

Significance
Flaviviruses transmitted by mosquitoes cause millions of infections annually. During the flavivirus infection, most of nonstructural proteins remain intracellular and become cytotoxic if accumulated over time. We identified that an evolutionarily conserved ubiquitin ligase HRD1 directly interacts with flavivirus NS4A and ubiquitylates a conserved lysine residue for ERAD-mediated degradation to facilitate flaviviruses infections in both mammalian hosts and mosquitoes. Moreover, LS-102 is the specific inhibitor of HRD1 and can effectively inhibit DENV2 infection in mice and Aedes aegypti mosquitoes. We revealed the mechanism, by which flaviviruses balance the homeostasis of viral protein and provided a potential therapeutic target to interrupt flavivirus infection and transmission.
Keywords: flavivirus, mosquito, NS4A, HRD1, ER-associated degradation
Abstract
Mosquito-borne flaviviruses such as dengue (DENV) and Zika (ZIKV) cause hundreds of millions of infections annually. The single-stranded RNA genome of flaviviruses is translated into a polyprotein, which is cleaved equally into individual functional proteins. While structural proteins are packaged into progeny virions and released, most of the nonstructural proteins remain intracellular and could become cytotoxic if accumulated over time. However, the mechanism by which nonstructural proteins are maintained at the levels optimal for cellular fitness and viral replication remains unknown. Here, we identified that the ubiquitin E3 ligase HRD1 is essential for flaviviruses infections in both mammalian hosts and mosquitoes. HRD1 directly interacts with flavivirus NS4A and ubiquitylates a conserved lysine residue for ER-associated degradation. This mechanism avoids excessive accumulation of NS4A, which otherwise interrupts the expression of processed flavivirus proteins in the ER. Furthermore, a small-molecule inhibitor of HRD1 named LS-102 effectively interrupts DENV2 infection in both mice and Aedes aegypti mosquitoes, and significantly disturbs DENV transmission from the infected hosts to mosquitoes owing to reduced viremia. Taken together, this study demonstrates that flaviviruses have evolved a sophisticated mechanism to exploit the ubiquitination system to balance the homeostasis of viral proteins for their own advantage and provides a potential therapeutic target to interrupt flavivirus infection and transmission.
Flaviviridae is a genus of single-stranded and positive-sense RNA viruses in which many members are transmitted by arthropod vectors, such as ticks and mosquitoes. Mosquito-borne flaviviruses, including dengue (DENV), Zika (ZIKV), Japanese encephalitis (JEV), and West Nile (WNV) viruses, cause hundreds of millions of infection cases annually and act as etiological agents of human hemorrhagic fever, encephalitis and meningitis (1–3). The genomes of flaviviruses are approximately 10 to 11 kbs, which are translated by cellular ribosomes into a single polyprotein. Subsequently, several cellular and viral proteases take charge of cleaving the viral polyprotein into seven nonstructural and three structural proteins that serve different roles throughout the viral replication cycle (4–6). Indeed, the translation-cleavage strategy exploited by flaviviruses shows an advantage in effectively synthesizing their adjacent proteins through the formation of the viral replication complex with a limited copy number of viral genomes (7, 8). Nonetheless, this strategy could be disadvantageous in controlling the amount of each viral protein. The cleavage of a polyprotein produces an equal amount of each structural or nonstructural viral protein. Since the structural proteins are packaged into progeny virions, while nonstructural proteins, many of which are enzymatic, participate in viral replication and can be reused many times, the required quantities of structural proteins are much greater than those of nonstructural proteins (9). Given the cytotoxicity of these nonstructural proteins accumulating in the endoplasmic reticulum (ER) (10), viruses may hijack some host cellular mechanism(s) to remove redundant nonstructural proteins, thus maintaining cellular fitness and facilitating viral infection and propagation. However, the machinery that maintains the homeostasis of viral proteins remains to be understood.
Ubiquitination is a regulatory system that governs a multitude of cellular functions through posttranslational modification, which can be catalyzed by a multienzyme cascade including E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugating enzyme), and E3 (ubiquitin ligase) enzymes (11–14). In contrast to the limited number of E1 and E2 enzymes, there are several hundred E3 ubiquitin ligases that confer substrate specificity to the ubiquitination process (15–17). Canonically, ubiquitination regulates protein levels by targeting substrates for proteasomal or lysosomal degradation through the ubiquitin-protease system (UPS) (18). Alternatively, the ubiquitination system can influence the function of substrate proteins by a nondegradative mechanism (19). Indeed, many viruses hijack the ubiquitination processes to enhance various steps of the infection lifecycle in host cells (20, 21). For example, the entry of dengue (DENV) and Zika (ZIKV) viruses (22, 23), the assembly of influenza A virus (24, 25) and Lassa virus (26), and the budding of Ebola and Marburg viruses (27, 28) all exploit ubiquitination. In this study, we report that flaviviruses have evolved to usurp the E3 ubiquitin ligase HRD1 for ubiquitination of NS4A, by which excess NS4A is removed by the ERAD pathway. This mechanism avoids the buildup of excessive NS4A that interrupts the expression of processed viral proteins in the ER. The application of an HRD1 antagonist may provide a practical strategy to simultaneously prevent both flavivirus infection in hosts and mosquitoes, thus reducing the disease prevalence worldwide.
Results
The Role of Potential E3 Ubiquitin Ligases in Flavivirus Infection of Mosquitoes.
Mosquito-borne flaviviruses maintain their lifecycles between vertebrate hosts and mosquitoes. Accumulating evidence indicates that they exploit the ubiquitination system throughout the stages of their replication cycle in host cells (22, 29–31). Nonetheless, quite a few studies have investigated the roles of the ubiquitination in flavivirus infection of mosquitoes. E3 ubiquitin ligases are evolutionarily conserved and can be generally categorized into the Really Interesting New Gene (RING) finger-containing ligases, homologous to the E6AP carboxyl terminus (HECT)-type ligases, and RING-between-RING (RBR) ligases (15). We identified 110 Aedes aegypti genes encoding 91 proteins with RING finger, 14 proteins with HECT, and five proteins with RBR domains (Fig. 1A and SI Appendix, Table S1), suggesting the function of these proteins as E3 ubiquitin ligases in mosquitoes. Double-stranded RNA (dsRNA)-mediated silencing in A. aegypti mosquitoes was then employed to assess the role of these genes in DENV2 infection. dsRNAs of these genes were individually microinjected into female A. aegypti mosquitoes. DENV2 was sequentially inoculated 3 d later, and the effect on viral load was assessed 6 d after infection. Compared to the GFP dsRNA inoculated control, knockdown of the AAEL001217, AAEL003248, AAEL003466, AAEL004697, and AAEL011413 genes significantly impaired the DENV2 burden in the mosquitoes, while the AAEL003489, AAEL008374, AAEL022272, and AAEL023019 genes increased it (Fig. 1B). Nonetheless, in these genes regulating DENV2 infection, silencing AAEL008374 and AAEL004697 rather than the others played significant regulatory roles in ZIKV infection of A. aegypti, in which deficiency of AAEL004697 presented the strongest phenotype (Fig. 1C). The AAEL004697 gene encodes a mosquito homologous protein of human hydroxymethylglutaryl reductase degradation protein 1 (HRD1), which acts as an E3 ubiquitin ligase with a cytosolic RING motif and takes charge of protein degradation from the endoplasmic reticulum (32). We therefore focused on AAEL004697 and then designated it A. aegypti HRD1 (AaHRD1) throughout the following investigation.
Fig. 1.
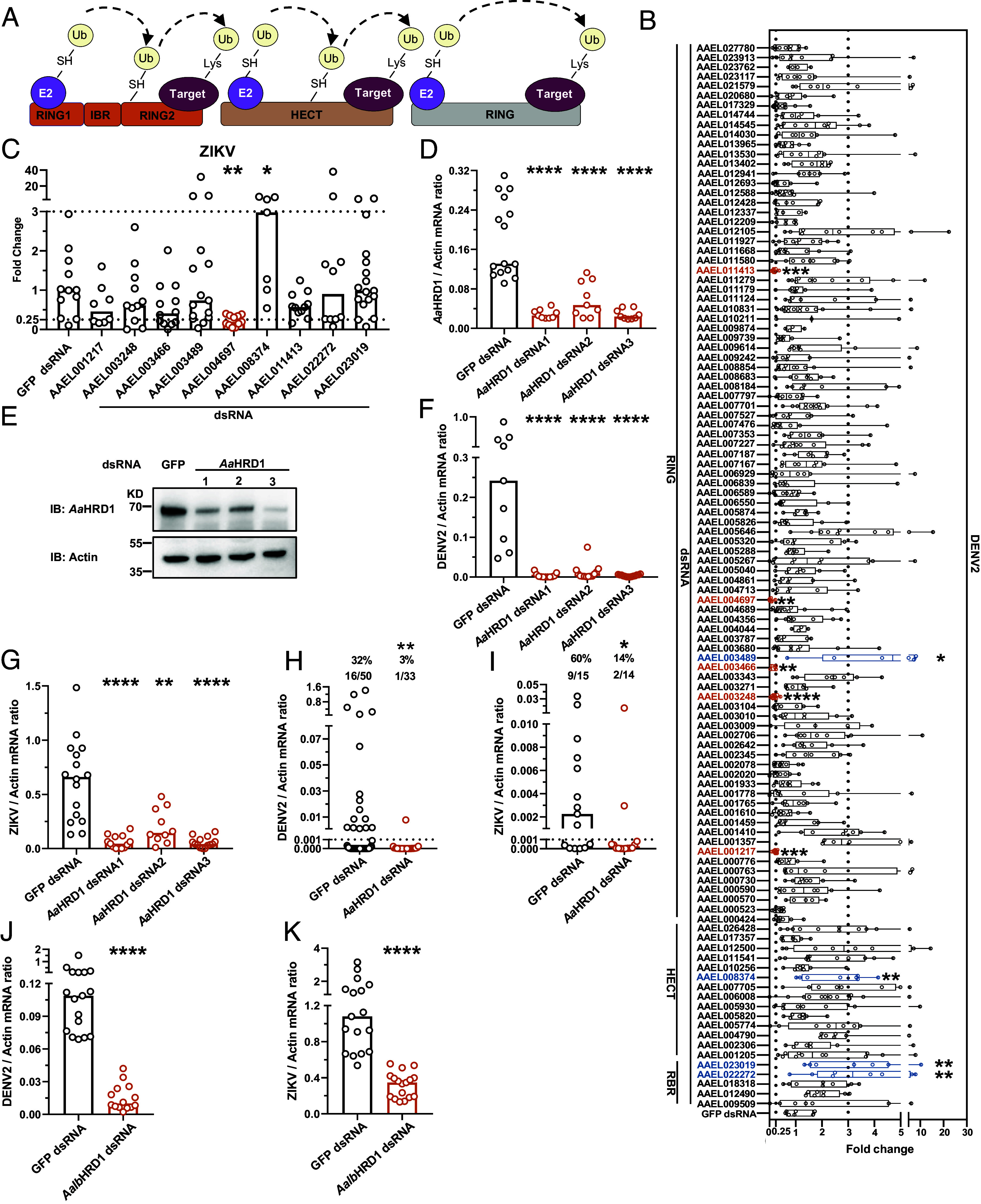
Identification of HRD1s facilitating flavivirus infection in the Aedes mosquitoes. (A) Schematic representation of E3 ligases. (B) Identification the roles of E3 ligases in DENV2 infection of A. aegypti. A. aegypti protein (GCF_002204515.2) was annotated by InterProScan using the iprscan5.py script from the European Bioinformatics Institute. Five RBR, 14 HECT and 91 RING E3 ligases have been identified in A. aegypti. A. aegypti mosquitoes were inoculated with dsRNAs of E3 ligases, and microinjected with GFP dsRNA served as a negative control. DENV2 was inoculated 3 d later, and viral loads were assessed via TaqMan qPCR and normalized against A. aegypti actin (AAEL011197). Compared with the control group, silencing these genes results in viral loads increasing by more than threefold (blue column) and decreasing by over fourfold (orange column), respectively. These genes were selected as candidates for further investigation. (C) The roles of selected genes in ZIKV infection of A. aegypti. A. aegypti were inoculated with dsRNAs of E3 ligases and thoracically infected with ZIKV 3 d later. Viral loads were determined via TaqMan qPCR. (D and E) Identification of the deficiency of three independent dsRNAs of AaHRD1. Indicated dsRNAs were microinjected into A. aegypti, and the expression of AaHRD1 was determined 3 d later via SYBR Green qPCR and normalized against A. aegypti actin (D) or western blot (E). (F and G) Silencing AaHRD1 inhibited DENV2 and ZIKV infections in A. aegypti. A. aegypti mosquitoes were inoculated with three independent dsRNAs of AaHRD1 and then thoracically infected with DENV2 or ZIKV 3 d later. Viral loads of DENV2 (F) and ZIKV (G) were assessed by qPCR. (H and I) Silencing AaHRD1 impaired DENV2 and ZIKV infections in A. aegypti via an in vitro membrane feeding system. Human blood (50% v/v) and supernatant from DENV2 (NGC)/ZIKV-infected Vero cells (50% v/v) were used to feed A. aegypti. The infectivity of DENV2 (H) and ZIKV (I) was measured 8 d later via qPCR. (J and K) Knockdown of AalbHRD1 reduced DENV2 and ZIKV infections in A. albopictus. A. albopictus were inoculated with AalbHRD1 dsRNA and then thoracically infected with DENV2/ZIKV. Viral loads of DENV2 (J) and ZIKV (K) were measured and normalized against A. albopictus actin (AALF010408). (E) The experiment was repeated three times with similar results. (B–D and F–K) Each dot represents a mosquito and the median of the results was shown. A nonparametric Mann–Whitney U test was used for statistical analyses of viral load. (H and I) The number at the top of each column represents the infected number/total number. The limit of detection is illustrated by the black dashed line. Differences in the infectivity ratio were compared using Fisher’s exact test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s., not significant (P > 0.05).
We next generated three individual dsRNAs against AaHRD1 to further validate the role of AaHRD1 in flavivirus infection. The three parallel dsRNAs all impaired the expression of AaHRD1 at the mRNA level, as detected by qPCR (Fig. 1D), and in mosquito lysates, as measured by western blotting (Fig. 1E). The anti-AaHRD1 murine polyclonal antibody was prepared by immunization with recombinant AaHRD1-ΔTM peptide expressed in the Escherichia coli BL21 DE3 strain (SI Appendix, Fig. S1A). The specificity of the antibody for AaHRD1 was validated in the Aag2 cells transfected with or without AaHRD1 dsRNA (SI Appendix, Fig. S1B). Both DENV2 (Fig. 1F) and ZIKV (Fig. 1G) loads were significantly suppressed in these AaHRD1-silenced mosquitoes, indicating the essential role of AaHRD1 in mosquito-flavivirus interactions. We also found that both DENV2 (Fig. 1H) and ZIKV (Fig. 1I) infectivity obviously decreased in AaHRD1-silenced mosquitoes via an in vitro blood-feeding system. dsRNA-mediated knockdown of AaHRD1 did not influence the survival of mosquitoes in the experimental settings (SI Appendix, Fig. S1C). Suppressor/enhancer of Lin-12-like (Sel1L), an adaptor protein of HRD1, resides on the membrane of the endoplasmic reticulum and controls HRD1 stability (SI Appendix, Fig. S1D) (32–34). We therefore identified the A. aegypti Sel1L homologous gene (AaSel1L) by sequence comparison. Consistently, silencing AaSel1L suppressed both DENV2 and ZIKV infections in A. aegypti (SI Appendix, Fig. S1 E and F). Aedes albopictus is the other native mosquito vector for DENV and ZIKV transmission (35). We identified homologs of AaHRD1 and AaSel1L with 82 to 84% identity in the A. albopictus genome (AalbHRD1 and AalbSel1L). dsRNA-mediated knockdown of AalbHRD1 (Fig. 1 J and K) and AalbSel1L (SI Appendix, Fig. S1 G and H) reduced both flavivirus infections in A. albopictus.
Some flaviviruses causing neurotropic infections, such as JEV, are naturally carried and transmitted by Culex mosquitoes (36, 37). We therefore assessed whether HRD1 may facilitate JEV infection in Culex quinquefasciatus. We identified a homolog of AaHRD1 with 77% identity in the C. quinquefasciatus, and then named C. quinquefasciatus HRD1 (CqHRD1). dsRNA-mediated silencing CqHRD1 impaired JEV infection in Culex mosquitoes (SI Appendix, Fig. S1 I and J). Overall, the mosquito HRD1 generally plays a proviral role in flavivirus infection.
HRD1s Ubiquitylate NS4As to Facilitate Flavivirus Infection in Mosquitoes.
Previous studies have demonstrated that multiple ER membrane-integrated nonstructural proteins of flaviviruses can be ubiquitinylated in infected cells and thus degraded by the ERAD pathway (10). HRD1 acts as a key ERAD component with the activity of E3 ubiquitin ligase (38). Since the recognition of substrate by an E3 ligase is necessary for the ubiquitination cascade (15), we therefore assessed the interactions between AaHRD1 and DENV2 nonstructural proteins. The recombinant plasmids expressing 7 DENV2 nonstructural proteins were cotransfected with the plasmid carrying the AaHRD1 gene. The cell lysates were then collected for a coimmunoprecipitation (co-IP) assay. Notably, AaHRD1 specifically interacted with DENV2 NS4A but not the other nonstructural proteins (SI Appendix, Fig. S2A). To investigate the interplay between the endogenous AaHRD1 and native viral proteins, A. aegypti Aag2 cells were infected with DENV2 for 72 h. Subsequently, the lysates of infected cells were subjected to immunoprecipitation with an anti-AaHRD1 murine antibody. There was NS4A, rather than other viral proteins, detected in the AaHRD1-mediated immune precipitates (Fig. 2A). Nonetheless, antibodies against DENV2 NS2A were not commercially available. Therefore, we ectopically expressed DENV2 NS2A in Aag2 cells. Immunoprecipitation with an anti-AaHRD1 antibody showed that endogenous AaHRD1 did not interact with DENV2 NS2A (SI Appendix, Fig. S2B). Overall, the present study indicated that endogenous AaHRD1 exclusively interacts with NS4A but not with other viral nonstructural proteins in DENV2-infected cells. Indeed, ectopically expressed DENV2 NS4A was highly ubiquitinated in A. aegypti Aag2 cells (Fig. 2B). dsRNA-mediated knockdown of AaHRD1 significantly suppressed the ubiquitination of DENV2 NS4A in Aag2 cells (Fig. 2C). Silencing AaHRD1 gene did not influence the viability of Aag2 cells (SI Appendix, Fig. S2C). A previous study indicated that the 329th substitution from cysteine (C) to serine (S) (C329S), which is located in the RING motif, resulted in the catalytic inactivation of human HRD1 (39). Indeed, the 329th residue of human HRD1 corresponds to the 328th cysteine in AaHRD1 according to the sequence alignment. The C328S mutation of AaHRD1 offset the ubiquitination of DENV2 NS4A (Fig. 2D). We next investigated the effect of endogenous AaHRD1 on ubiquitination of NS4A in DENV2-infected mosquito cells. A. aegypti Aag2 cells were transfected with AaHRD1 dsRNA and then infected with DENV2 for 72 h. The lysates of infected cells were used for an immunoprecipitation assay with anti-NS4A antibody and result showed that silencing the AaHRD1 gene largely impaired the ubiquitination of DENV2 NS4A in the lysates of DENV2-infected Aag2 cells (Fig. 2E), indicating that the endogenous AaHRD1 can recognize and thus ubiquitinate NS4A in mosquito cells with flavivirus infection. As an ER-located E3 ligase, the classical physiological role of HRD1 is to ubiquitinate substrates and degrade them via the proteasome. We therefore assessed the outcome of AaHRD1-mediated ubiquitination in DENV2 NS4A. In the cycloheximide (CHX) chase assay, AaHRD1 effectively reduced the amount of ectopically expressed DENV2 NS4A; nonetheless, the AaHRD1-C328S mutant did not modulate the abundance of NS4A (Fig. 2F). Thus, AaHRD1-mediated ubiquitination caused instability of DENV2 NS4A. We next assessed the role of CqHRD1 in ubiquitination of JEV NS4A. Both CqHRD1 and JEV NS4A were coexpressed in C. quinquefasciatus Cxq-1 cells (40). CqHRD1 specifically interacted with JEV NS4A in a co-IP assay (SI Appendix, Fig. S3A). Notably, ectopically expressed JEV NS4A was ubiquitinylated; nonetheless, dsRNA-mediated knockdown of CqHRD1 largely impaired the ubiquitylation of JEV NS4A in Cxq-1 cells (SI Appendix, Fig. S3 B and C). In consistence with that of AaHRD1, the C328S mutation of CqHRD1 disabled the ubiquitination of JEV NS4A (SI Appendix, Fig. S3D). The results indicate the specificity of mosquito HRD1 catalysis in ubiquitination of flavivirus NS4As.
Fig. 2.

AaHRD1 ubiquitylates and causes the instability of NS4A thus facilitating DENV2 infection in mosquitoes. (A) The interaction of NS4A with endogenous AaHRD1 in DENV2-infected Aag2 cells. Aag2 cells were infected with DENV2 at 0.1 MOI for 3 d and then harvested. IP was performed with anti-AaHRD1 murine polyclonal antibodies and analyzed via WB. (B) NS4A is ubiquitinated in Aag2 cells. A plasmid encoding NS4A was transfected into Aag2 cells. Cell lysates were subjected to IP with anti-Flag antibodies, and the level of NS4A ubiquitination was measured with anti-ubiquitin (Ub). (C) Silencing AaHRD1 impaired the ubiquitination of NS4A. Aag2 cells were transfected with AaHRD1 dsRNA 3 d later, transfected with NS4A. The level of NS4A ubiquitination was determined with anti-Ub. (D) AaHRD1 enhanced the ubiquitination of NS4A. Aag2 cells were transfected with indicated plasmids. IP was performed with anti-Flag antibodies and the level of NS4A ubiquitination was measured. (E) Silencing AaHRD1 impaired the ubiquitination of NS4A in DENV2-infected Aag2 cells. Aag2 cells were transfected with AaHRD1 dsRNA 3 d later, infected with DENV2 at 0.1 MOI for 3 d, and then harvested. Because knockdown of AaHRD1 inhibited viral replication, the input of NS4A was normalized and then for IP with anti-NS4A rabbit antibodies, and the level of NS4A ubiquitination was measured. (F) AaHRD1 resulted in the instability of NS4A. S2 cells were transfected with indicated plasmids and then subjected to CHX (10 μg/mL) treatment for the indicated time. The amount of NS4A was analyzed via WB. (G) AaHRD1 ubiquitylated the 80th lysine residue of DENV2 NS4A. S2 cells were transfected with the indicated plasmids. IP was conducted, and then the level of NS4A ubiquitination was measured. (H and I) The K80R substitution in NS4A reduced DENV2 replication in A. aegypti. A. aegypti were thoracically infected with 300 nL (2,000 f.f.u. mL−1) of NS4A or NS4A-K80R strain. Viral loads were assessed at the indicated times via qPCR (H). (I) Either NS4A strain or NS4A-K80R strain (500 μL, 2 × 106 f.f.u. mL−1) and 500 μL fresh human blood were mixed and then fed on A. aegypti via an in vitro blood-feeding system. Viral infectivity was determined 8 d later by qPCR. (A–G) These experiments were repeated three times with similar results. (H) Viral loads were normalized against A. aegypti actin and analyzed statistically using the Mann–Whitney U test. (I) The limit of detection is illustrated by the black dashed line. The data were analyzed statistically using Fisher’s exact test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s., not significant (P > 0.05).
We next investigated the details of the AaHRD1-NS4A interplay. AaHRD1 is an integral ER membrane protein with a transmembrane (TM) domain and a cytosolic RING motif (SI Appendix, Fig. S4A). Based on these defined domains, we ectopically expressed two truncated peptides, AaHRD1-ΔRING (Δ290aa to 328aa) and AaHRD1-ΔTM (Δ4aa to 232aa), with DENV2 NS4A in Drosophila S2 cells and found that the peptides with the TM domain truncation completely abolished the interaction between AaHRD1 and NS4A (SI Appendix, Fig. S4B). Consistently, the AaHRD1-ΔTM truncated peptide was unable to ubiquitinate NS4A in S2 cells ectopically expressing both proteins (SI Appendix, Fig. S4C), indicating that the TM domain of AaHRD1 is indispensable for the AaHRD1-NS4A interplay. There are 3 lysine (K) residues in DENV2 NS4A (Lys20, Lys80 and Lys125). To determine the site(s) of ubiquitination by AaHRD1, we replaced each of the lysine residues noted above individually with arginine (R) in NS4A. The immunoprecipitation assay demonstrated that the K80R substitution, rather than the others, significantly decreased the ubiquitination of NS4A by ectopic expression of AaHRD1 (Fig. 2G).
The aforementioned studies showed that AaHRD1 facilitates flavivirus infection, while AaHRD1 ubiquitylates the 80th lysine residue of DENV2 NS4A. We next assessed whether the AaHRD1-mediated ubiquitination of NS4A may be essential for DENV2 infection in mosquitoes. A DENV2 16681 strain infectious clone in which the 80th lysine of NS4A was replaced by arginine. DENV2-16681-NS4A-K80R was constructed, and the mutant viruses were rescued in Vero cells. In a serial passage of DENV2-16681-NS4A-K80R, the NS4A-K80R substitution was stable in all sequenced samples in the mutant (SI Appendix, Fig. S5A). DENV2-16681-NS4A-K80R showed much less replication than its parental 16681 strain in both Aag2 and C6/36 cells (SI Appendix, Fig. S5 B and C); nonetheless, they displayed no difference in the replication of AaHRD1-silenced Aag2 cells (SI Appendix, Fig. S5D). Subsequently, we thoracically microinjected both DENV2-16681-NS4A-K80R and its parental strain into A. aegypti mosquitoes. The K80R substitution resulted in a nearly 10-fold reduction in the replication of 16681 strain in mosquitoes over the time course of infection (Fig. 2H). The blood meal mixture of fresh human blood (50% v/v) and 16681 strain-containing supernatants from infected cells (50% v/v) were used to feed A. aegypti via an in vitro blood-feeding system. The prevalence of mosquito infection was largely reduced by feeding the DENV2-16681-NS4A-K80R mutant compared to that of the parental strain (Fig. 2I).
As the specific residue is ubiquitinylated by HRD1, the 80th lysine of DENV NS4A is conserved across flaviviruses (SI Appendix, Fig. S5E), which corresponds to the 79th lysine of JEV NS4A, as a result of JEV NS4A encoding one fewer amino acid than DENV NS4A. To assess the role of the 79th lysine of JEV NS4A in CqHRD1-mediated ubiquitination, we replaced the 79th lysine residue with arginine in JEV NS4A (JEV-NS4A-K79R). The ubiquitination of JEV NS4A-K79R was significantly impaired in mosquito cells ectopically expressing CqHRD1 (SI Appendix, Fig. S6A). Subsequently, we investigated the role of the CqHRD1-mediated NS4A ubiquitination in JEV infection in Culex mosquitoes. A JEV (SA14 strain) infectious clone with the substitution of the NS4A 79th residue from lysine to arginine was constructed (JEV-SA14-NS4A-K79R), and the mutant JEV was rescued in Vero cells. The NS4A-K79R substitution was stable in a serial of passage of JEV-SA14-NS4A-K79R mutant (SI Appendix, Fig. S6B). C. quinquefasciatus mosquitoes were orally infected by an in vitro blood-feeding system. The prevalence of mosquito infection was largely impaired by feeding the JEV-SA14-NS4A-K79R mutant compared to the parental control (SI Appendix, Fig. S6C). Overall, the results demonstrated that mosquito HRD1 ubiquitylates a lysine residue of NS4A and resulted in the NS4A instability, thus facilitating flavivirus infection in mosquitoes.
NS4A Inhibits the Expression of Processed Viral Proteins Thus Interrupting DENV Replication.
Flavivirus nonstructural proteins are essential for viral replication, while some act as virulence factors in host cells (10). We therefore assessed the role of NS4A in DENV2 infection of Aag2 and C6/36 cells. DENV2 replication significantly decreased over a time course in NS4A-expressing compared to GFP-expressing C6/36 and Aag2 cells (Fig. 3A and SI Appendix, Fig. S7 A and B), indicating that overexpression of NS4A inhibited flaviviral replication. Notably, a previous study revealed that flavivirus NS4As specifically interact with human and mosquito Sec61 translocon subunits (41). Accumulating evidence indicates that the Sec61 translocon orchestrates the cotranslational insertion of transmembrane proteins into the ER membrane (42, 43). Indeed, a membrane protein such as the flavivirus polyprotein can be translocated across the ER membrane through the heterotrimeric Sec61 complex comprising α, β, and γ subunits, which occurs via two distinct pathways: either the signal recognition particle (SRP)-dependent cotranslational pathway or the SRP-independent Sec62-dependent posttranslational translocation pathway (Fig. 3B) (44). We therefore assessed the role of these factors in DENV2 infection of mosquitoes. The dsRNAs corresponding to these genes were individually microinjected into A. aegypti mosquitoes. DENV2 was sequentially inoculated 3 d later, and viral load was assessed 6 d after infection. The genes encoding AaSRα, AaSec62, and AaSec61-α/β/γ were silenced in mosquitoes (SI Appendix, Fig. S8 A–F). Compared to the GFP dsRNA-inoculated controls, knockdown of the genes encoding the A. aegypti SRP receptor α subunit (AaSRα) and Sec61 complex subunits (AaSec61-α/β/γ), rather than that of the Sec62 gene (AaSec62), almost abolished DENV2 infection in mosquitoes (Fig. 3C and SI Appendix, Table S2), suggesting that the flavivirus polyprotein exploits the SRP-dependent pathway to translocate to the ER membrane. We next assessed the interplay between the subunits of the SRP-dependent Sec61 translocon and DENV2 NS4A. The co-IP assay showed that AaSRα specifically interacted with NS4A in the ectopic expression system (Fig. 3D). Indeed, SRP54 serves as the key component of SRP in the recognition of transmembrane proteins for transportation to SRα in the ER membrane (45). A binding competition assay indicated that DENV2 NS4A interrupted the recognition of AaSRα to A. aegypti SRP54 (AaSRP54) (Fig. 3E). Subsequently, we cloned three truncations of NS4A (NS4A-ΔN40, NS4A-Δ41-87 and NS4A-ΔC40) based on its structure (SI Appendix, Fig. S9A). These truncations of NS4A were ectopically expressed with AaSRα in S2 cells, and the co-IP assay showed that the NS4A-ΔN40 truncated peptide lost its interaction with AaSRα (SI Appendix, Fig. S9B), and a comparable result was reached via an immunofluorescence assay (SI Appendix, Fig. S9C), demonstrating that the N-terminal 40 amino acids of NS4A mediate its association with AaSRα. The truncation of the 41 to 87th residues, rather than the ΔN40 or ΔC40 residues of NS4A, abolished the colocalization of NS4A with the ER (SI Appendix, Fig. S9D). Moreover, we found that NS4A-ΔN40 is unable to inhibit the recognition of AaSRα to AaSRP54 (SI Appendix, Fig. S9E). To further investigate the role of NS4A in the processing of flaviviral polyproteins in the ER, we ectopically expressed DENV2 NS4A in C6/36 cells and subsequently infected the transfected cells with DENV2. The ER from infected cells was stained by an antibody against the KDEL sequence, a specific C-terminal sequence (K/HDEL) in the ER-resident proteins (46) and an antibody against an ER-located ATPase A. albopictus TER94 (AalbTER94) (47). The production of processed viral proteins in the ER, which was detected by an NS2B antibody, was significantly reduced by the expression of DENV2 NS4A. Nonetheless, coexpression of AaHRD1, rather than AaHRD1-C328S, offset the NS4A-mediated suppression of viral protein expression in the ER, as detected by an immunofluorescence assay (Fig. 3 F and G). Moreover, the ER from infected cells was isolated and then the abundance of ER-associated flavivirus proteins was further measured by an immunoblotting assay (Fig. 3H). Altogether, NS4A interrupts the expression of processed flavivirus proteins in the ER, thus inhibiting viral replication. Therefore, AaHRD1 can cause the instability of flavivirus NS4A by ubiquitination system, thus facilitating flavivirus infection in mosquitoes.
Fig. 3.

NS4A suppresses viral replication by interrupting the expression of processed flavivirus proteins. (A) Overexpression of NS4A inhibited DENV2 infection in C6/36 cells. C6/36 cells stably expressing NS4A or GFP were infected with DENV2 at 0.001 MOI for the indicated time. Viral loads were assessed via qPCR. (B) Schematic of proteins involved in endoplasmic reticulum (ER) translocation in eukaryotes. (C) Silencing the components of AaSRα-Sec61 complex inhibited DENV2 infection in A. aegypti. Mosquitoes were inoculated with dsRNAs against the ER translocation complex, followed by inoculation with DENV2 3 d later. Viral loads were determined via qPCR. (D) NS4A interacted with AaSRα. S2 cells were cotransfected with NS4A and AaSRα. Cell lysates were subjected to IP with anti-V5 antibodies and then analyzed via WB. (E) NS4A interrupted the association of AaSRα with AaSRP54. S2 cells were transfected with the indicated plasmids. Cell lysates were subjected to IP with anti-V5 antibodies and analyzed via WB. (F–H) AaHRD1 offset the NS4A-mediated inhibition of expression of processed viral proteins in the ER. C6/36 cells were transfected with indicated plasmids and then infected with DENV2 at 0.1 MOI. At 48 h postinfection, viral protein NS2B and ER markers were stained with Alexa Fluor 488-conjugated anti-rabbit IgG and Alexa Fluor 594-conjugated anti-mouse IgG, respectively. (Scale bars, 10 μm.) (F) Representative immune fluorescence images from four biological replicates were shown. NS2B-positive cells on ER were counted by ImageJ, and data were analyzed statistically using the unpaired t test (G). C6/36 cells were transfected with indicated plasmids and then infected with DENV2 at 0.1 MOI. ER was extracted and then the abundance of ER-associated flavivirus protein was analyzed via WB (H). (D, E, and H) These experiments were repeated three times with similar results. (A and G) Data are presented as the mean ± SEM and were analyzed statistically using the unpaired t test. (C) Viral loads were statistically analyzed via the Mann–Whitney U test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s., not significant (P > 0.05).
HRD1 Facilitates Flavivirus Infection in Mammalian Hosts.
HRD1 is highly evolutionarily conserved between arthropod vectors and mammalian hosts (SI Appendix, Fig. S10A). We therefore assessed the role of HRD1 in flavivirus infections of mammals. The Homo sapiens HRD1 (HsHRD1) gene was knocked down in human lung carcinoma A549 cells by a siRNA transfection (Fig. 4A and SI Appendix, Fig. S10B). Knockdown of the HsHRD1 gene did not influence the cell viability in A549 cells (SI Appendix, Fig. S10C). Thus, silencing HsHRD1 significantly impaired the infection of DENV2 (Fig. 4B) and ZIKV (Fig. 4C), indicating a proviral role of HsHRD1 in flavivirus infection. We next assessed the interaction between HsHRD1 and DENV2 NS4A. The plasmids encoding NS4A and HsHRD1 were cotransfected into HEK293T cells. An immunoprecipitation assay demonstrated that HsHRD1 interacted with DENV2 NS4A in an ectopic expression system (SI Appendix, Fig. S10D). The interplay of endogenous proteins was further validated in the lysates of DENV2-infected A549 cells (Fig. 4D). Consistent with the findings in mosquitoes, ectopically expressed DENV2 NS4A was highly ubiquitinylated in human cells. The 329th substitution from cysteine (C) to serine (S) (C329S) caused the catalytic inactivation of HsHRD1 (39). The C329S mutation of HsHRD1 (HsHRD1-C329S) offset the increase in ubiquitination on DENV2 NS4A (Fig. 4E). In the CHX chase assay, HsHRD1 reduced the amount of ectopically expressed DENV2 NS4A in human HEK293T cells; nonetheless, HsHRD1-C329S lost the activity to induce the ubiquitination-mediated instability of NS4A (Fig. 4F). The aforementioned results indicated the specificity of HsHRD1 in the instability of flavivirus NS4A protein by ubiquitination system. Consistently, the 80th lysine residue of NS4A was the ubiquitination site of HsHRD1 (Fig. 4G). The DENV2-16681-NS4A-K80R mutant showed much lower replication than its parental 16681 strain in A549 cells (SI Appendix, Fig. S10E). Subsequently, a type I and II interferon receptor-deficient (ifnagr−/−) C57BL/6 (AG6) mouse model was footpad infected with DENV2-16681-NS4A-K80R or its parental 16681 strain. The K80R substitution resulted in lower DENV2 viremia (Fig. 4H), significant viral attenuation in multiple mouse tissues (Fig. 4 I and J), and much lower mortality of infected animals (Fig. 4K). Subsequently, the replication of either JEV-SA14-NS4A-K79R or parental strains was assessed in the AG6 mice. Similar results were found in the JEV infection (SI Appendix, Fig. S11 A–D). The results further validated the evolutionarily conserved role of HRD1-mediated NS4A instability in flavivirus infection and pathogenesis.
Fig. 4.

HsHRD1 enhances flavivirus infection in mammalian hosts by ubiquitylating NS4A and causing its instability. (A) Identification of HsHRD1 siRNA deficiency via WB. A549 cells were transfected with indicated siRNAs for 36 h. Gene expression was measured via WB. (B and C) Silencing HsHRD1 suppressed DENV2/ZIKV infections in A549 cells. A549 cells were transfected with indicated siRNAs and then infected with DENV2 or ZIKV at 0.01 MOI. The DENV2 (B) or ZIKV (C) viral load was assessed at 48 h postinfection via qPCR. (D) NS4A interacted with endogenous HsHRD1 in DENV2-infected A549 cells. A549 cells were infected with DENV2 (MOI = 0.1) for 3 d, and cell lysates were subjected to IP with anti-HsHRD1 antibodies and analyzed via WB. (E) HsHRD1 enhanced the ubiquitination of NS4A. HEK293T cells were transfected with indicated plasmids. IP was conducted with anti-Flag antibodies, and NS4A ubiquitination levels were measured. (F) HsHRD1 caused the instability of NS4A. HEK293T cells were transfected with indicated plasmids and treated with CHX (10 μg/mL) for the indicated time, and cell lysates were subjected to WB. (G) HsHRD1 ubiquitylated the 80th lysine residue of DENV2 NS4A. HEK293T cells were transfected with indicated plasmids. IP was conducted with anti-Flag antibodies, and the level of NS4A ubiquitination was determined. (H–K) The K80R substitution in DENV2 NS4A reduced pathogenicity in AG6 mice. Four-week-old male AG6 mice were infected with 5 × 104 f.f.u. of NS4A or NS4A-K80R strain by footpad inoculation. Blood samples were collected for DENV2 viremia assessment from Day 1 to Day 7 (H). Infected mice were killed at 2 d postinfection, and the viral load in tissues was determined by qPCR (I and J). The survival rates of the mice were recorded daily (K). (A and D–G) These experiments were repeated three times with similar results. (H) n = 4 (NS4A) and n = 5 (NS4A-K80R) biological replicates. (I and J) n = 5 (NS4A) and n = 4 (NS4A-K80R) biological replicates. (K) n = 10 biological replicates. (B and C) Data are presented as the mean ± SEM and were analyzed statistically using the unpaired t test. (H–J) Data were analyzed statistically using the Mann–Whitney U test. (K) The data were analyzed statistically using the log-rank (Mantel–Cox) test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s., not significant (P > 0.05).
Pharmacological Inhibition of HRD1 Interrupts DENV2 Infection in Mammalian Hosts and Mosquitoes.
The ultimate goal of this study was to inhibit flavivirus infections in both mammalian hosts and mosquitoes by pharmacologically targeting HRD1. A small molecule named LS-102 was identified by high-throughput screening to specifically inhibit the E3 ligase activity of HRD1. LS-102 showed high safety in testing animals and thus has been proposed as a drug candidate in mouse models against HRD1-related diseases, such as rheumatoid arthritis, liver cirrhosis, and sarcoglycanopathy (48–51). Since HRD1 has been identified as an evolutionarily conserved factor for flavivirus infection, we therefore assessed whether pharmacological inhibition of HRD1 by LS-102 treatment may interrupt DENV2 infection in hosts and mosquitoes. As an HRD1 inhibitor, incubation with LS-102 offset the NS4A ubiquitination mediated by AaHRD1 and HsHRD1 (SI Appendix, Fig. S12 A and B) and maintained NS4A stability in both experimental settings through a CHX chase assay (SI Appendix, Fig. S12 C and D), suggesting the antagonistic activity of LS-102 on both mosquito and mammalian HRD1s. Subsequently, we measured the EC50 of LS-102 in C6/36 cells and A549 cells and the EC50 of LS-102 were 2.757 μM and 1.391 μM, respectively (SI Appendix, Fig. S13 A and B). We further investigated the effect of LS-102 on DENV2 infection of A. aegypti mosquitoes. Thoracic microinjection of LS-102 with DENV2 impaired infection in mosquitoes in a dose-dependent manner (Fig. 5A). Oral feeding of LS-102 with a blood meal largely reduced the DENV2 prevalence in A. aegypti (Fig. 5B). The survival of mosquitoes was not influenced by inoculation of LS-102 by either thoracic microinjection or oral feeding (SI Appendix, Fig. S13 C and D). We next assessed the pharmacological effect of LS-102 in DENV2 infection and the pathogenesis of AG6 mice. After being infected with 5 × 104 f.f.u. DENV2 16681 strain, the animals were intraperitoneally inoculated with LS-102 (15 mg/kg) every day post infection. Notably, the daily treatment of LS-102 resulted in a significant reduction in DENV2 viremia (Fig. 5C) and much lower DENV2 burdens in mouse tissues (SI Appendix, Fig. S13 E–H), thus reducing the mortality of infected AG6 mice (Fig. 5D). Subsequently, we assessed the potential prophylactic effect of LS-102 on DENV2 infection and the pathogenesis of AG6 mice. The animals were intraperitoneally inoculated with LS-102 (15 mg/kg) every day beginning 7 d before infection. After being infected with the 5 × 104 f.f.u DENV2 16681 strain, the animals were continuously treated daily with LS-102 (15 mg/kg) for 7 d. Notably, LS-102 treatment resulted in a 10-fold reduction in DENV2 viremia and viral burden in animal tissues (SI Appendix, Fig. S13 I–M), thus enhancing the survival of infected AG6 mice (SI Appendix, Fig. S13N).
Fig. 5.

The specific inhibitor of HRD1 LS-102 impairs DENV2 infection in mammalian hosts and mosquitoes. (A and B) LS-102 interrupted DENV2 infection in mosquitoes. Mixture of DENV2 and dose gradient LS-102 was microinjected into A. aegypti. Viral loads were measured via qPCR (A). LS-102 mixed with human blood (50% v/v) and supernatant from DENV2-infected Vero cells (50% v/v) were used to feed A. aegypti via an in vitro blood-feeding system. After a blood meal for 8 d, viral load was determined via qPCR (B). (C and D) LS-102 inhibited DENV2 (16681 strain) infection in AG6 mice. Four-week-old female AG6 mice were infected with 5 × 104 f.f.u. of the DENV2 16681 strain by footpad inoculation and intraperitoneally inoculated with LS-102 (15 mg/kg) dissolved in corn oil every day postinfection. Blood samples were collected from infected mice for viremia assessment from Day 1 to Day 7 using qPCR, Data were combined with two independent experiments (C). The survival rates of mice were recorded daily (D). (E) Schematic of the DENV2 acquisition model. (F–H) DENV2-infected mice treated daily with LS-102 reduced the prevalence of DENV2 infection in blood-fed mosquitoes. Four-week-old female AG6 mice were treated as described in (E), blood samples were collected for viremia detection from Day 1 to Day 6 by qPCR (F). Fed mosquitoes were maintained for an additional 8 d and then the DENV2 loads (G) and prevalence (H) were determined. Data were combined with two independent experiments. (G) The number at the top of each column represents the infected number/total number. d.p.i., days post inoculation. (C) n = 4 (Control) and n = 5 (LS-102) biological replicates. (D) n = 8 biological replicates. (F) n = 6 biological replicates. (A, B, and G) Viral load was analyzed statistically using the Mann–Whitney U test. (B and G) The limit of detection is illustrated by the black dashed line. (C and F) Data are presented as the mean ± SEM and were analyzed statistically using the Mann–Whitney U test. (D) The data were analyzed statistically using the log-rank (Mantel–Cox) test. (H) The data were analyzed statistically using Fisher’s exact test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s., not significant (P > 0.05).
We finally assessed the pharmacological effect of LS-102 on the DENV2 lifecycle via an “infected host-mosquito” transmission model (Fig. 5E). The AG6 mice were footpad infected with 5 × 104 f.f.u. DENV2 16681 strain and then treated daily with LS-102 (15 mg/kg) via intraperitoneal injection. Consistently, treatment with LS-102 reduced DENV2 viremia (Fig. 5F). The infected mice were subjected to daily biting for 30 min from 1 to 6 d post infection (Fig. 5E). The fed mosquitoes were reared for an additional 8 d and then collected to detect DENV2 loads. Notably, the daily treatment of LS-102 in DENV2-infected hosts largely reduced the prevalence of DENV2 infection in blood-fed mosquitoes (Fig. 5 G and H). Overall, our data suggest that the molecular compound LS-102 acts as a potential drug candidate to synchronically interrupt HRD1, which is evolutionarily conserved in both hosts and mosquitoes, can effectively inhibit DENV infection in hosts and mosquitoes.
Discussion
Many positive-stranded RNA viruses, such as flaviviruses, encode polyproteins, which are then cleaved into individual functional proteins by host proteases and viral NS2B/NS3 protease (6). Cleavage produces an equal amount of each viral protein; nonetheless, the needs of structural proteins are much higher than those of nonstructural proteins (9, 10). Thus, viruses must exploit a strategy to remove extra nonstructural proteins because of the high toxicity of nonstructural proteins to the normal status of host cells. Homeostasis of viral protein levels is essential for viral propagation (10). The endoplasmic reticulum (ER)-associated degradation (ERAD) pathway is an evolutionarily conserved quality control system that recognizes misfolded ER proteins and transports them to the cytoplasm for proteasomal degradation (52). During this process, the targeted substrates are ubiquitylated by ER-associated E3 ubiquitin ligases such as HRD1. Subsequently, ubiquitylated protein substrates are extracted from the membrane by valosin-containing protein (VCP)/p97 and then delivered to the 26S proteasome for degradation (47, 53). Indeed, previous studies have identified that some ER membrane-integrated flaviviral nonstructural proteins can be ubiquitylated and selectively degraded in infected cells (10). In addition, several ERAD components are important for flavivirus replication (54, 55), as disruption of this pathway significantly impaired viral genome replication. However, the molecular mechanisms underlying the roles of these ERAD components in viral genome replication remain largely unknown. In this study, we identified that HRD1 played an evolutionarily conserved role in facilitating flaviviruses infections in both mammalian hosts and mosquitoes. Flavivirus NS4A inhibits the expression of processed viral proteins in the ER. HRD1s can directly interact with flavivirus NS4A and ubiquitylate a conserved lysine residue of NS4A for instability to facilitate flavivirus infection. The conserved lysine residue substitution largely reduced both prevalence of infection in mosquitoes and the viral loads in AG6 transgenic mice. Nonetheless, this lysine residue may be involved in other aspects of NS4A function independent of HRD1-mediated ubiquitylation, which might affect the replication of the mutant. The comprehensive role of this lysine residue remains to be understood in further investigation. Overall, this study demonstrated that flaviviruses have evolved a sophisticated mechanism to exploit the ubiquitination system to balance the homeostasis of viral proteins for their own advantage.
As a nonstructural protein, flavivirus NS4A can induce the remodeling of the cellular membrane and hijack various cellular signaling pathways, including counteracting the interferon response and modulating the unfolded protein response (UPR) to facilitate viral replication (56). Nonetheless, the accumulation of NS proteins during the later stage of infection can interrupt the ER homeostasis. For example, the NS2 of HCV inhibited its own replication by triggering ER stress (57). Accumulation of the NS proteins of JEV suppressed viral genome replication independent of the UPR, and overexpression of ZIKV NS proteins also impaired viral replication (10, 58). According to our results, NS4A plays a “two-edged sword” in flaviviral replication, e.g., NS4A may facilitate viral replication in the early stage; nonetheless, its accumulation plays an opposite role in the later stage of infection. Intriguingly, the precise mechanism switching the role of NS4A in flaviviral infection remains to be further investigated.
Indeed, flaviviruses exploit the ubiquitination system throughout the stages of their replication cycle in both host and mosquito cells, thereby enhancing their replication and increasing their pathogenicity. For example, ubiquitination of structural proteins is important for the entry process of many flaviviruses. The ubiquitination on the lysine 38 residue (K38) of the envelope (E) protein of ZIKV, which is conserved in Flavivirus members, plays a direct role in viral entry, suggesting that flaviviruses may use ubiquitinated E proteins to promote interactions with receptors, thus enhancing virus entry, replication and pathogenesis (22). NS4A of tick-borne encephalitis virus (TBEV) requires ubiquitination to interact with STAT1/2, thus interrupting the activation of infection signaling by STAT1/2 phosphorylation and dimerization (59). In addition to the roles of ubiquitylated viral proteins in flavivirus replication and pathogenesis, flaviviral components also regulate the ubiquitination of host factors to facilitate viral infection. Subgenomic flavivirus RNA (sfRNA) binds to TRIM25 and prevents its deubiquitination (60). Tripartite motif-containing protein 5 (TRIM25) is well established as an E3 ubiquitin ligase that ubiquitinates RIG-I for downstream signaling to induce interferon (61). Since TRIM25 can be repressed at a ubiquitinated status, the expression of sfRNA maintains TRIM25 repression, impairing the activation of RIG-I and interferon production (60). In addition to mammalian hosts, accumulating evidence indicates that ubiquitination plays an important role in flavivirus-mosquito interactions. For example, many genes of the ubiquitination proteasome system are up-regulated during WNV infection of Culex mosquito cells, including Culex Cul-4 and the orthologs Cullin 4A and Cullin 4B. Culex Cul-4 may function as a negative regulator of the mosquito JAK/STAT pathway by mediating STAT degradation (62), suggesting evolutionarily conserved roles of the ubiquitination system in flavivirus-host interactions.
Identification of the specific ubiquitination factors that are commonly required for viral infection may provide intervention targets for the development of broad-spectrum antiviral drugs. Essential components of the ubiquitin-proteasomal system have been successfully targeted in cancer therapeutics by proteasomal inhibitors such as bortezomib, carfilzomib, and ixazomib (63–65). Halofuginone, as a potential drug, has been identified to induce the proteasomal degradation of TMPRSS2 via the E3 ubiquitin ligase complex DDB1-CUL4-associated factor DCAF (66). Since TMPRSS2 is a serine protease that promotes SARS-CoV entry by proteolytic cleavage of the spike proteins (67, 68), the study emphasized the potential of an E3 ubiquitin ligase as a therapeutic target for the prevention of coronaviral diseases. In this study, we found that the HRD1 antagonist LS-102, which was initially identified as a potential drug compound for the treatment of rheumatoid arthritis, effectively interrupted DENV2 infection in both AG6 mice and A. aegypti mosquitoes. This study provides insight into the role of the ubiquitination system in flaviviral infection and offers a therapeutic drug candidate to combat mosquito-borne flaviviral diseases.
Materials and Methods
Acquisition of Human Blood.
The blood used for mosquito oral feeding was collected from healthy donors who provided written informed consent. The full study protocol was approved by the local ethics committee at Tsinghua University (69), and ethics permit numbers are 20200057.
Mice, Mosquitoes, and Viruses.
C57BL/6 mice deficient in type I and II interferon receptors (AG6 mice) were donated by the Institute Pasteur of Shanghai, Chinese Academy of Sciences. They were bred and maintained in a pathogen-free environment and all animal operations were approved by the Institutional Animal Care and Use Committee of Tsinghua University. A. aegypti (the Rockefeller strain), C. quinquefasciatus and A. albopictus (the Jiangsu strain) were reared in a sugar solution at 28 °C and 80% humidity following published standard rearing procedures. Female mosquitoes were subjected to further investigation. DENV2 (New Guinea C strain, AF038403.1; 16681 strain, U87411) and ZIKV (PRVABC59 strain, KU501215) were used in this study. DENV2 with a mutation in the NS4A 80th residue from lysine to arginine was generated in an infectious clone of 16681, named DENV2-16681-NS4A-K80R; a JEV SA14 strain infectious clone with the substitution in the NS4A 79th residue from lysine to arginine was constructed (JEV-SA14-NS4A-K79R), as described previously (62). The viral titers were determined by a plaque formation assay or focus forming assay (FFA) and the 50% mosquito infection dose (M.I.D.50), as described previously (70–72).
Cells and Reagents.
Drosophila melanogaster S2 cell and A. aegypti Aag2 cells were cultured at 28 °C in Schneider’s Drosophila medium (SDM, 21720-024, Gibco). A. albopictus C6/36 cells, Vero cells, A549 cells and HEK293T cells were cultured in DMEM (11965118, Gibco). C6/36 cell lines stably expressing NS4A or GFP were maintained in DMEM with puromycin (20 μg/mL, MCE). Cxq-1 cells were cultured at 28 °C in M199 complete medium (12350039, Gibco). All kinds of medium were supplemented with 10% heat-inactivated fetal bovine serum and 1% antibiotic-antimycotic. All cell lines were authenticated by the American Type Culture Collection (ATCC) and were free of mycoplasma contamination. LS-102 and Cycloheximide (CHX) were dissolved in 100% dimethyl sulfoxide (DMSO) and then diluted in DMEM or SDM for in vitro studies. They were purchased from Sigma-Aldrich and MCE, respectively.
Gene Silencing and Viral Infection in Mosquitoes.
Female mosquitoes were first anesthetized on a cold tray, and 1 μg/300 nL of dsRNA was microinjected into their thoraxes. A more detailed process for gene silencing has been described elsewhere (71). A total of 10 M.I.D50/300 nL virus was intrathoracically microinjected into per mosquito 3 d later. The gene silencing efficiency was assessed by qRT-PCR and western blot. The primers used for dsRNA synthesis and gene detection are shown in SI Appendix, Table S3.
Membrane Blood Feeding.
Fresh human blood was collected in heparin-coated tubes (367884, BD Vacutainer) at the Hospital of Tsinghua University. The blood (50% v/v) was mixed with viruses (50% v/v) for mosquito oral feeding via a Hemotek membrane feeding system (6W1, Hemotek). Engorged female mosquitoes were selected and transferred into a new container. These mosquitoes were maintained for an additional 8 d and then killed for detection of the infection rate via qPCR.
qPCR Detection.
Total RNA was extracted from homogenized mosquitoes, cell lysates, or homogenized mouse tissue samples using the Multisource RNA miniprep kit (AP-MN-MS-RNA-250, Axygen) and reverse transcribed into complementary DNA via an iScript cDNA synthesis kit (170-8890, Bio-Rad). A TaqMan or SYBR Green qPCR amplification kit was used to quantify viral genomes. The sequences of the primers and probes used in this study are shown in SI Appendix, Table S3.
SDS-PAGE and Western Blotting Analysis.
Mosquitoes and cell samples were collected and lysed with lysis buffer (87787, Thermo Scientific) and then incubated on ice, centrifuged, and finally boiled. The prepared samples were separated by 12% SDS-PAGE and then transferred onto polyvinylidene fluoride membranes (1620177, Bio-Rad). The membrane was blocked with 5% skim milk for 30 min, and then incubated with primary antibody, followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit (AS014, ABclonal) or anti-mouse IgG (AS003, ABclonal) for 1 h. The primary antibody were used as follows: mouse anti-AaHRD1, anti-VCP (AalbTER94, 2649S, CST), rabbit anti-V5 (13202S, CST), mouse anti-Ubiquitin (3936 T, CST), mouse anti-KDEL ER marker (sc-58774, Santa Cruz), rabbit anti-HsHRD1 (A2605, ABclonal), mouse anti-GFP (AE012, ABclonal), rabbit anti-Ubiquitin (A19686, ABclonal), rabbit anti-Myc (AE009, ABclonal), rabbit anti-HA (AE036, ABclonal), rabbit anti-Flag (AE004, ABclonal), rabbit anti-Actin (A2066, Sigma), mouse anti-GAPDH (AC033, ABclonal), rabbit anti-NS1, anti-NS2B, anti-NS3, anti-NS4A, anti-NS4B, anti-NS5 (GTX639322, GTX124246, GTX124252, GTX132069, GTX124250, GTX103350, GeneTex). Goat anti-rabbit IgG and goat anti-mouse IgG secondary antibody were used.
Expression and Purification of Recombinant Protein.
The genes encoding DENV2 nonstructural proteins were amplified from cDNA of the DENV2 NGC strain and then cloned and inserted into a pXJ40 vector. After codon optimization, full-length DENV2 NS4A/mutants, NS2A, and JEV NS4A/mutants were then cloned and inserted into insect vectors for expression in insect cells. C6/36 cells were transfected with recombinant plasmids encoding NS4A or GFP and then selected and maintained in DMEM with puromycin (20 μg/mL) to generate stable cells. Plasmids encoding AaHRD1/mutants, AaSRα, AaSRP54, CqHRD1/mutants, and HsHRD1/mutants were cloned and inserted into insect vectors and pXJ40 vector, respectively. The gene encoding AaHRD1 was codon optimized and then cloned and inserted into the pXJ40 vector for expression in HEK293T cells. AaHRD1-ΔTM (deficient in transmembrane domains) was cloned and inserted into the pET28a(+) vector and expressed in the E. coli BL21 DE3 strain using 500 μM IPTG for 4 h at 37 °C to generate inclusion bodies. The inclusion bodies were broken by supersonic lysis buffer (5 mM Tris-HCl, 1 mM EDTA, 30 mM NaCl), washed with Buffer I (20 mM Tris-HCl, 5 mM EDTA, 100 mM NaCl) with or without Triton X-100 three times, and then dissolved in 8 M urea. Sequences of primes for plasmids’ construction are shown in SI Appendix, Table S4.
Antiserum.
The recombinant AaHRD1-ΔTM protein was emulsified in complete Freund’s adjuvant and intraperitoneally injected into 6-wk-old female BALB/c mice at 100 μg/animal/injection. Animals were boosted three times with 80 μg/animal/injection in incomplete Freund’s adjuvant, twice at 2-wk intervals and the last time at 1 wk. Sera were collected 5 d after the last boost and then used for western blot and immunoprecipitation analyses.
Plaque Formation Assay.
The titers of DENV2 (NGC strain) and ZIKV were determined by a plaque formation assay. Culture supernatants from infected cells were collected and then filtered through a 0.22-μm filter (SLGP033RB, Millipore) to remove cell debris. More details about the plaque assay have been described previously (71).
FFA.
FFA was conducted as described previously (73). In brief, Vero cells were cultured in 24-well plates overnight and then infected with 250 μL of 10-fold serially diluted viruses at 37 °C for 2 h. The cells were then overlaid with overlay medium and cultured continuously. Subsequently, cells were washed with 1× PBS after removing the overlay medium, followed by fixation with 4% (w/v) fixative solution (P1110, Solarbio), and then permeabilized and incubated with 4G2 at 4 °C overnight. After incubation with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (H + L) for 1 h, the focus-forming spots were examined and counted on the microscope after staining with True Blue (Sera Care, 5510-0053).
Immunoprecipitation Assays.
For coimmunoprecipitation (Co-IP) assays, human cells or insect cells were transfected with indicated plasmids. Cell lysates were prepared in 500 μL IP lysis buffer as described for WB analysis 48 h later. First, 40 μL lysates were removed for the input assay, and the remaining lysates were incubated with anti-NS4A antibodies or antibodies coupled with magnetic beads [anti-Flag (P2215, Beyotime), anti-HA (HY-K0201, MCE), anti-Myc (M047-11, MBL) or anti-V5 (P2141, Beyotime)] at 4 °C. Then, the beads were collected by DynaMag™-2 (12321D, Invitrogen) and washed three times with wash buffer. For detection the interaction of viral proteins with endogenous AaHRD1 or HsHRD1 in DENV2-infected cells, cell lysates were incubated with HRD1s antibodies overnight at 4 °C. Then, protein A/G magnetic beads (HY-K0202, MCE) were added for 2 h of incubation. After washing with buffer, the samples were boiled and subjected to WB analysis.
Immunofluorescence Analysis.
C6/36 cells and Aag2 cells were cultured overnight and then transfected with indicated plasmids, dsRNAs or infected with DENV2. Cells were harvested and fixed at room temperature and then permeabilized. Subsequently, the cells were blocked with 3% BSA for 30 min and then incubated with the primary antibody at 4 °C overnight, followed by incubating with fluorescence-conjugated secondary antibodies for 1 h at room temperature. The nuclei were stained with DAPI (10236276001, Roche) for 5 min. Cells were finally mounted with Prolong™ Diamond Antifade Mountant (P36941, Invitrogen) and examined on a Leica SP8 STED confocal microscope. The primary antibody was used as follows: mouse anti-AaHRD1, mouse anti-4G2, rabbit anti-NS2B, mouse anti-KDEL, mouse anti-Flag, rabbit anti-Myc. Alexa Fluor 488-conjugated anti-rabbit IgG (A11034, Invitrogen) and Alexa Fluor 594-conjugated anti-mouse IgG (ab150116, abcam) secondary antibody was used.
ER Isolation.
C6/36 cells were transfected with indicated plasmids and then infected with DENV2 at 0.1 MOI for 24 h. The cells were harvested and subjected to ER isolation according to the protocol supplied by the Minute™ ER Enrichment Kit (Invent, ER-036). Equivalent amounts of protein from the ER were analyzed by WB.
siRNA Transfection.
siRNA oligos were ordered from Shanghai GenePharma. The sequences of siRNAs are shown in SI Appendix, Table S3. siRNA oligos were transfected into A549 cells using Lipofectamine™ RNAiMAX (13778075, Invitrogen) transfection reagent following the manufacturer’s instructions.
Mosquitoes Feeding on Infected Mice.
Female mosquitoes were selected and separated into a new container covered by a net and then starved for 24 h before feeding on mice. DENV2-infected AG6 mice were intraperitoneally inoculated with LS-102 and then anesthetized. Subsequently, anesthetized mice were placed on the top of the containers and bitten by mosquitoes for 30 min in the dark. After anesthetization on ice, the engorged mosquitoes were transferred into new containers and maintained for 8 d and then killed for viral detection.
Statistical Analysis.
The animals were randomly divided into different groups. Mosquitoes that died before detection were discarded. The investigators were not blinded to the allocation during the experiments or to the outcome assessment. Descriptive statistics are provided in the figure legends. The results were pooled for further comparison in case of no detection of significant variation. Given the nature of the experiments and the types of samples, nonparametric Mann–Whitney U tests or unpaired t tests were used to assess the differences between continuous variables. Differences in mosquito infection rates were analyzed via Fisher’s exact test. The survival rates of the infected mice were statistically analyzed using the log-rank (Mantel–Cox) test. All analyses were conducted using GraphPad Prism statistical software.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank the core facilities of the Center for Life Sciences and Center of Biomedical Analysis for technical assistance (Tsinghua University). We thank Bin Liang (Center of Biomedical Analysis, Tsinghua University) for providing technical help. We also thank Xianwen Zhang (Shenzhen Bay Laboratory) and professor Feng Cui (Institute of Zoology, Chinese Academy of Sciences) for providing pXJ40 vector and Cxq-1 cells, respectively. This study was supported by grants from the National Key Research and Development Plan of China (2021YFC2300200, 2021YFC2302405, 2022YFC2303200, and 2022YFC2303400), the National Natural Science Foundation of China (32188101, 31825001, 81961160737, and 82102389), Shenzhen San-Ming Project for Prevention and Research on Vector-borne Diseases (SZSM202211023), Science and Technology Project of Southwest United Graduate School of Yunnan (202302AO370010). The New Cornerstone Science Foundation through the New Cornerstone Investigator Program, and the Xplorer Prize from Tencent Foundation.
Author contributions
G.C. designed research; L.W., L.Z., S.F., L.C., G.W., Y.Z., and P.W. performed research; C.L. contributed new reagents/analytic tools; L.W. analyzed data; and G.C. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Daep C. A., Munoz-Jordan J. L., Eugenin E. A., Flaviviruses, an expanding threat in public health: Focus on dengue, West Nile, and Japanese encephalitis virus. J. Neurovirol. 20, 539–560 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weaver S. C., Reisen W. K., Present and future arboviral threats. Antiviral. Res. 85, 328–345 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elrefaey A. M. E., Hollinghurst P., Reitmayer C. M., Alphey L., Maringer K., Innate immune antagonism of mosquito-borne flaviviruses in humans and mosquitoes. Viruses 13, 2116 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ng W. C., Soto-Acosta R., Bradrick S. S., Garcia-Blanco M. A., Ooi E. E., The 5’ and 3’ untranslated regions of the flaviviral genome. Viruses 9, 137 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mukhopadhyay S., Kuhn R. J., Rossmann M. G., A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 3, 13–22 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Neufeldt C. J., Cortese M., Acosta E. G., Bartenschlager R., Rewiring cellular networks by members of the Flaviviridae family. Nat. Rev. Microbiol. 16, 125–142 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia-Blanco M. A., Vasudevan S. G., Bradrick S. S., Nicchitta C., Flavivirus RNA transactions from viral entry to genome replication. Antiviral Res. 134, 244–249 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Yost S. A., Marcotrigiano J., Viral precursor polyproteins: Keys of regulation from replication to maturation. Curr. Opin. Virol. 3, 137–142 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lobigs M., Lee E., Inefficient signalase cleavage promotes efficient nucleocapsid incorporation into budding flavivirus membranes. J. Virol. 78, 178–186 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tabata K., et al. , Endoplasmic reticulum-associated degradation controls virus protein homeostasis, which is required for flavivirus propagation. J. Virol. 95, e0223420 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deshaies R. J., Joazeiro C. A., RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Swatek K. N., Komander D., Ubiquitin modifications. Cell Res. 26, 399–422 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rotin D., Kumar S., Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell. Biol. 10, 398–409 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Smit J. J., Sixma T. K., RBR E3-ligases at work. EMBO Rep. 15, 142–154 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berndsen C. E., Wolberger C., New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 21, 301–307 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Sherpa D., Chrustowicz J., Schulman B. A., How the ends signal the end: Regulation by E3 ubiquitin ligases recognizing protein termini. Mol. Cell 82, 1424–1438 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muller L., Kutzner C. E., Balaji V., Hoppe T., In vitro analysis of E3 ubiquitin ligase function. J. Vis. Exp. 171, e62393 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Cowan A. D., Ciulli A., Driving E3 ligase substrate specificity for targeted protein degradation: Lessons from nature and the laboratory. Annu. Rev. Biochem. 91, 295–319 (2022). [DOI] [PubMed] [Google Scholar]
- 19.Tracz M., Bialek W., Beyond K48 and K63: Non-canonical protein ubiquitination. Cell. Mol. Biol. Lett. 26, 1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valerdi K. M., Hage A., van Tol S., Rajsbaum R., Giraldo M. I., The role of the host ubiquitin system in promoting replication of emergent viruses. Viruses 13, 369 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mukherjee R., Dikic I., Regulation of host-pathogen interactions via the ubiquitin system. Annu. Rev. Microbiol. 76, 211–233 (2022). [DOI] [PubMed] [Google Scholar]
- 22.Giraldo M. I., et al. , Envelope protein ubiquitination drives entry and pathogenesis of Zika virus. Nature 585, 414–419 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dejarnac O., et al. , TIM-1 ubiquitination mediates dengue virus entry. Cell Rep. 23, 1779–1793 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Su W. C., Yu W. Y., Huang S. H., Lai M. M. C., Ubiquitination of the cytoplasmic domain of influenza A virus M2 protein is crucial for production of infectious virus particles. J. Virol. 92, e01972-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Su W. C., et al. , Pooled RNAi screen identifies ubiquitin ligase Itch as crucial for influenza A virus release from the endosome during virus entry. Proc. Natl. Acad. Sci. U.S.A. 110, 17516–17521 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baillet N., et al. , E3 ligase ITCH interacts with the Z matrix protein of lassa and mopeia viruses and is required for the release of infectious particles. Viruses 12, 49 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han Z., et al. , ITCH E3 ubiquitin ligase interacts with ebola virus VP40 to regulate budding. J. Virol. 90, 9163–9171 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yasuda J., Nakao M., Kawaoka Y., Shida H., Nedd4 regulates egress of Ebola virus-like particles from host cells. J. Virol. 77, 9987–9992 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang K., et al. , Interferon-stimulated TRIM69 interrupts dengue virus replication by ubiquitinating viral nonstructural protein 3. PLoS Pathog. 14, e1007287 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Byk L. A., et al. , Dengue virus genome uncoating requires ubiquitination. mBio 7, e00804-16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taylor R. T., et al. , TRIM79alpha, an interferon-stimulated gene product, restricts tick-borne encephalitis virus replication by degrading the viral RNA polymerase. Cell Host Microbe 10, 185–196 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schoebel S., et al. , Cryo-EM structure of the protein-conducting ERAD channel Hrd1 in complex with Hrd3. Nature 548, 352–355 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iida Y., et al. , SEL1L protein critically determines the stability of the HRD1-SEL1L endoplasmic reticulum-associated degradation (ERAD) complex to optimize the degradation kinetics of ERAD substrates. J. Biol. Chem. 286, 16929–16939 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun S., et al. , Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc. Natl. Acad. Sci. U.S.A. 111, E582–E591 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lwande O. W., et al. , Globe-trotting Aedes aegypti and Aedes albopictus: Risk factors for arbovirus pandemics. Vector. Borne. Zoonotic. Dis. 20, 71–81 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu K., et al. , Mosquito defensins enhance Japanese encephalitis virus infection by facilitating virus adsorption and entry within the mosquito. J. Virol. 94, e01164-20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Charrel R., et al. , Differential infectivities among different Japanese encephalitis virus genotypes in Culex Quinquefasciatus mosquitoes. PLoS Negl. Trop. Dis. 10, e0005038 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu X., et al. , Structural basis of ER-associated protein degradation mediated by the Hrd1 ubiquitin ligase complex. Science 368, eaaz2449 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu Y., et al. , ER-localized Hrd1 ubiquitinates and inactivates Usp15 to promote TLR4-induced inflammation during bacterial infection. Nat. Microbiol. 4, 2331–2346 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Luo L., Cui F., Establishment and identification of a new cell line from Culex pipiens quinquefasciatus (Diptera: Culicidae). Acta Entomol. Sinica 61, 79–85 (2018). [Google Scholar]
- 41.Shah P. S., et al. , Comparative flavivirus-host protein interaction mapping reveals mechanisms of dengue and zika virus pathogenesis. Cell 175, 1931–1945.e1918 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Denks K., et al. , The Sec translocon mediated protein transport in prokaryotes and eukaryotes. Mol. Membr. Biol. 31, 58–84 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Linxweiler M., Schick B., Zimmermann R., Let’s talk about Secs: Sec61, Sec62 and Sec63 in signal transduction, oncology and personalized medicine. Signal. Transduct. Target. Ther. 2, 17002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jadhav B., et al. , Mammalian SRP receptor switches the Sec61 translocase from Sec62 to SRP-dependent translocation. Nat. Commun. 6, 10133 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saraogi I., Shan S. O., Molecular mechanism of co-translational protein targeting by the signal recognition particle. Traffic 12, 535–542 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Denecke J., De Rycke R., Botterman J., Plant and mammalian sorting signals for protein retention in the endoplasmic reticulum contain a conserved epitope. EMBO J. 11, 2345–2355 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakatsukasa K., Brodsky J. L., Kamura T., A stalled retrotranslocation complex reveals physical linkage between substrate recognition and proteasomal degradation during ER-associated degradation. Mol. Biol. Cell. 24, S1761–S1768 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fujita H., et al. , The E3 ligase synoviolin controls body weight and mitochondrial biogenesis through negative regulation of PGC-1beta. EMBO J. 34, 1042–1055 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yagishita N., et al. , RING-finger type E3 ubiquitin ligase inhibitors as novel candidates for the treatment of rheumatoid arthritis. Int. J. Mol. Med. 30, 1281–1286 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fujita H., Aratani S., Yagishita N., Nishioka K., Nakajima T., Identification of the inhibitory activity of walnut extract on the E3 ligase Syvn1. Mol. Med. Rep. 18, 5701–5708 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Bianchini E., Fanin M., Mamchaoui K., Betto R., Sandona D., Unveiling the degradative route of the V247M alpha-sarcoglycan mutant responsible for LGMD-2D. Hum. Mol. Genet. 23, 3746–3758 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Preston G. M., Brodsky J. L., The evolving role of ubiquitin modification in endoplasmic reticulum-associated degradation. Biochem. J. 474, 445–469 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Christianson J. C., et al. , Defining human ERAD networks through an integrative mapping strategy. Nat. Cell. Biol. 14, 93–105 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marceau C. D., et al. , Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 535, 159–163 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma H., et al. , A CRISPR-based screen identifies genes essential for west-nile-virus-induced cell death. Cell Rep. 12, 673–683 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klaitong P., Smith D. R., Roles of non-structural protein 4A in flavivirus infection. Viruses 13, 2077 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.von dem Bussche A., et al. , Hepatitis C virus NS2 protein triggers endoplasmic reticulum stress and suppresses its own viral replication. J. Hepatol. 53, 797–804 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu Y., et al. , Intrinsic features of Zika Virus non-structural proteins NS2A and NS4A in the regulation of viral replication. PLoS Negl. Trop. Dis. 16, e0010366 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang Q., et al. , Tick-borne encephalitis virus NS4A ubiquitination antagonizes type I interferon-stimulated STAT1/2 signalling pathway. Emerg. Microbes. Infect. 9, 714–726 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Manokaran G., et al. , Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 350, 217–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gack M. U., et al. , TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446, 916–920 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Chen L., et al. , Neighboring mutation-mediated enhancement of dengue virus infectivity and spread. EMBO Rep 23, e55671 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kisselev A. F., Site-specific proteasome inhibitors. Biomolecules 12, 54 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gandolfi S., et al. , The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 36, 561–584 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Clemens J., et al. , Bortezomib, carfilzomib and ixazomib do not mediate relevant transporter-based drug-drug interactions. Oncol. Lett. 14, 3185–3192 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen Y., et al. , A high-throughput screen for TMPRSS2 expression identifies FDA-approved compounds that can limit SARS-CoV-2 entry. Nat. Commun. 12, 3907 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hoffmann M., et al. , SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e278 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shang J., et al. , Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. U.S.A. 117, 11727–11734 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu X., et al. , A mutation-mediated evolutionary adaptation of Zika virus in mosquito and mammalian host. Proc. Natl. Acad. Sci. U.S.A. 118, e2113015118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu Y., et al. , Transmission-blocking antibodies against mosquito C-type lectins for dengue prevention. PLoS Pathog. 10, e1003931 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cheng G., et al. , A C-type lectin collaborates with a CD45 phosphatase homolog to facilitate West Nile virus infection of mosquitoes. Cell 142, 714–725 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Diamond M. S., et al. , Complement-related proteins control the flavivirus infection of Aedes aegypti by inducing antimicrobial peptides. PLoS Pathog. 10, e1004027 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hammarlund E., et al. , A flow cytometry-based assay for quantifying non-plaque forming strains of yellow fever virus. PLoS One 7, e41707 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.