-binding box (34). Pseudobipolar, multipolar spindles, and polar chromosomes are modeled in MCF10A cells with inducible expression of polo-like kinase 4 (PLK4-WT-tetOn). Pseudobipolar spindles develop from multipolar spindles whose centrosomes have clustered together to form an apparent bipolar mitotic spindle, a process that is partially dependent on the kinesin-like protein HSET. We model these by inducing centrosome amplification via tetracycline-induction of PLK4 which, in MCF10A cells often cluster their centrosomes prior to anaphase. We model multipolar spindles similarly, only we challenge the centrosome amplified MCF10A cells with an inhibitor of HSET to prevent the clustering of centrosomes observed in the pseudobipolar model. Last, we model polar chromosomes via a sequential chemical treatment strategy in the same MCF10A cell line (albeit without PLK4 overexpression). Here, cells are partially synchronized in prometaphase then released into CENP-E inhibitor so they incompletely align their chromosomes to the metaphase plate. We then bypass the spindle assembly checkpoint using MPS1 inhibitor, forcing the cells into anaphase. This sequential chemical treatment induces anaphase onset with multiple polar chromosomes with high penetrance (35). After induction of each CIN model, we performed imaging (fixed immunofluorescence and 4+ h time lapse fluorescence microscopy), cytogenetic (mitotic chromosome counts and cenFISH), and sequence-based assays (bRNAseq, bDNAseq, and scDNAseq) to measure CIN.
Fig. 1.
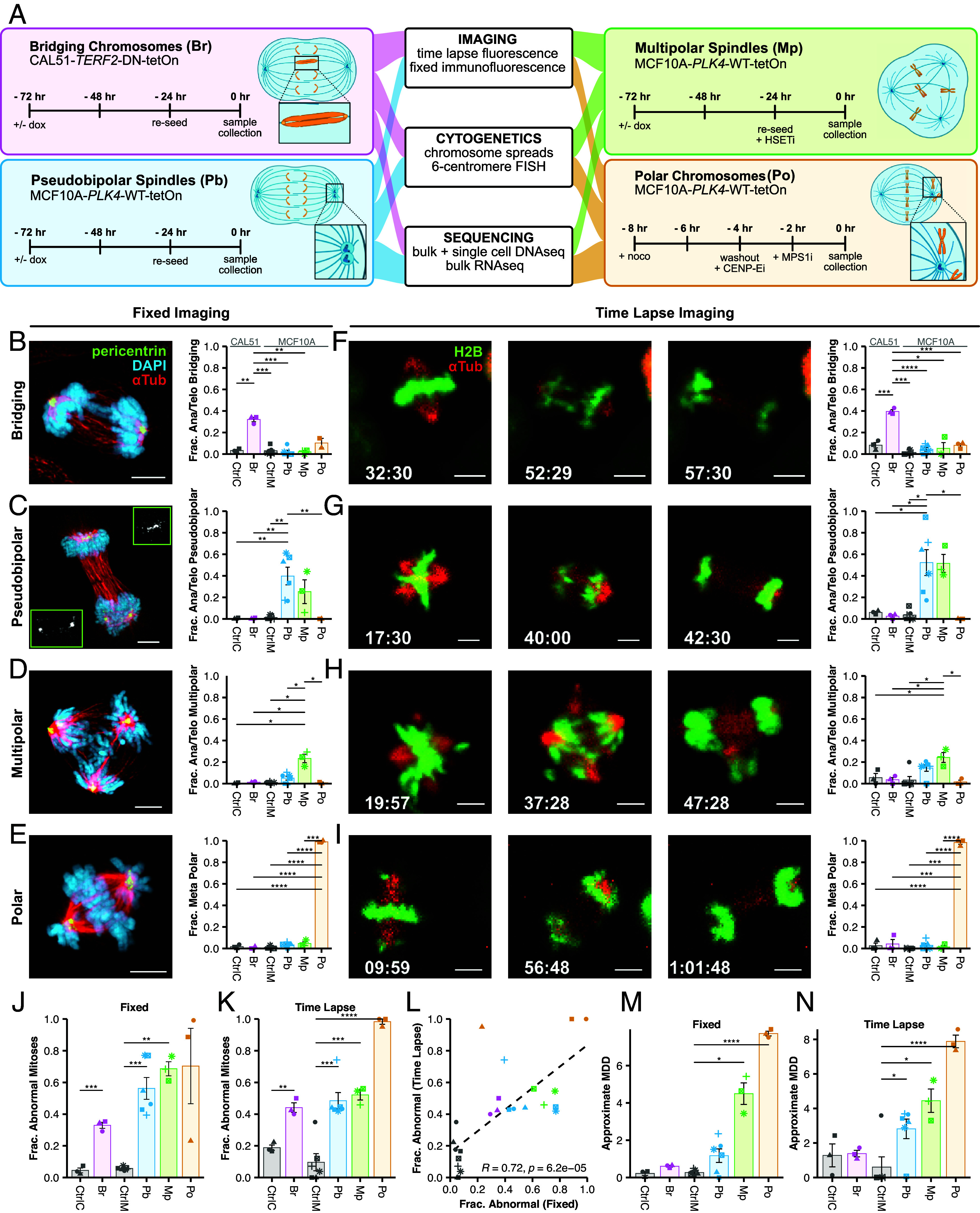
Inducible CIN models validated by imaging. (A) Inducible models of CIN phenotypes. Bridging chromosomes (Br), Pseudobipolar (Pb), Multipolar spindles (Mp), and Polar chromosomes (Po). Br, Pb, and Mp were treated with doxycycline for 72 h before harvest. Po cells used for mitotic chromosome spreads and centromeric cenFISH were washed out at T-0 and harvested 24 h later after a 2 h 50 ng/mL colcemid treatment. (B–E) Representative images of specific CIN phenotypes and observed incidence by fixed immunofluorescence or (F–I) time lapse fluorescence imaging. (J) Fraction of abnormal mitoses by fixed immunofluorescence or (K) time lapse fluorescence imaging. P < 0.05 (*), <0.01 (**), <0.001 (***), and <0.0001 (****). (L) Correlation between fixed immunofluorescence and time lapse imaging with Pearson correlation coefficient; α = 0.01. (M) MDD estimates from fixed imaging. (N) MDD estimates from time lapse. MDD values were approximated as the sum of phenotype specific MDD values for each condition (SI Appendix, Table S2). Colors represent the biological replicate; bars and error bars indicate mean and SE. Significance values are from a two-tailed, two-sample Student’s t tests corrected by the Benjamini–Hochberg method. For time lapse imaging, N ≥ 20 cells for each of ≥3 biological replicates, except single replicates of CtrlC and Pb. For fixed imaging, N ≥ 50 metaphase and ≥anaphase/telophase cells for each of ≥3 biological replicates, except single replicates of CtrlM, Pb.
Visualization of Mitosis Sensitively Detects CIN.
We first verified that our models induced CIN by microscopy. Using fixed and time lapse imaging, we found the expected CIN phenotypes for each model, indicating successful induction (Fig. 1 B–N). As expected, the Br model induced anaphase bridges in ~30 to 40% cells after doxycycline, visible on fixed and live analysis (Fig. 1 B and F). For the Pb model, we often observed multipolar spindles that focus to pseudobipolar spindles by anaphase onset such that a single spindle pole often has two pericentrin foci (Fig. 1C). The Pb model displayed pseudobipolar anaphases in 40 to 50% of cells (Fig. 1 C and G), though about 5 to 15% of cells exhibited multipolar anaphase (Fig. 1 D and H). The Mp model induced ~25% multipolar anaphases after doxycycline + CW-069. The Po model induced high penetrance of CIN with polar chromosomes found in virtually all cells (Fig. 1 E and I).
To evaluate measures of CIN, we quantified the total mitotic aberrations by microscopy using fixed and time lapse imaging (Fig. 1 J and K). As expected, all had statistically elevated CIN over controls and the two methods were significantly correlated (R = 0.72, P < 0.001) (Fig. 1L). To directly compare the relative levels of CIN imparted by each model, we quantified a standardized measure of the fundamental feature of CIN, the mis-segregation rate.
While direct observation of rate (or penetrance) of abnormal mitosis indicates CIN, it does not necessarily equate to the mis-segregation rate. Mis-segregation rate is also a function of the number of chromosomes involved in erroneous segregation (which we call magnitude), the rate at which those chromosomes actually segregate to the correct cell (which we call resolution), and the total number of chromosomes available to be segregated (ploidy). We previously described a metric, Mis-segregations per Diploid Division (MDD), which accounts for these factors (36). Thus, MDD measures the severity of CIN in terms of mis-segregation rate by approximating the total number of chromosomes that are likely to be mis-segregated in a given division. This is then normalized to the background ploidy of the sample to ensure the metric scales by the severity of CIN rather than extent of karyotype heterogeneity produced, which is a function of ploidy and change depending on the study (Materials and Methods).
To approximate MDD from mitotic observations, we assume that apparent bridging chromosomes produce 1 mis-segregation (1 chromosome at 0% resolution as chromosomes must be broken) and lagging chromosomes produce <1 mis-segregation (1 chromosome at 90% resolution). We estimate that cells with polar chromosomes, in our model, produce 7.8 mis-segregations (7.8 chromosomes at 0% resolution) based on quantitative immunofluorescence imaging of polar centromere foci (SI Appendix, Fig. S1A). Based on previously published scDNAseq data (27), we know cells dividing on multipolar spindles produce an average of 18 mis-segregations (18 chromosomes at 0% resolution). Last, while hidden merotelic attachments may occur within the main chromosome masses of pseudobipolar spindles, the rate at which this occurs is unknown. For the purposes of this approximation, we assume pseudobipolar spindles do not produce chromosome mis-segregation in and of themselves (0 chromosomes at 0% resolution) (SI Appendix, Table S1 and Materials and Methods). By this approximation, the mis-segregation rates (MDD) observed for each model are 0.22 ± 0.1 for CtrlC, 0.60 ± 0.04 for Br, 0.27 ± 0.06 for CtrlM, 1.17 ± 0.36 for Pb, 4.49 ± 0.58 for Mp, and 7.72 ± 0.13 for Po by fixed imaging (Fig. 1M). By time lapse imaging, these approximations are 1.28 ± 0.66 for CtrlC, 1.39 ± 0.19 for Br, 0.6 ± 0.6 for CtrlM, 2.82 ± 0.56 for Pb, 4.45 ± 0.67 for Mp, and 7.88 ± 0.36 for Po (Fig. 1N). We also performed a sensitivity analysis to understand how the assumed magnitude of CIN, the number of chromosomes involved, in each model affected the overall approximation (SI Appendix, Fig. S2). As expected, MDD were most sensitive to changes in the assumed magnitude for each phenotype. For example, changing the assumption of the number aberrant chromosomes associated with multipolar spindles more drastically effects approximated MDD in the Mp model than for other mitotic defects. Importantly, to infer the MDD from microscopy, assumptions are made about resolution of mis-segregation based on data and previous studies. In summary, these findings validate our CIN models and demonstrate low levels of CIN with Br and Pb, intermediate levels with Mp, and high levels with Po. These distinct models and mechanisms of CIN confirm they are suitable models to compare quantitative measures of CIN.
Short-lived CIN Phenotypes Are Underestimated in Fixed Imaging.
While the two imaging methods significantly correlated (Fig. 1L), time lapse imaging was more sensitive to certain CIN phenotypes (SI Appendix, Fig. S1B). For example, we detected more multipolar metaphases and anaphases with lagging chromosomes using time lapse imaging. These differentially detected defects are transient in nature, suggesting that the differences are not an artifact of live imaging. Together, these results indicate that measurement of CIN using fixed imaging, as is common in retrospective clinical analyses of CIN, may underestimate the incidence of some mitotic defects.
Cytogenetic Methods Are Less Sensitive to Ongoing CIN than Fluorescence Imaging.
Mitotic chromosome counts and centromeric FISH are commonly used cytogenetic approaches to measure CIN (17, 37–39). Importantly, these measures depend on the association between ongoing CIN and the resultant cell-to-cell variation in chromosome copy numbers. Chromosome counts detected variation around the modal chromosome count (46 for CAL51s and 47 for MCF10As) in all CIN models and their controls (Fig. 2A). In several models, in addition to near-diploid aneuploidy, there were small fractions of counts consistent with triploid (n = 69) or tetraploid (n = 92) cells. However, the variation observed was not statistically significant for any of our phenotypic models (Fig. 2B). The cenFISH probe counts can estimate CIN as nonmodal counts (Fig. 2 C and D). In some cases, CIN measures were similar to controls, likely reflecting the difficulty of measuring CIN by sampling only 6 chromosomes after only 1 to 2 aberrant divisions. However, nonmodal probe counts were elevated for the models with the highest CIN: twofold for Mp and threefold for Po, though only the former was statistically significant (Fig. 2D). Recent data suggest a bias for mis-segregation of larger chromosomes, though this can vary based on the insult that causes chromosome missegregation (21, 22, 40). If there is a bias, it may be important to survey more than a few chromosomes.
Fig. 2.
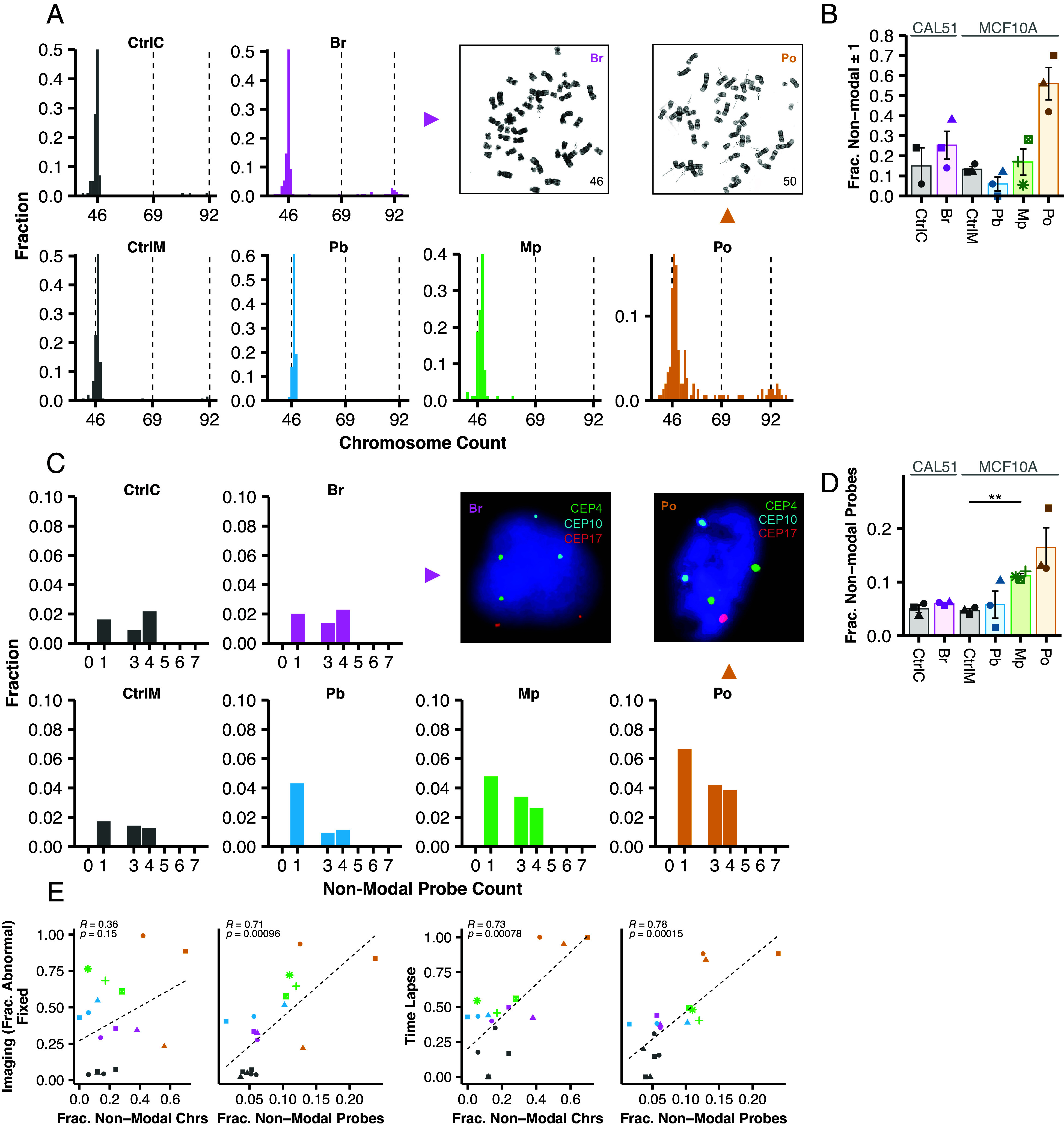
Cytogenetic methods are less sensitive to ongoing CIN than fluorescence imaging. (A) Representative mitotic spreads and histograms. (B) Fraction of mitotic spreads where chromosome counts differ from the mode ±1. N ≥ 50 mitotic spreads for each of three biological replicates, except CtrlC with two biological replicates, and Mp. (C) Representative cenFISH images from each phenotypic CIN model with probe count histograms. Diploid counts are excluded from histograms. (D) Fraction of cells whose cenFISH probe counts differ from the mode. Shapes of individual points indicate the biological replicate. (E) Correlations between each cytogenetic method and fixed immunofluorescence or time lapse imaging with Pearson correlation coefficients. For correlations, α = 0.01. N ≥ 200 cells for each of three biological replicates. Bars and error bars indicate mean and SE. Symbolic significance thresholds are 0.05 (*) and 0.01 (**).
Although these methods did not detect statistically significant copy number variation (CNV), they largely correlate with microscopic detection of mitotic defects (R > 0.71, P < 0.001) (Fig. 2E). One exception is that nonmodal mitotic chromosome counts did not correlate with fixed imaging but did with time lapse (R = 0.36, P = 0.15). Together, these data suggest that cytogenetics methods are less sensitive than inspection of mitosis at detecting ongoing CIN.
Bulk Transcriptomic and Genomic CIN Signatures Do Not Reflect Ongoing CIN.
Transcriptional signatures of CIN such as CIN70 and HET70 are used as proxy measures to assess CIN from bulk transcriptomic data in tumor samples (41, 42). These are derived indirectly by identifying gene expression that correlates with aneuploidy in tumor samples (CIN70) (30) and karyotype heterogeneity in cell lines (HET70) (41). Neither CIN70 nor HET70, to our knowledge, has been tested in inducible models of CIN. To determine whether these directly measure induced CIN. We performed bulk RNA sequencing and measured CIN70 and HET70 signatures in our models (SI Appendix, Table S3). To validate our results, we verified that doxycycline addition caused a fourfold increase of TERF2 expression in the Br condition and a 32-fold increase in PLK4 expression in Pb and MP. In each case, these were among the top differentially expressed genes (SI Appendix, Fig. S3 A and B). Turning to CIN scores, we plotted the distribution of all 70 genes (Fig. 3 A and B) with the mean representing the score. As illustrated, neither CIN70 nor HET70 was increased in any of the CIN models. In fact, CIN70 decreased slightly in Pb and Mp models (Fig. 3A), likely due to a decrease in cell proliferation after centrosome amplification (43). Likewise, neither CIN70 nor HET70 significantly correlated with microscopic detection of mitotic defects (Fig. 3C). This suggests that these bulk transcriptional scores do not detect ongoing CIN.
Fig. 3.
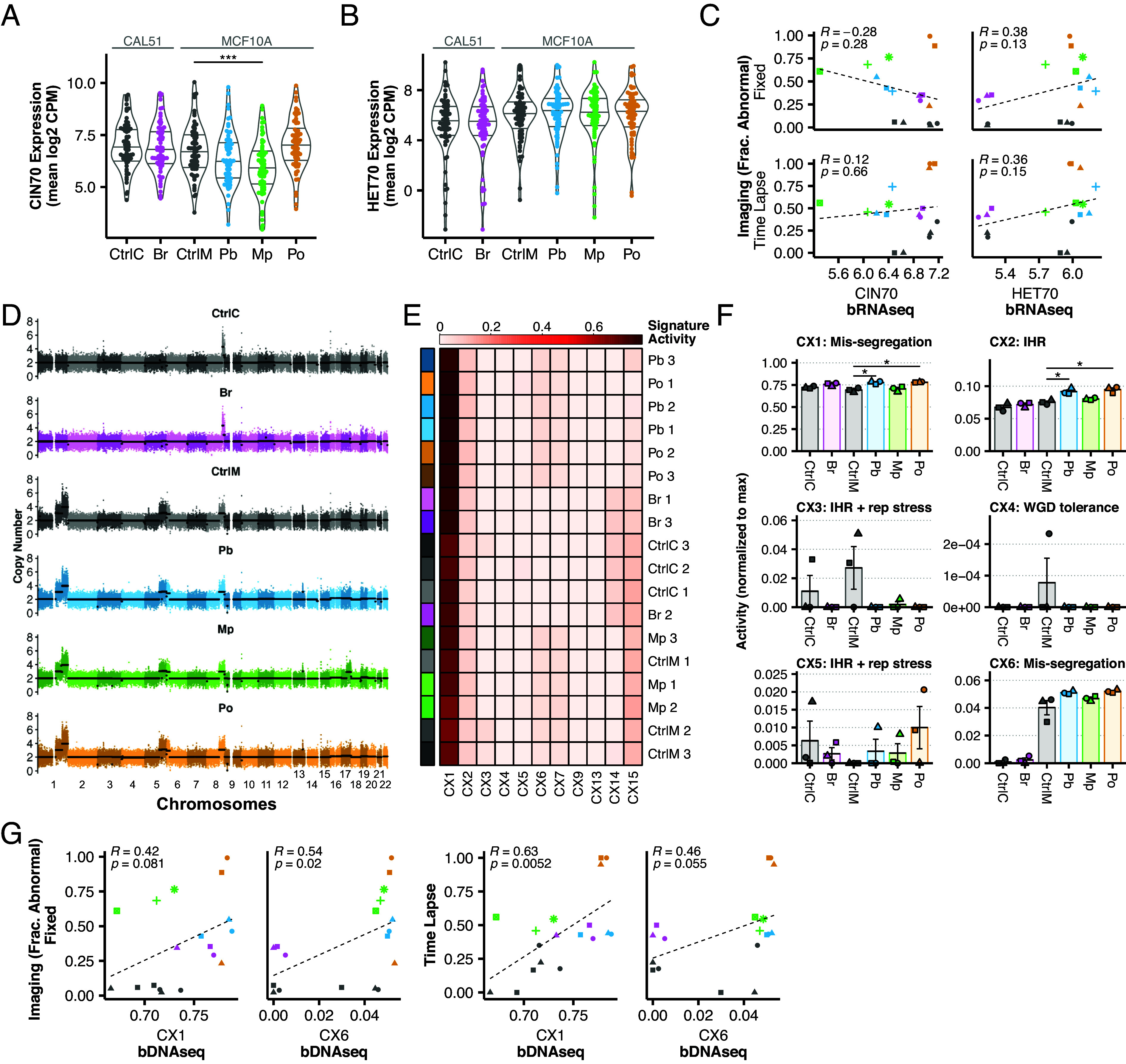
Bulk transcriptomic and genomic CIN signatures do not reflect ongoing CIN. (A) Expression [mean log2 counts per million (CPM)] of CIN70 and (B) HET70 genes from bRNAseq data. Each point indicates the average expression level across three biological replicates. (C) Correlations between bulk transcriptional scores and fixed immunofluorescence or time lapse imaging with Pearson correlation coefficients. (D) Representative whole-genome copy number profiles of 500 cells from each CIN model at ~10× depth. Points indicate 30 Kb bins with average segment copy number in black. (E) Normalized CIN signatures. Color annotations on left indicate models and shades indicate replicates. Rows are clustered by similarity signatures. (F) Normalized signature activities of all CIN signatures whose mechanistic etiologies were classified as “high confidence.” Shapes indicate the biological replicates. Mean and SE are shown. (G) Correlations between each “mis-segregation” signature and fixed immunofluorescence or time lapse imaging with Pearson correlation coefficients, with α = 0.01.
In addition to RNA, bulk genomic DNA measures of CIN are proposed to detect characteristic signatures of CNV from SNP array and genome sequencing data—essentially measuring patterns of aneuploidy (44). These measures characterize the final state of the tumor, which could either arise from an early event in oncogenesis, or through continuous CIN with selection for certain aneuploid clones. In the latter circumstance, bulk DNA would potentially measure “historical CIN.” While these signatures are not designed to measure ongoing CIN, it is worth testing that possibility as they are often described as CIN signatures. Whole-genome sequencing (~10× coverage) and copy number calling in our models revealed nearly identical copy number profiles between control and CIN-induced groups (Fig. 3D). We next analyzed recently published CIN signatures (44) in each replicate and clustered the models and controls by signature (Fig. 3E). CX1 is the predominant CIN signature in all groups (~70 to 75%) and in control cells. CX1 corresponds to large-scale copy number alterations consistent with whole chromosome or chromosome arm mis-segregation. CX1 is significantly increased in Pb and Po models than in control cells, but not Mp. CX6 did not differ between controls and induced CIN models, other than Pb, even though it similarly is reported to represent whole chromosome and chromosome arm mis-segregation and makes up no more than 6% of the CIN signature activity. CX2, CX3, and CX5 correspond to impaired homologous recombination (IHR). CX2 is elevated within the Pb and Po models, though CX3 and CX5 exhibit no statistically significant differences (Fig. 3F). Last, CX4, a putative signature of WGD tolerance, was only found in a single replicate of control MCF10As, which are not whole-genome doubled. Since our models of CIN are induced over 8 to 72 h, they may not provide sufficient time for extensive cellular selection, a process that is likely required for these signatures to appear as they are based on the averaged CNVs of the population. Nevertheless, we conclude that the DNA genomic signatures of CIN do not directly measure ongoing CIN. Accordingly, neither CX1 nor CX6 correlated with mitotic defects via fixed microscopy (P > 0.01) and only CX1 correlated with time lapse microscopy (R = 0.63, P < 0.01) (Fig. 3G). In sum, bulk genomic measures of CIN, whether transcriptomic or genomic, do not directly measure ongoing CIN, even in an ideal context where tumor purity is not at issue.
scDNAseq Detects Ongoing Numerical CIN and Enables Inference of Mis-segregation Rates.
The analyses above suggest that optimal CIN measures would i) detect all chromosomes, ii) directly detect CNV variation across cells, and iii) have high throughput. scDNAseq meets these characteristics. Even low coverage of reads across the genomes are sufficient to infer copy numbers across all chromosomes. Thus, scDNAseq has been employed to measure cell–cell variation and to infer CIN (27, 45). We therefore evaluated scDNAseq as a measure of CIN.
To evaluate scDNAseq, we sampled 32 single cells per replicate and included a bulk sample of 500 cells to infer average karyotype (SI Appendix, Fig. S4). We filtered for quality and inferred large-scale chromosome copy number alterations at 1Mb resolution, resulting in 378 high-quality single-cell copy number profiles (Fig. 4A). Bulk analyses reveal CAL51 as diploid with a focal 8q amplification; MCF10A cells also had 8q plus gains of 1q, 5q, and recurring subclonal gains of Xq. To quantify chromosomal deviations, we evaluated the absolute difference between each single-cell karyotype and the modal karyotype inferred from bulk samples. Both controls—CAL51 and MCF10A—had a small number of deviations from modal karyotype (Fig. 4B). We did not detect a clear increase in chromosomal deviations in the Br model. This could be due to resolution of chromatin bridges, chromosome breakages resulting in structural variation, but conserved copy number, which would be undetectable by low coverage scDNAseq, or segmental copy number changes smaller than 50% of the chromosome. However, we ruled out the last possibility using an alternative analysis which failed to detect telomere proximal breaks (SI Appendix, Fig. S5). We found an increase in nonmodal chromosomes with the Pb model and significant increases in the Mp and Po models, which average about five to six deviations per cell respectively (Fig. 4B). The percentage of cells with deviations from the mode followed a similar trend, though also detects a small increase in the Br model that is not statistically significance (Fig. 4C). As expected, the Mp model displayed a significant bias of chromosome losses over gains (Fig. 4D)—this is expected since division of duplicated chromosomes into 3+ daughter cells reduces chromosome number. Other models showed no such bias with gains/losses being roughly equal. Taken as a whole, these data support scDNAseq as a sensitive method for detecting CIN.
Fig. 4.
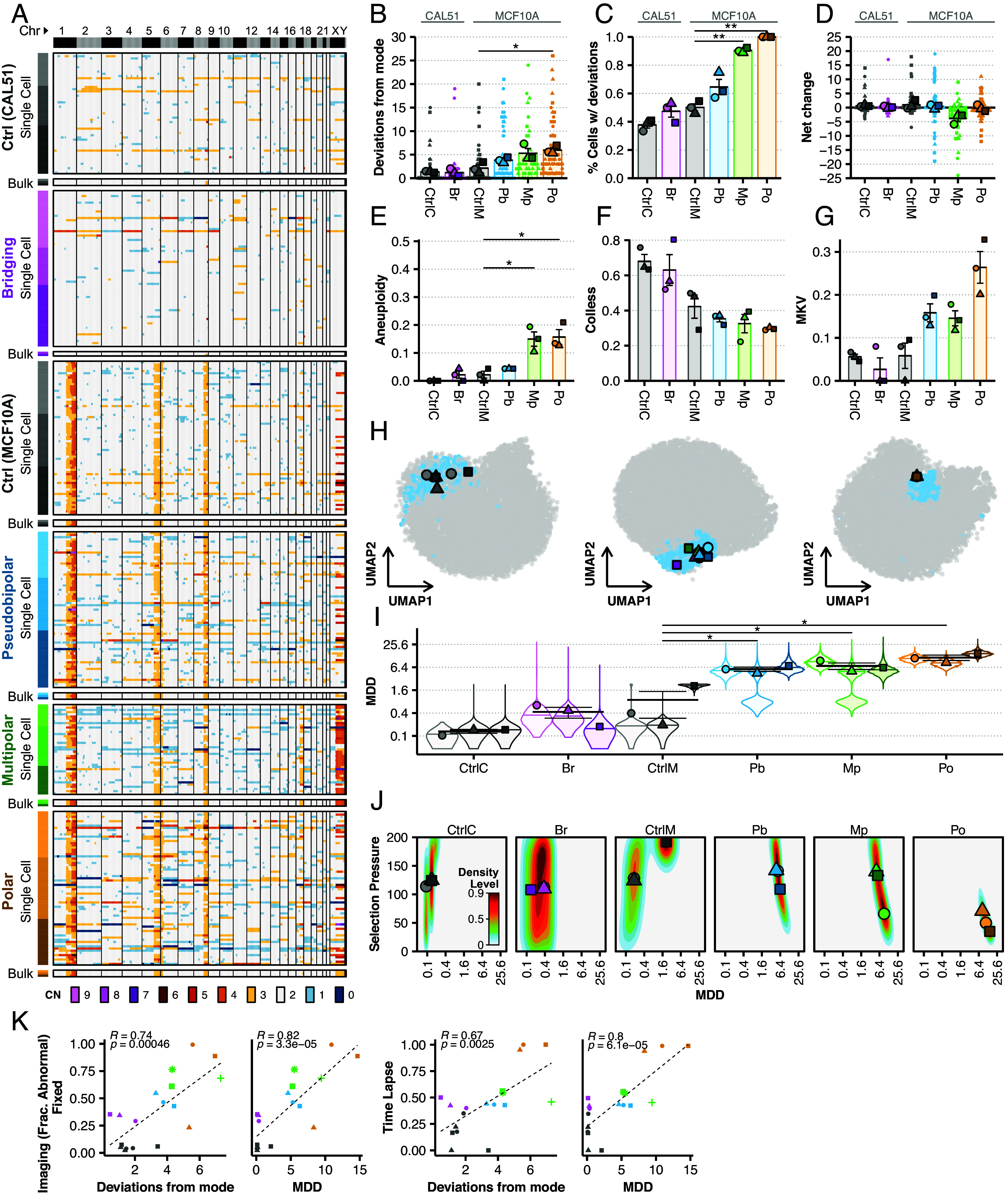
scDNAseq detects CIN and enables inference of mis-segregation rates. (A) Heatmaps of unsorted single cell and bulk (500 cells) copy number profiles from scDNAseq with copy number (CN) indicated by color. Color annotations on left of heatmap indicate models and shades indicate replicates. Vertical lines separate chromosomes. Whole chromosome CNs are used to quantify, for each phenotypic model, (B) the absolute number of whole chromosome deviations from its modal karyotype, (C) the percentage of cells with whole chromosome deviations, and (D) the net change of total chromosomes in each cell. (E) Aneuploidy (the sub-clonal mean variance within karyotypes), (F) Colless indices (i.e., phylogenetic imbalance), and (G) mean karyotype variance (MKV, the mean variance of each chromosome) were used as summary statistics for ABC. (H) Space of summary statistics of independent prior simulation datasets used for ABC projected (small data points) with summary statistics measured in each phenotypic CIN model (large data points) using uniform manifold approximation and projection. Blue points indicate accepted simulations for at least one model and replicate, whereas gray were rejected. (I) Posterior distributions (violin plots) of mis-segregation rates (MDD) across all replicates for each phenotypic CIN model (individual points) inferred using ABC. A log2 scale is used to better illustrate the data. (J) Joint posterior density distributions of accepted mis-segregation rates and selective pressure from ABC for all replicates of each phenotypic CIN model. Data points are mean values for each replicate. Parameter values for prior simulation datasets are as follows: CtrlC and CtrlM—MDD = [0 … 2.3], S = [0 … 200], Time Steps = [30 … 50]; Br, Pb, and Mp — MDD = [0 … 46], S = [0 … 200], Time Steps = [0 … 4]; Po—MDD = [0 … 46], S = [0 … 200], Time Steps = [0 … 2]. (K) Correlations between the whole-chromosome deviations or inferred MDD and fixed immunofluorescence or fluorescence time lapse imaging with Pearson correlation coefficients, with α = 0.01. Shapes of individual points indicate the biological replicate. Bars and error bars indicate mean and SE.
Single-cell resolution may enable detection of CIN signatures (44), so we repeated our previous analysis using the single-cell copy number profiles. Again, the proposed whole/arm mis-segregation signature, CX1, was predominant among the groups (SI Appendix, Fig. S6). However, the relative activity of this signature between groups did not correspond to CIN as observed by microscopy or by directly measuring whole-chromosome copy number alterations. CX6, the other proposed whole/arm mis-segregation signature, had much lower activity among groups and did not correspond to observed CIN. Interestingly, despite their low activity, the only signatures that seemed to reflect the observed trend were decreasing CX4 and increasing CX8, proposed signatures of whole-genome doubling and replication stress, respectively. Thus, these measures of CNV at single-cell resolution do not well characterize ongoing CIN.
Because the fitness levels imparted by different karyotypes can be acted on over time by natural selection, the absolute number of chromosomal deviations in a population may not capture CIN in its entirety. We recently addressed this issue and developed a computational framework to infer mis-segregation rates from scDNAseq datasets using ABC, a method to statistically relate biological and simulated data (27). In short, this approach uses a large prior dataset of evolutionary trajectories simulated under varying rates of chromosome mis-segregation and selection pressures (27). The simulated prior dataset consists of a series of population measurements taken over time: aneuploidy (average sub-clonal variance within karyotypes), karyotype heterogeneity (MKV; average variance within chromosomes across the population), and the Colless index, a measure of asymmetry in the shape of the reconstructed copy-number-based phylogeny, indicative of ongoing selection (46, 47). We repeat these measures in our experimental data and perform parameter inference with ABC to find simulation parameters that produce similar populations.
As expected, our CIN models increased the median aneuploidy and MKV in the populations, though only the aneuploidy observed in Mp and Po was statistically significant (Fig. 4 E and G). Further, Colless index decreased indicating a low level of selection after CIN induction (Fig. 4F). This likely reflects the lack of selection over the short-time of the experiment (8 to 72 h); by contrast, the control population has been under long-term selection and has higher Colless. Together, the summary statistics enable inference of mis-segregation rates by ABC.
We next applied ABC to infer chromosome mis-segregation rates. Dimensionality reduction verified that the biological data falls within the “summary space” of the simulated data (Fig. 4H). Inferring mis-segregation rates (taken as the average of the posterior rate distributions) revealed a wide range across models from 0.3 to 11.5 MDD (36, 48). There was only a twofold increase in mis-segregation rate in the Br model of bridging chromosomes: CtrlC and Br showed mis-segregation rates of 0.12 ± 0.02 and 0.28 ± 0.07 MDD, respectively. CtrlM cells had inferred mis-segregation rates at 0.83 ± 0.64 MDD while the Pb, Mp, and Po models had much higher mis-segregation rates of 5.4 ± 0.49, 6.73 ± 1.35, and 11.3 ± 1.84 MDD, respectively (Fig. 4I). In comparison, the approximated mis-segregation rates of in RPE1 cells is 0.01 to 0.05 MDD and for U2OS, 0.33 to 0.46 MDD (36). These rates correlate with the number of deviations measured directly from whole chromosome copy number data, particularly when accounting for the partial induction of CIN over 8 h with the Po model (Fig. 4B and SI Appendix, Fig. S7). They also correlate with MDD values approximated for each model by imaging methods (SI Appendix, Fig. S7). Joint posterior distributions reflect lower apparent evidence of ongoing selection in the Po model, as compared to the other models, in concordance with the short time-span of CIN induction (Fig. 4J). The mis-segregation rate observed in the Mp model, when taking into account the penetrance of the phenotype (~25% penetrance via imaging), is similar to previously observed mis-segregation rates caused by multipolar divisions (~18 MDD at ~100% penetrance) (27). Overall, both the inferred MDD and the number of chromosomal deviations significantly correlate the fraction of cells with mitotic defects at imaging (R > 0.67, P < 0.01) (Fig. 4K).
We conclude that CIN can be measured several ways using single cell copy number profiles. Copy number alterations can be calculated directly, particularly if the period of CIN is relatively short, in which case karyotype selection is not a significant factor. Inferring chromosome mis-segregation rates by comparing to simulated data works in both short and long time periods, though it is ideal for long-time periods when karyotype selection becomes a strong confounder.
Concordance and Performance of CIN Measures.
To assess and summarize the performance of CIN measures, we performed standardized effect size and pairwise correlation analyses. As expected, fixed immunofluorescence, time lapse, nonmodal mitotic chromosome, and centromeric probe counts were significantly correlated (α = 0.01), excepting the pair of fixed imaging and chromosome counts (Fig. 5A and SI Appendix, Fig. S7). By contrast, bulk transcriptional CIN signatures (CIN70, HET70) did not correlate to imaging, nor cytogenetics, nor between themselves. Considering genomic signatures on bulk (CX1, CX6 bDNAseq), and with single-cell data (CX1 scDNAseq; CX6 scDNAseq), these also correlated poorly with one another with the exception of single-cell CX1 and CX6. Interestingly, the CIN signature CX1, when measured in bDNAseq data, did significantly correlate with time-lapse imaging, but not fixed imaging. HET70 negatively correlated with the CX1 and CX6 signatures in single cells. This seemed to be a cell line–dependent effect as MCF10A cells had relatively high HET70 expression and low CX1 and CX6 signature activity than CAL51, regardless of CIN model (SI Appendix, Figs. S6 and S7). scDNAseq chromosomal analyses, regardless of whether they considered whole-chromosome deviations from mode or inferred MDD from ABC, correlate with each other as well as fixed imaging, time lapse, and FISH analyses. We conclude that the single-cell analyses perform well for measuring rates of ongoing CIN.
Fig. 5.
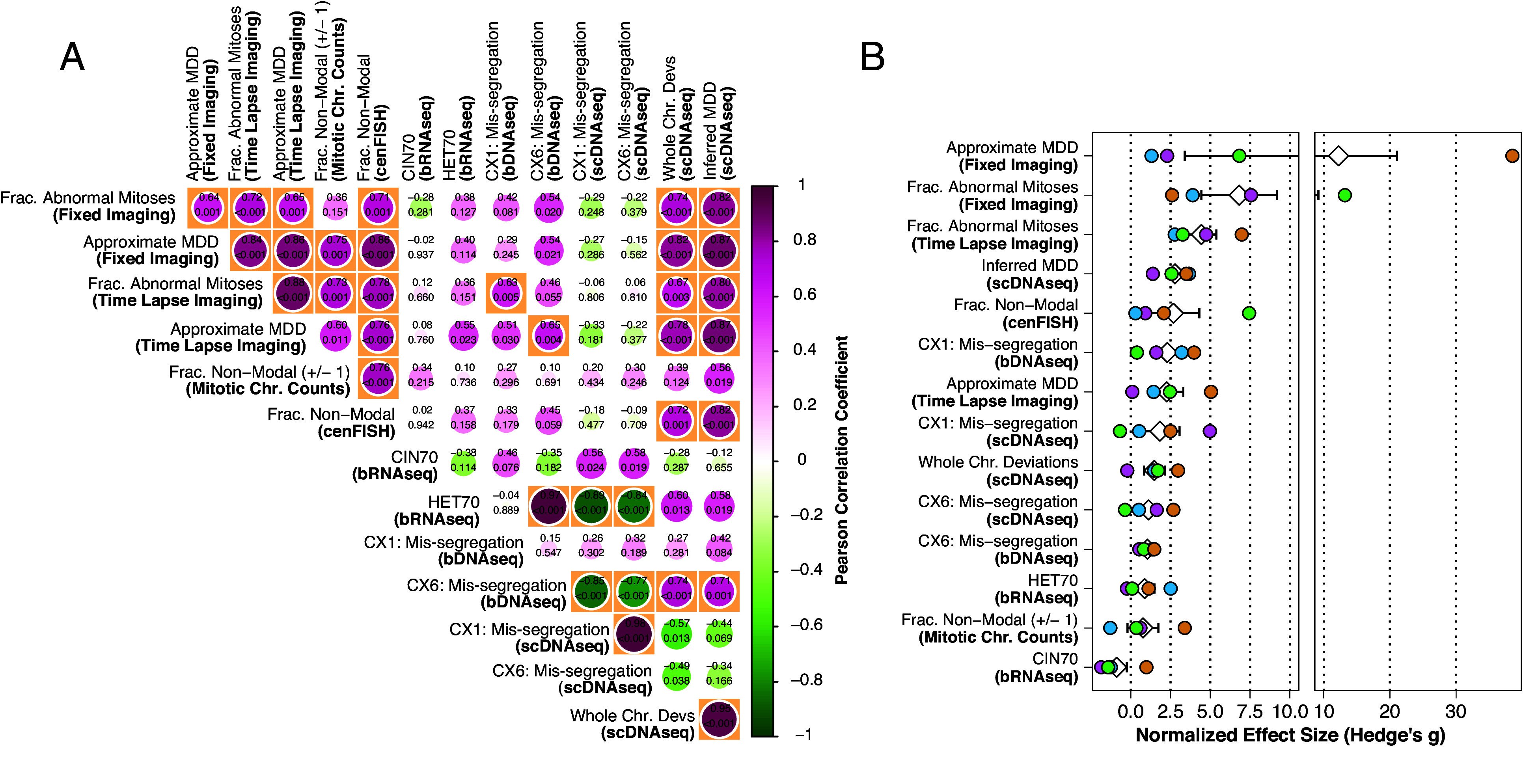
Concordance and performance of CIN measures. (A) Pairwise Pearson correlation coefficients of CIN measurements. Significant correlations (α = 0.01) are highlighted in orange boxes. Size of circles represents degree of correlation. (B) Standardized effect sizes (Hedge’s g) of each CIN measurement for each model. Effect sizes are calculated from comparison of each phenotypic model with its respective control. White diamond and error bars indicate mean and SE.
We next compared the sensitivity of different measures. Standardized effect sizes are analogous to the signal-to-noise ratio. For instance, a Hedge’s g statistic of two represents an effect twice that of the pooled SD for a given comparison. Thus, we looked at the standardized effect sizes produced using each measure in each phenotypic model compared to its control. Average effect sizes across all CIN phenotypes were high among the imaging methods (Fig. 5B). Inferred MDD from scDNAseq was high, as expected from sampling chromosomes and accounting for cell selection. Next, CX1 measured by bDNAseq was also high though, as noted previously, these scores do not reflect observed CIN levels by imaging or single-cell sequencing. The effect size for cenFISH was similar to whole chromosome deviations in scDNAseq. The CIN signature of mis-segregation (CX6), as measured by bulk or single cell DNAseq was middling. Transcriptional measures of CIN (CIN70 and HET70) and CIN measured by mitotic chromosome counts exhibited the lowest effect sizes. We conclude that highly sensitive quantitative measures of CIN can be achieved by direct microscopy or by scDNAseq combined with inference of rates by ABC.
Because the primary purpose of this study is to identify which CIN measures are most sensitive under equivalent conditions, there may be cases where a particular measure, while often used, lacked the power to achieve a statistically significant result under our strategy. We performed a power sensitivity analysis to identify these borderline cases (SI Appendix, Fig. S8). Measures which significantly correlated (α = 0.01) with the fraction of mitotic defects apparent overall in both imaging techniques but weren’t themselves sensitive enough to independently detect CIN in some phenotypic models include cenFISH and scDNAseq. scDNAseq failed to significantly detect elevated CIN (fraction nonmodal chromosomes or inferred MDD) in the Br model while cenFISH failed to detect elevated CIN (fraction nonmodal FISH probes) in the Br, Pb, and Po models.
Given effect sizes observed in the Br model with scDNAseq methods (fraction nonmodal chromosomes or inferred MDD), power levels less than 0.5 would be required to deem the effects statistically significant at α = 0.05. In other words, the likelihood of detecting a real significant event would be less than 50%. It is unlikely that additional replicates would have aided this measure. Rather, scDNAseq, used in this way, cannot detect CIN in this model. A potential cause is that bridging chromosomes produce segmental alteration too small for our approach which calculates the modal copy number of the whole chromosome, though our previous breakpoint analysis would refute this explanation (SI Appendix, Fig. S5). Another explanation is the complete resolution of chromosome bridges, without breakage, which would be undetectable by scDNAseq. Though chromosome bridge breakage resulting in large segmental alterations in the very same daughter cells were previously observed in this model using Look-Seq, a more discrete scDNAseq approach (49). Nevertheless, it is clear that scDNAseq performed here was insufficient to detect this CIN phenotype.
The same applies in the Br and Pb models as detected by cenFISH. The effect sizes for both are below the critical effect size for a significant result at a power of 0.2. This is not surprising for the Br model as the likelihood that a chromosome breakage would include a centromere that is probed, particularly given the lack of apparent effect in the scDNAseq data, is small. Although we did observe a significant effect with scDNAseq for the Pb model, this could be due to the relatively low resolution of cenFISH. It is likely that, with an additional biological replicate or a better signal-noise ratio, elevated CIN in the Po model could have been significantly detected with cenFISH. As it stands, the effect size observed in the Po model using cenFISH could be considered significant at a power level of ~0.6.
Here, we employ MDD as a standardized and quantitative measure of CIN. Two systems with the same MDD have the same per chromosome rate of mis-segregation, regardless of their chromosome content. Whereas MDD is a quantitative measure of CIN, it is not comprehensive. It does not account for segmental chromosome aberrations, systematic or correlated chromosome gains or losses. It does not address the rate of micronuclei generation, or downstream effects. Different mechanisms of CIN could generate the same MDD in some cases. Nevertheless, MDD is a single quantitative measure of whole-chromosome genomic instability that can be standardized and compared across studies.
Discussion
Ongoing CIN is defined by an elevated rate of chromosome mis-segregation, which varies across tumors and depends on the penetrance and magnitude of specific CIN mechanisms. The intrinsic rate of mis-segregation in a cell population produces functional consequences for tumorigenesis, cancer progression, and treatment response. Very low mis-segregation rates limit tumorigenesis, presumably due to reduced adaptive potential, while high mis-segregation rates have the same effect through loss of necessary genetic material. Intermediate mis-segregation rates can promote tumorigenesis. Once a tumor has formed, reports conflict on whether patients whose tumors exhibit moderate to high intrinsic rates of mis-segregation tend to have poorer clinical outcomes (8, 10, 14–20), likely due to limitations of current methods for measuring CIN in patient tumors. Breast cancer patients whose tumors exhibit high mis-segregation tend to have improved response to taxanes, likely because CIN sensitizes to the multipolar mitotic spindles produced by these drugs. Despite the clinical significance of CIN and its variability across tumors, its measurement is not currently used to guide patient care (8, 12, 13, 17, 50).
The reason CIN is not measured for clinical use is ultimately due to three factors: accessibility, scalability, and sensitivity. In reviewing the CIN measures tested in this study, we find varying levels of accessibility and scalability (Table 1). Some measures are clinically accessible in terms of necessary equipment and reagents, but they do not easily scale because of time required for sample preparation or data acquisition. For example, fixed imaging is commonplace for pathological assessment of tumors. In this study, fixed immunofluorescence imaging was among the most sensitive methods to detect ongoing CIN. However, the quantification of CIN phenotypes requires substantial time and a sufficient number of mitoses from highly proliferative tumors (8). Karyotypes and chromosome counts require a large number of mitotic cells, not easily obtained by culturing tumor tissues. Likewise, time lapse imaging is not possible directly in patient tumors and is laborious, time-consuming, and expensive even with ex vivo culture, fluorescent labeling, and imaging of patient organoids. Direct observation of mitotic defects, considered the gold standard measure of CIN, is indicative of but does not necessarily equate to the occurrence of mis-segregations.
Table 1.
Characteristics of CIN measurement methods
| Method | Accessibility | Scalability | Sensitivity | Imaging concordance |
|---|---|---|---|---|
| Fixed imaging | +++ | No | (4/4) bridges/pseudobipolar/multipolar/polar | – |
| Time lapse imaging | – | No | (4/4) bridges/pseudobipolar/multipolar/polar | – |
| Chromosome spread | – | No | (0/4) | No |
| 6-centromere FISH | +++ | Yes | (1/4) multipolar | Yes |
| Bulk RNA seq | ++ | Yes | (0/4) | No |
| Bulk DNA seq | +++ | Yes | (0/4) | No |
| scDNAseq | + | Yes | (3/4) pseudobipolar/multipolar/polar | Yes |
Categories for clinical accessibility are (–) insurmountable barriers, (+) requires special equipment and/or reagents and time-consuming and/or laborious, (++) requires special equipment and/or reagents or time-consuming and/or laborious, or (+++) commonly performed clinical assays use the same equipment and reagents. Scalability is determined by the pre-existence of established protocols and/or platforms for high throughput sample preparation and data acquisition in a clinical or nonclinical setting. Sensitivity to a specific mechanism is determined by a significant difference between the control and CIN-induced groups. Imaging concordance is determined by significant correlation to both imaging methods.
Measures of CIN based on bulk genomic and transcriptomic sequencing are attractive due to the wide availability of shared high-throughput sequencing data and because they are already in routine clinical use. However, bulk sequencing failed to detect ongoing CIN in any of our phenotypic models. This is likely because bulk sequencing measures CNVs and RNA levels averaged across many cells without the ability to detect cell-to-cell differences. One challenge in the field of genomic integrity is that CIN and aneuploidy are often conflated. To be fair, aneuploidy is a product of prior mis-segregation events, providing a rationale to infer a degree of CIN from the current degree of aneuploidy in a tumor. This is also described as historical CIN, which represents an accumulation of prior chromosome gains/losses and cellular selection. However, this does not permit inference of ongoing chromosome gains or losses because any degree of aneuploidy could, in fact, be a product of a single event that was not repeated. By contrast, ongoing CIN, described here, is a process in which there are ongoing aberrations over multiple cell divisions, which is associated with metastasis and response to microtubule-targeted therapies (8, 12, 13, 17, 50).
The de novo karyotype heterogeneity in cell populations with ongoing CIN does not significantly alter the predominant karyotype and is not detected by bulk genomic CIN signatures (e.g., CX1 and CX6). This demonstrates that these signatures detect historical aneuploidy rather than ongoing CIN. Nor do the gene expression states of new aneuploid clones significantly alter the predominant transcriptional phenotype with respect to previously established transcriptomic CIN scores (CIN70 and HET70). MCF10A cells scored slightly higher on HET70 than CAL51. While this may represent cell line–specific gene expression, we note that the scDNAseq data show MCF10A cells exhibiting higher karyotype heterogeneity than CAL51 at baseline, despite exhibiting similar rates of mitotic errors during imaging. Thus, while HET70 scores did not respond to induced CIN and karyotype heterogeneity, we cannot rule out that HET70 detects a pre-existing transcriptional phenotype that is tolerant of aneuploidy and increased karyotype heterogeneity. However, while HET70 did not detect induced, ongoing CIN in any of our models, MCF10A cells did score higher on HET70 than CAL51. While this may represent cell line specific gene expression, the scDNAseq data show MCF10A cells exhibiting higher karyotype heterogeneity than CAL51 at baseline. Although HET70 scores did not increase with induced CIN and karyotype heterogeneity HET70 may detect a transcriptional phenotype that correlates with aneuploidy tolerance.
As a clinically accessible alternative to imaging, scDNAseq provides the best measure of CIN in terms of sensitivity and correlation to both fixed and time lapse imaging. CIN can be measured by quantifying the absolute number of chromosomal deviations from the modal (i.e., clonal) karyotype of a population. Additionally, the rate of mis-segregation resulting in a given population of single cell copy number profiles can be measured by pairing computational modeling and statistical inference. Both analysis methods performed well in measures of ongoing induced CIN over relatively few cell divisions. However, we have previously found that the latter, inference of CIN, performs better in the context of longer time scales, such as the growth of a tumor (27). While single-cell sequencing is not currently used in clinical care, major advances in ultra-high-throughput sequencing will likely make low-coverage scDNAseq accessible at clinical capacity (51–53). Toward this end, we estimated that ~200 cells are needed for accurate measurement of CIN (27). Although we did not detect increased segmental copy number alterations caused by bridging chromosomes in our inducible model, further advancement of sequencing technology and scDNAseq methods may enable more robust detection of copy-neutral structural variation in single cells. Importantly, the measurement of structural CIN using scDNAseq would require relatively uniform coverage of the entire genome, which is not provided by mutation panels. For intact tumors, it would be important to distinguish the genome structures of tumor cells from stromal cells, which could be accomplished by simultaneously detecting cancer-specific mutations.
One limitation of this study is that our models had a short time of CIN induction. This approach ensures ongoing CIN, but methods that failed to detect CIN here could correlate with ongoing CIN after cellular selection or could indicate a cellular context permissive of ongoing CIN. For example, although cenFISH only detects the highest levels of CIN this may become more sensitive over several generations with elevated CIN, which would increase the probability that one of a limited set of probes would detect an alteration. While our bulk genomics and transcriptomics methods are not reliable measures of ongoing CIN, further work will be required to determine how the clinically accessible methods that did detect CIN (cenFISH and scDNAseq) perform in longitudinal models such as murine tumors or patient-derived tumor spheroids.
We did not test downstream effects of CIN. The incidence of micronuclei apparent by microscopy reflects ongoing CIN and is reported to promote metastasis (3). We did not track micronuclei in this study, though their incidence would likely be phenotype-specific, as reported elsewhere (54). We also did not evaluate single-cell RNAseq (scRNAseq) as a method of measuring CIN. Currently, there are no widely used scRNAseq-based measures of CIN and we did not seek new methods of CIN measurement. However, given the preponderance of evidence of the transcriptional consequences of CIN and aneuploidy (26, 55–59), it is conceivable that robust transcriptional CIN signatures could be derived at single-cell resolution. Whether these would reflect gene dosage, a general response to CIN or would be phenotype/mechanism-dependent is unclear. In any case, the reliability of these measures could be limited by dosage compensation (60–64). Although we could select specific genes minimally affected by compensation, the sparse data with current scRNAseq platforms remains a challenge. On the other hand, large-scale DNA copy number alterations can be inferred from scRNAseq data (65, 66), which would provide an additional dimension of single cell genomic data and perhaps increase the reliability of a measure of CIN based on single-cell chromosome copy number data.
This work provides a thorough empirical analysis of the relative capability of current CIN measures to detect ongoing CIN across specific, inducible phenotypic models of CIN. We find current measures differ in their ability to detect ongoing CIN and that some fail to reliably detect CIN at all. Imaging approaches are the most sensitive and reliable. Cytogenetic approaches have low sensitivity, only significantly detecting the model which had among the highest mis-segregation rates. Bulk genomic and transcriptomic measures do not reflect ongoing CIN while single-cell genomic methods, particularly the inference of mis-segregation rates, offer both sensitivity and potential for clinical accessibility. In light of these conclusions, we recommend single-cell genomics with Bayesian inference as the best method for further development of a clinically accessible measure of CIN. Importantly, the models and data generated here serve as a resource for investigators seeking to validate innovative measures of ongoing CIN from bulk and single-cell sequencing.
Materials and Methods
Cell lines, cultivation, and fixed and time-lapse microscopy, and cytogenetics used standard approaches, and are described in further detail in SI Appendix, Methods.
scDNAseq and Analysis.
Cells were reseeded in six-well plates at 40% and grown to ~70 to 80% density over 18 to 24 h prior to harvest. Standard procedures are described in SI Appendix, Methods. Copy number calls were performed in a local installation of Ginkgo (67) with a variable bin size of ~2.5 Mb using global segmentation. Minimum ploidy was set to 1.35 and maximum ploidy to 3 to reflect our flow cytometric gating strategy for FACS. Whole chromosome copy numbers were estimated by taking the mode of copy numbers across genomic bins for each chromosome. Thus, even sub-chromosomal gains and losses of greater than 50% of a chromosome should remain detectable.
Quantification of single cell CIN signatures.
We quantified previously published putative CIN signatures from Drews et al. (44) using resources made publicly available in a github repository by the authors (https://web.archive.org/web/20220615195321/https://github.com/markowetzlab/CINSignatureQuantification). We used the function “quantifyCNSignatures” in the available R package CINSignatureQuantification (44) to call signatures from unrounded segment copy number matrices derived using Ginkgo, as described above and report the normalized, unscaled signature activity levels to avoid masking the relative activity of each signature.
Inference of mis-segregation rates.
We used agent-based simulation and approximate Bayesian computation to infer mis-segregation rates from scDNAseq data. Agent-based simulation of CIN and karyotype selection in growing populations was performed in NetLogo (v6.0.4) (68) and approximate Bayesian computation was performed using the R (v4.2.2) (69) using the abc package (v2.1) (70) as previously described (27). Additional details are described in SI Appendix, Methods.
Bulk DNA Sequencing and Analysis.
Sample preparation, sorting, and bulk DNA library preparation were prepared in parallel with and in the same manner as single-cell DNA samples. Then, 500 cells were sorted for each bulk DNA sample. Standard procedures are described in SI Appendix, Methods. Bulk DNA copy numbers were called in R using QDNAseq (v1.34.0) (71) and a bin size of 30 Kb. Segment copy numbers were called using bin copy numbers smoothed over two bins and Anscombe transformed (transformFun = “sqrt” in the “segmentBins” function). Putative CIN signatures were derived from bulk copy number profiles and reported in the same manner as for single-cell DNA copy number profiles as described above.
Bulk RNA Sequencing.
Cells were reseeded in six-well plates at 40% and grown to ~70 to 80% density over 18 to 24 h prior to harvest. Standard procedures are described in SI Appendix, Methods. Linear modeling and differential expression analysis were performed in limma (v3.46) (72, 73). Imaging-based approximation of MDD and Statistical methods are described in SI Appendix, Methods.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank the University of Wisconsin Carbone Cancer Center (UWCCC) Shared Resources funded by the UWCCC Support Grant P30 CA014520—Flow Cytometry Core Facility (1S10RR025483-01), Cancer Informatics Shared Resource, Small Molecule Screening Facility. Thanks also to Dr. Rob Lera for microscopy assistance, and members of the laboratories of Drs. M.E.B., B.A.W., and Aussie Suzuki. This work was supported by NIH grants 1R01CA234904 (B.A.W. and M.E.B.) and P30CA014520 (UWCCC Support Grant). A.R.L. was supported by F31CA254247, and A.S.Z. was supported by T32GM008688.
Author contributions
A.R.L., B.A.W., and M.E.B. designed research; A.R.L., S.B., K.O., and L.H. performed research; A.S.Z. and V.L.H. contributed new reagents/analytic tools; A.R.L. and S.B. analyzed data; V.L.H., B.A.W., and M.E.B. study supervision; and A.R.L., S.B., A.S.Z., B.A.W., and M.E.B. wrote the paper.
Competing interests
M.E.B. declares all interests without adjudicating relationship to the published work. He is on the medical advisory board of Strata Oncology, receives research funding from Abbvie, Genentech, Puma, Arcus, Apollomics, Loxo Oncology/Lilly, and Elevation Oncology, and holds patents on a microfluidic device for drug testing and for homologous recombination and super-resolution microscopy technologies. All other authors report no conflicts of interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
Sequencing data were deposited to NCBI SRA (PRJNA992891) (74). Data and code for analysis were deposited to OSF (https://osf.io/aqvyx/) (75).
Supporting Information
References
- 1.Boveri T., Zur frage der entstehung maligner tumoren (Fischer, 1914). [Google Scholar]
- 2.von Hansemann D. P., Ueber asymmetrische Zelltheilung in Epithel-krebsen und deren biologische Bedeutung. Virchows Arch. Pathol. Anat. Physiol. Klin. Med. 119, 299–326 (1890). [Google Scholar]
- 3.Bakhoum S. F., et al. , Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vasudevan A., et al. , Single-chromosomal gains can function as metastasis suppressors and promoters in colon cancer. Dev. Cell, 52, 413–428.e6 (2020), 10.1016/j.devcel.2020.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ippolito M. R., et al. , Gene copy-number changes and chromosomal instability induced by aneuploidy confer resistance to chemotherapy. Dev. Cell 56, 2440–2454.e6 (2021), 10.1016/j.devcel.2021.07.006. [DOI] [PubMed] [Google Scholar]
- 6.Lee A. J. X., et al. , Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 71, 1858–1870 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lukow D. A., et al. , Chromosomal instability accelerates the evolution of resistance to anti-cancer therapies. Dev. Cell 56, 2427–2439.e4 (2021), 10.1016/j.devcel.2021.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bakhoum S. F., Danilova O. V., Kaur P., Levy N. B., Compton D. A., Chromosomal instability substantiates poor prognosis in patients with diffuse large B-cell lymphoma. Clin. Cancer Res. 17, 7704–7711 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denu R. A., et al. , Centrosome amplification induces high grade features and is prognostic of worse outcomes in breast cancer. BMC Cancer 16, 1–13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jamal-Hanjani M., et al. , Tracking the evolution of non–small-cell lung cancer. N. Engl. J. Med. 376, 2109–2121 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Funk L. C., et al. , p53 is not required for high CIN to induce tumor suppression. Mol. Cancer Res. 19, 112–123 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoevenaar W. H. M., et al. , Degree and site of chromosomal instability define its oncogenic potential. Nat. Commun. 11, 1501 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silk A. D., et al. , Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc. Natl. Acad. Sci. U.S.A. 110, E4134–E4141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jamal-Hanjani M., et al. , Extreme chromosomal instability forecasts improved outcome in ER-negative breast cancer: A prospective validation cohort study from the TACT trial. Ann. Oncol. 26, 1340–1346 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Birkbak N. J., et al. , Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 71, 3447–3452 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roylance R., et al. , Relationship of extreme chromosomal instability with long-term survival in a retrospective analysis of primary breast cancer. Cancer Epidemiol. Biomarkers Prev. 20, 2183–2194 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scribano C. M., et al. , Chromosomal instability sensitizes patient breast tumors to multipolar divisions induced by paclitaxel. Sci. Transl. Med. 13, eabd4811 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaki B. I., Suriawinata A. A., Eastman A. R., Garner K. M., Bakhoum S. F., Chromosomal instability portends superior response of rectal adenocarcinoma to chemoradiation therapy. Cancer 120, 1733–1742 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Lashen A., et al. , The characteristics and clinical significance of atypical mitosis in breast cancer. Mod. Pathol. 35, 1341–1348 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohashi R., et al. , Prognostic utility of atypical mitoses in patients with breast cancer: A comparative study with Ki67 and phosphohistone H3. J. Surg. Oncol. 118, 557–567 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Dumont M., et al. , Human chromosome-specific aneuploidy is influenced by DNA-dependent centromeric features. EMBO J. 39, e102924 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Worrall J. T., et al. , Non-random Mis-segregation of human chromosomes. Cell Rep. 23, 3366–3380 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cimini D., Moree B., Canman J. C., Salmon E. D., Merotelic kinetochore orientation occurs frequently during early mitosis in mammalian tissue cells and error correction is achieved by two different mechanisms. J. Cell Sci. 116, 4213–4225 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Santaguida S., et al. , Chromosome Mis-segregation generates cell-cycle-arrested cells with complex karyotypes that are eliminated by the immune system. Dev. Cell 41, 638–651.e5 (2017), 10.1016/j.devcel.2017.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Torres E. M., et al. , Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317, 916–924 (2007), 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- 26.Stingele S., et al. , Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol. Syst. Biol. 8, 608 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lynch A. R., Arp N. L., Zhou A. S., Weaver B. A., Burkard M. E., Quantifying chromosomal instability from intratumoral karyotype diversity using agent-based modeling and Bayesian inference. ELife 11, e69799 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bolhaqueiro A. C. F., et al. , Ongoing chromosomal instability and karyotype evolution in human colorectal cancer organoids. Nat. Genet. 51, 824–834 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Cross W., et al. , Stabilising selection causes grossly altered but stable karyotypes in metastatic colorectal cancer. bioRxiv [Preprint] (2020). 10.1101/2020.03.26.007138 (Accessed 23 March 2024). [DOI]
- 30.Carter S. L., Eklund A. C., Kohane I. S., Harris L. N., Szallasi Z., A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 38, 1043–1048 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Sheltzer J. M., A transcriptional and metabolic signature of primary aneuploidy is present in chromosomally-unstable cancer cells and informs clinical prognosis. Cancer Res. 73, 6401–6412 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knouse K. A., Wu J., Whittaker C. A., Amon A., Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc. Natl. Acad. Sci. U.S.A. 111, 13409–13414 (2014), 10.1073/pnas.1415287111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Navin N., et al. , Tumour evolution inferred by single-cell sequencing. Nature 472, 90–95 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brunori M., et al. , TRF2 inhibition promotes anchorage-independent growth of telomerase-positive human fibroblasts. Oncogene 25, 990–997 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Bennett A., et al. , Cenp-E inhibitor GSK923295: Novel synthetic route and use as a tool to generate aneuploidy. Oncotarget 6, 20921–20932 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lynch A. R., Shermineh B., Burkard M. E., The reckoning of chromosomal instability: Past, present, future. Chromosome Res. 32, 2 (2024). [DOI] [PubMed] [Google Scholar]
- 37.Zasadil L. M., et al. , Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci. Transl. Med. 6, 229ra43 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lengauer C., Kinzler K. W., Vogelstein B., Genetic instability in colorectal cancers. Nature 386, 623–627 (1997). [DOI] [PubMed] [Google Scholar]
- 39.Levan A., Biesele J. J., Role of chromosomes in cancerogenesis, as studied in serial tissue culture of mammalian cells. Ann. N.Y. Acad. Sci. 71, 1022–1053 (1958). [DOI] [PubMed] [Google Scholar]
- 40.Klaasen S. J., et al. , Nuclear chromosome locations dictate segregation error frequencies. Nature 607, 604–609 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sheltzer J. M., A transcriptional and metabolic signature of primary aneuploidy is present in chromosomally unstable cancer cells and informs clinical prognosis. Cancer Res. 73, 6401–6412 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Venet D., Dumont J. E., Detours V., Most random gene expression signatures are significantly associated with breast cancer outcome. PLoS Comput. Biol. 7, e1002240 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holland A. J., et al. , The autoregulated instability of Polo-like kinase 4 limits centrosome duplication to once per cell cycle. Gene Dev. 26, 2684–2689 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drews R. M., et al. , A pan-cancer compendium of chromosomal instability. Nature 606, 976–983 (2022), 10.1038/s41586-022-04789-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bakker B., et al. , Predicting CIN rates from single-cell whole genome sequencing data using an in silico model. bioRxiv [Preprint] (2023). 10.1101/2023.02.14.528596 (22 February 2023) [DOI]
- 46.Colijn C., Gardy J., Phylogenetic tree shapes resolve disease transmission patterns. Evol. Med. Public Health 2014, 96–108 (2014), 10.1093/emph/eou018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mooers A., Heard S. B., Inferring evolutionary process from phylogenetic tree shape. Q. Rev. Biol. 72, 31–54 (1997), 10.1086/419657. [DOI] [Google Scholar]
- 48.Lynch A., Burkard M., CINFER: An interactive, web-based platform for inference of chromosome mis-segregation rates from scDNAseq data. Shinyapps.io. https://burkardlab.shinyapps.io/CINFER/. Accessed 23 March 2024.
- 49.Umbreit N. T., et al. , Mechanisms generating cancer genome complexity from a single cell division error. Science 368, eaba0712 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu Y., et al. , Paclitaxel induces micronucleation and activates pro-inflammatory cGAS–STING signaling in triple-negative breast cancer. Mol. Cancer Ther. 20, 2553–2567 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Laks E., et al. , Clonal decomposition and DNA replication states defined by scaled single-cell genome sequencing. Cell 179, 1207–1221.e22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Minussi D. C., et al. , Breast tumours maintain a reservoir of subclonal diversity during expansion. Nature 592, 302–308 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yin Y., et al. , High-throughput single-cell sequencing with linear amplification. Mol. Cell 76, 676–690.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tucker J. B., et al. , Misaligned chromosomes are a major source of chromosomal instability in breast cancer. Cancer Res. Commun. 3, 54–65 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sheltzer J. M., Torres E. M., Dunham M. J., Amon A., Transcriptional consequences of aneuploidy. Proc. Natl. Acad. Sci. U.S.A. 109, 12644–12649 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Donnelly N., Passerini V., Durrbaum M., Stingele S., Storchova Z., HSF 1 deficiency and impaired HSP 90-dependent protein folding are hallmarks of aneuploid human cells. EMBO J. 33, 2374–2387 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chunduri N. K., et al. , Systems approaches identify the consequences of monosomy in somatic human cells. Nat. Commun. 12, 5576 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pavelka N., et al. , Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 468, 321–325 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garribba L., et al. , Short-term molecular consequences of chromosome mis-segregation for genome stability. Nat. Commun. 14, 1353 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schukken K. M., Sheltzer J. M., Extensive protein dosage compensation in aneuploid human cancers. Genome Res 32, 1254-1270 (2021), 10.1101/2021.06.18.449005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheng P., et al. , Proteogenomic analysis of cancer aneuploidy and normal tissues reveals divergent modes of gene regulation across cellular pathways. ELife 11, e75227 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gasch A. P., et al. , Further support for aneuploidy tolerance in wild yeast and effects of dosage compensation on gene copy-number evolution. ELife 5, e14409 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Torres E. M., Springer M., Amon A., No current evidence for widespread dosage compensation in S. cerevisiae. ELife 5, e10996 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hose J., et al. , Dosage compensation can buffer copy-number variation in wild yeast. ELife 4, e05462 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gao R., et al. , Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat. Biotechnol. 39, 599–608 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.De Falco A., Caruso F., Su X.-D., Iavarone A., Ceccarelli M., A variational algorithm to detect the clonal copy number substructure of tumors from scRNA-seq data. Nat. Commun. 14, 1074 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Garvin T., et al. , Interactive analysis and assessment of single-cell copy-number variations. Nat. Methods 12, 1058–1060 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wilensky U., NetLogo (Center for Connected Learning and Computer-Based Modeling, Northwest University, Evanston, IL, 1999). http://ccl.northwestern.edu/netlogo/. Accessed 23 March 2024.
- 69.Core R. Team, R: A language and environment for statistical computing (2021). https://www.R-project.org. Accessed 23 March 2024.
- 70.Csillery K., Francois O., Blum M. G. B., Abc: An R package for approximate Bayesian computation (ABC). Methods Ecol. Evol. 3, 475–479 (2012), 10.1111/j.2041-210x.2011.00179.x. [DOI] [PubMed] [Google Scholar]
- 71.Scheinin I., et al. , DNA copy number analysis of fresh and formalin-fixed specimens by shallow whole-genome sequencing with identification and exclusion of problematic regions in the genome assembly. Genome Res. 24, 2022–2032 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ritchie M. E., et al. , limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Law C. W., Chen Y., Shi W., Smyth G. K., voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lynch A. R., et al. , A survey of CIN measures across mechanistic models. NCBI Short Read Archive. https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA992891. Deposited 9 July 2023.
- 75.Lynch A. R., et al. , A survey of CIN measures across mechanistic models. Open Science Framework. https://osf.io/aqvyx/. Deposited 23 March 2024.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
Sequencing data were deposited to NCBI SRA (PRJNA992891) (74). Data and code for analysis were deposited to OSF (https://osf.io/aqvyx/) (75).