

Abstract
INTRODUCTION
Synaptic loss is an early prominent feature of Alzheimer's disease (AD). The recently developed novel synaptic vesicle 2A protein (SV2A) PET‐tracer UCB‐J has shown great promise in tracking synaptic loss in AD. However, there have been discrepancies between the findings and a lack of mechanistic insight.
METHODS
Here we report the first extensive pre‐clinical validation studies for UCB‐J in control (CN; n = 11) and AD (n = 11) brains using a multidimensional approach of post‐mortem brain imaging techniques, radioligand binding, and biochemical studies.
RESULTS AND DISCUSSION
We demonstrate that UCB‐J could target SV2A protein with high specificity and depict synaptic loss at synaptosome levels in AD brain regions compared to CNs. UCB‐J showed highest synaptic loss in AD hippocampus followed in descending order by frontal cortex, temporal cortex, parietal cortex, and cerebellum. 3H‐UCB‐J large brain‐section autoradiography and cellular/subcellular fractions binding studies indicated potential off‐target interaction with phosphorylated tau (p‐tau) species in AD brains, which could have subsequent clinical implications for imaging studies.
Highlights
Synaptic positron emission tomography (PET)–tracer UCB‐J could target synaptic vesicle 2A protein (SV2A) with high specificity in Alzheimer's disease (AD) and control brains.
Synaptic PET‐tracer UCB‐J could depict synaptic loss at synaptosome levels in AD brain regions compared to control.
Potential off‐target interaction of UCB‐J with phosphorylated tau (p‐tau) species at cellular/subcellular levels could have subsequent clinical implications for imaging studies, warranting further investigations.
Keywords: Alzheimer's disease, biomarkers, PET ligands, SV2A, synaptic loss, UCB‐J
1. BACKGROUND
Synapses are complex and dynamic molecular entities crucial for neuronal communication and brain homeostasis, with their activity involving specialized machineries regulated by synaptic vesicles (SVs). 1 It has been shown that synaptic complexity can lead to great alterations in neuronal function and activity during healthy and disease states. 2 Synaptic loss, which is one of the early prominent features of Alzheimer's disease (AD), could result from intrinsic mechanistic alterations or be due to the influence of extrinsic factors such as amyloid beta (Aβ) and tau, specifically their soluble forms causing synaptotoxicity. 3 , 4 , 5 , 6 , 7 , 8 , 9 The molecular cascades underlying synaptic loss in AD are still a topic of debate, with studies proposing the involvement of multiple pathways including complement system–mediated inflammatory mechanisms such as synaptic tagging followed by microglial/astrocytic phagocytosis. 10 , 11 Considering the strong correlation of synaptic loss/degeneration with cognitive decline, 12 , 13 there has been a strong focus recently on developing novel synaptic positron emission tomography (PET)‐tracers/biomarkers that could be used clinically to trace synaptic integrity early in the AD continuum for diagnostic and therapeutic interventions. In this context, fluorine‐18 labeled fluorodeoxyglucose (18F‐FDG) glucose metabolism PET is available and used routinely in clinics to monitor neuronal/functional changes in the brain. 14 18F‐FDG‐PET is a valuable tool; however, the emphasis still has been put into looking for more specific markers of synaptic density because the metabolism of 18F‐FDG is not limited to neurons but can also be observed in glial cells. It has been shown that the 18F‐FDG PET signal is sensitive to astroglial metabolism. 15 Moreover, 18F‐FDG PET in general is an indicator of neurodegeneration or neuronal injury (see Aβ, tau, and neurodegeneration (AT(N)) biomarkers criteria), 16 and in principle does not reflect how synaptic function and proteins are affected during disease pathogenesis in the brain.
A novel potential PET‐ligand called UCB‐J was developed by Nabulsi et al. 17 It specifically targets synaptic vesicle 2A protein (SV2A), expressed ubiquitously by SVs throughout all brain areas and, therefore, seems like a viable candidate for tracing early synaptic loss. 17 , 18 SV2A expression also seems to be independent of neurotransmitter phenotype or other synaptic proteins. 19 UCB‐J has recently been heavily in the spotlight with several in vivo 20 , 21 , 22 , 23 and post‐mortem studies 24 , 25 testing its potential as an ideal direct biomarker for synaptic integrity in AD and other proteinopathies. These initial exploratory studies clearly showed the clinical potential of UCB‐J but at the same time demonstrated the complexity of tracing synapses as highlighted by the variation between these studies. For instance, Metaxas et al. 24 showed no difference in UCB‐J binding between AD and control (CN) cases in the frontal cortex autoradiography, whereas Patel et al. 25 demonstrated a 57% reduction in UCB‐J binding in AD cortical homogenate as compared to the CN cases. These contradicting results clearly highlight the immense need of in‐depth validation studies for UCB‐J to cement its status as a standout synaptic density biomarker and imaging tool clinically. Hence, in this study, we performed extensive preclinical validation studies using post‐mortem brain‐imaging techniques (large frozen brain‐section autoradiography) and radioligand binding studies (saturation, competition, and regional distribution) alongside biochemical analyses in AD and CN brains to explore the future clinical potential of UCB‐J as a specific synaptic biomarker for AD. We also analyzed and compared the UCB‐J binding behavior in brain homogenates (BHs) and subcellular P1‐nuclear and P2‐synaptosomal/membrane fractions prepared from AD and CN brains to better understand its binding mechanism in context of in vivo imaging/binding.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional (e.g., PubMed) sources and meeting abstracts and presentations. Although the recently developed novel synaptic positron emission tomography (PET)–tracer UCB‐J, which targets the ubiquitously expressed synaptic vesicle protein 2A (SV2A), has shown great promise in tracking synaptic loss in Alzheimer's disease (AD) in a handful of post‐mortem and in vivo studies, there have been discrepancies between the findings and a lack of mechanistic insight. These relevant citations are appropriately cited.
Interpretation: Our findings demonstrated the high specificity of UCB‐J for SV2A and superiority of subcellular synaptosomal P2 fractions in tracing synaptic loss in post‐mortem AD brains compared to controls. Of interest, our studies also suggested potential off‐target interaction of UCB‐J at the subcellular level with nuclear phosphorylated tau (p‐tau) species in AD brains, which could have subsequent clinical implications for in vivo imaging studies.
Future directions: Further studies are needed to establish the reliability of UCB‐J as a definite marker of synaptic density/loss. This will contribute significantly to understanding the role of synaptic loss in AD and related dementia disorders and reinforce the existing and future diagnostic and therapeutic strategies.
2. METHOD
2.1. Chemicals
3H‐UCB‐J (specific activity [SA] = 76–82 Ci/mmol), 3H‐MK6240 (SA = 45.7 Ci/mmol) and unlabeled UCB‐J and MK6240 were custom synthesized by Novandi Chemistry AB (Södertälje, Sweden). Levetiracetam (#L8668) was purchased from Sigma‐Aldrich AB, Sweden. Unlabeled AV‐1451 was custom synthesized by He Tian (Institute of Fine Chemicals, East China University of Science and Technology, Shanghai, China). All other chemicals were purchased from VWR and Sigma‐Aldrich AB, Sweden.
2.2. Autopsy material
Human frozen brain tissues from CN and AD cases were acquired from the Netherlands Brain Bank (Amsterdam, The Netherlands) and the Brain Bank at Karolinska Institutet (Stockholm, Sweden). The BH stocks (250 mg tissue/mL) were prepared in 1× phosphate‐buffered saline (PBS) buffer (pH 7.4) containing protease/phosphatase inhibitors and were kept frozen at −80°C in aliquots until used for the experiments.
Large frozen whole‐hemisphere brain tissue from one CN and one sporadic AD (sAD) patient were provided by Prof. Bernardino Ghetti, the Neuropathology Core of Dementia Laboratory, Indiana University School of Medicine (Indianapolis, IN, USA). Large frozen whole‐hemisphere brain tissue from one patient with sAD was provided by the Brain Bank at Karolinska Institutet. Please refer to Table 1 for the clinical demographic data on all cases used in this study.
TABLE 1.
Clinical demographic data for subjects used in this study.
| Sex (M/F) | Age (years) | Braak stage | APOE (E/E) | Onset | DS (years) | PMD (h:min) | Regions used | |
|---|---|---|---|---|---|---|---|---|
| For binding and immunoreactivity studies | ||||||||
| Control | ||||||||
| CN1 | F | 55 | 1 | 3/3 | 7:30 | FC, TC, PC, Cb | ||
| CN2 | M | 62 | 1 | 3/3 | 7:20 | FC, TC, PC, Hipp | ||
| CN3 | M | 72 | 2 | N/A | 4:20 | FC, TC, PC, Cb | ||
| *CN4 | M | 79 | 2 | 3/3 | 9:00 | FC | ||
| *CN5 | M | 81 | 2 | 3/3 | 7:55 | FC, TC, PC, Hipp, Cb | ||
| #CN6 | M | 82 | 2 | N/A | 5:55 | FC, TC, PC, Hipp, Cb | ||
| CN7 | F | 82 | 1 | 3/3 | 7:45 | FC, TC, PC, Cb | ||
| CN8 | M | 82 | 2 | N/A | 5:45 | FC, TC, PC, Cb | ||
| *CN9 | F | 84 | 1 | 3/3 | 6:55 | FC | ||
| #CN10 | F | 89 | 1 | N/A | 13:00 | FC, TC, PC, Cb | ||
| 6 M/4F | 76.8 ± 10.6 | 1—2 | 6 E3 0 E4 | 7.3 ± 2.4 | ||||
| AD | ||||||||
| AD1 | M | 62 | 6 | 4/3 | EOAD | 7–9 | 6:45 | FC, TC, PC, Cb |
| AD2 | F | 62 | 6 | 4/3 | EOAD | 7 | 4:45 | FC, TC, Cb |
| #AD3 | F | 64 | 6 | 4/3 | EOAD | 12 | 5:30 | FC |
| #AD4 | M | 68 | 6 | 4/3 | EOAD | 12 | 5:20 | FC |
| #AD5 | M | 77 | 6 | 4/4 | LOAD | 5 | 6:35 | FC |
| AD6 | M | 78 | 5 | 4/4 | LOAD | 7 | 6:35 | FC, TC, PC, Hipp, Cb |
| *AD7 | F | 79 | 5 | 4/4 | LOAD | N/A | 16:00 | FC |
| *AD8 | M | 81 | 5–6 | 4/4 | LOAD | N/A | 17:00 | FC |
| AD9 | F | 85 | 4 | 3/3 | LOAD | 5 | 6:00 | FC, TC, PC, Hipp |
| 5 M/ 4F | 72.8 ± 8.8 | 4–6 | 1 E3 8 E4 | 4EOAD 5LOAD | 8.0 ± 2.7 | 8.1 ± 4.8 | ||
| For large frozen section autoradiography studies | ||||||||
| Control a | F | 56 | N/A | 3/4 | 2:56 | |||
| sADa | F | 57 | N/A | 3/3 | EOAD | 8 | 2:48 | |
| sAD a | F | 79 | 5 | 4/4 | LOAD | N/A | 16:00 |
Note: All data are shown as means ± standard deviation (SD). * and # show CN and AD cases in pooled 1 and 2 samples used for saturation, competition, and immunoblot studies, respectively.
Abbreviations: APOE, apolipoprotein E; Cb, cerebellum; DS, symptom duration based on AD patient's clinical course information; EOAD, early‐onset Alzheimer's disease; FC, frontal cortex; F, female; Hipp, hippocampus; LOAD, late‐onset Alzheimer's disease; M, male, N/A, information not available; PC, parietal cortex; PMD, post‐mortem delay, sAD, sporadic Alzheimer's disease; TC, temporal cortex.
2.3. Subcellular fractionation
Subcellular fractionation protocol is illustrated after Figure 1. Briefly, BH prepared from different brain regions of CN and AD cases were centrifuged at 2800–3200 rpm for 10 min to separate nuclear fraction pellet (P1) and supernatant. The supernatant was again subjected to centrifugation at 11,000–12,000 rpm for 20–25 min to get synaptosomal/membrane pellet (P2) and supernatant (S2; microsomes and cytosolic proteins). The P1 and P2 pellets were resuspended in cold 1× PBS buffer (pH 7.4) containing protease/phosphatase inhibitors. The samples were kept on ice throughout the procedure, and centrifugation was done at 4–7°C. The protein concentration in P1, P2, and S2 fractions were determined using standard Biorad DC protein assay kit (product #5000111) and protocol.
FIGURE 1.
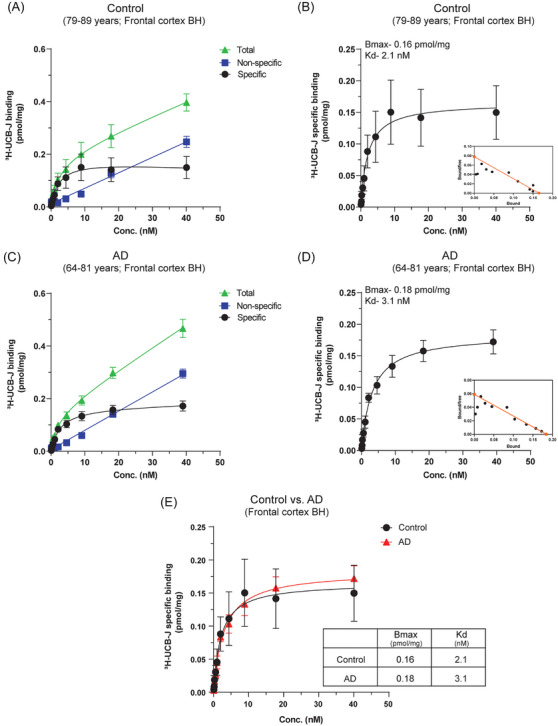
3H‐UCB‐J saturation‐binding studies in CN and AD BHs. 3H‐UCB‐J saturation‐binding studies were performed in the pooled frontal cortex brain tissue homogenates from CN (Pools 1 and 2; 79–89 years) and AD patients (Pools 1 and 2; 64–81 years) using increasing concentrations of 3H‐UCB‐J (0–40 nM). (A–B) and (C–D) show the saturation‐binding curves for the CN and AD cases, respectively, together with the corresponding Scatchard plots (inset). (E) Shows the comparison of 3H‐UCB‐J specific binding in CN and AD cases. Data are presented as means ± SEM. AD, Alzheimer's disease; CN, control; BHs, brain homogenates; Bmax, density of binding sites; Kd, dissociation constant.
2.4. Pooled samples for binding studies
The brain tissues from the frontal cortex of CN and AD cases were pooled to generate large quantities of BH, P1, and P2 fractions needed for the extensive saturation and competition binding studies. Two independent frontal cortex pooled samples were prepared for both CN and AD cases. The clinical information regarding cases used for the pooled samples is presented in Table 1.
CN Pool 1 CN4 + CN5 + CN9 (79–84 years); CN Pool 2 CN6 + CN10 (82–89 years). Pool 1 and 2 combined age range: 79–89 years.
AD Pool 1 AD7 + AD8 (79–81 years); AD Pool 2 AD3 + AD4 + AD5 (64–77 years). Pool 1 and 2 combined age range: 64–81 years.
If the experiments were performed on both Pool 1 and 2, then the combined data along with the combined age ranges was presented in the figures. In cases where the experiments were performed in only Pool 1 or 2, this has been specified in the figure or figure legends.
2.5. Saturation‐binding assays
The saturation‐binding assay was performed as published previously. 26 , 27 , 28 Briefly, BH (0.1 mg tissue) and P1 and P2 fractions (0.1 mg protein) prepared from two, independent pooled frontal cortex samples of CN and AD patients as described above and incubated with increasing concentration of 3H‐UCB‐J (0–40 nM) in 50 mM Tris‐HCl binding buffer pH 7.4 (50 mM Tris‐base, 140 mM NaCl, 2 mM MgCl2) at room temperature for 2 h. Unlabeled (1 μM) UCB‐J was used to determine non‐specific (NSP) binding and to calculate specific binding. The binding reaction was stopped by filtering through glass fiber filters (pre‐soaked for 2–3 h in 0.3% polyethylenimine), followed by three quick washes with cold binding buffer and overnight incubation of the filter in the scintillation liquid. On the next day, the radioactivity in the tubes incubated with reaction filters was counted with a beta scintillation counter (PerkinElmer Tri‐Carb 2910TR). The saturation‐binding curves (total, NSP, specific) were fitted and analyzed to calculate the dissociation constant (Kd) and maximum number of binding sites (Bmax) using the non‐linear regression function of GraphPad Prism 9.0 software. 29 Specific binding data were transformed with GraphPad Prism 9.0 software to prepare the Scatchard plots. For tau‐PET ligands blocking 3H‐UCB‐J saturation‐binding studies, the AD P1 fraction was pre‐blocked with 10 μM of unlabeled AV‐1451 and MK‐6240 on ice for 1 h. After 1 h, the P1 fraction was centrifuged at 2800 rpm for 10–15 min at 4°C to remove excess of free AV‐1451 and MK‐6240. The resulting pellet was resuspended in binding buffer and used for saturation‐binding studies as described above. The AV‐1451 blocking experiment was performed only on AD Pool 1 P1 fraction, whereas MK‐6240 blocking experiment was performed on both AD Pool 1 and Pool 2 P1 fractions.
2.6. Competition binding assays
Competition binding assays for 3H‐UCB‐J were performed on the P1 and P2 fractions (0.05 mg protein) prepared from the pooled frontal cortex of CN (Pool 1; 79–84 years) and AD patients (Pool 1; 79–81 years). The P1 and P2 fractions were incubated with a single concentration of 3H‐UCB‐J (5 nM) along with increasing concentrations of unlabeled UCB‐J and MK‐6240 (10−14 to 10−4) in 50 mM Tris‐HCl binding buffer pH 7.4 (50 mM Tris‐base, 140 mM NaCl, 2 mM MgCl2) for 2 h at room temperature. After the incubation period a similar protocol as the one used for the saturation‐binding assay was done, and the binding was quantified using a scintillation counter.
3H‐MK6240 competition binding assays with unlabeled UCB‐J in the CN and AD Pool 1 P1 fractions (0.05 mg protein) were also performed using the same protocol using 0.5 nM concentration of 3H‐MK6240 against increasing concentrations of unlabeled UCB‐J (10−14 to 10−4). The competition binding data were presented as % of total binding and analyzed using the non‐linear regression competitive‐binding function of GraphPad Prism 9.0 software to determine IC50 (half‐maximal inhibitory concentration) values.
2.7. 3H‐UCB‐J brain regional binding studies
Regional binding studies were performed on the frontal cortex [FC], temporal cortex [TC], and parietal cortex [PC], hippocampus [Hipp], and cerebellum [Cb] BHs (0.1 mg tissue), P1 and P2 fractions (0.01 mg protein) prepared from individual (not pooled) CN (CN1–3, CN5–8, and CN10; mean age 75.6 ± 11.6 years) and AD patients (AD1–2, AD6, and AD9; mean age 71.7 ± 11.6 years). BHs, P1, and P2 fractions from different brain regions were incubated with a single concentration of 3H‐UCB‐J (5 nM) for 2 h at room temperature in 50 mM Tris‐HCl binding buffer pH 7.4 (50 mM Tris‐base, 140 mM NaCl, 2 mM MgCl2). Following 2 h incubation, a similar protocol as the saturation‐binding assay was done, and the binding was quantified using a scintillation counter. NSP binding was determined with 1 μM unlabeled UCB‐J. The binding data was analyzed using GraphPad Prism 9.0 software and presented in terms of 3H‐UCB‐J specific binding (pmol/mg).
2.8. In vitro autoradiography studies
3H‐UCB‐J autoradiography studies on large frozen post‐mortem brain sections from one CN (56 years) and two patients with sAD (57 and 79 years) were performed as reported in our previous studies. 26 , 30 , 31 The large frozen sections were allowed to dry at room temperature for 30–45 min, followed by 1 h incubation with 3H‐UCB‐J (2–5 nM) at room temperature. Afterwards, the sections were rinsed for 5 min, three times in cold 50 mM Tris‐HCl binding buffer pH 7.4 (50 mM Tris‐base, 140 mM NaCl, 2 mM MgCl2) followed by two rinses in cold MiliQ water. The sections were allowed to dry together with a tritium standard (Larodan Fine Chemicals AB, Sweden) for 24 h at room temperature. After 24 h, the dried sections and standards were exposed against a phosphor‐plate for 7 days and then imaged using a BAS‐2500 phosphor imager (Fujifilm, Tokyo, Japan). NSP binding was determined with 1 μM unlabeled UCB‐J and 10 μM Levetiracetam. For semi‐quantitative analyses, the gray matter of regions of interest (ROI) such as frontal and temporal lobe, insula, and hippocampus was manually drawn on the autoradiogram using multigauge software, and the resulting photostimulated luminescence per square millimeter (PSL/mm2) was transformed into fmol/mg using the standard curve to determine the total, NSP, and specific binding of 3H‐UCB‐J in the ROI.
2.9. Immunoblot analysis of CN and AD BHs, P1, and P2 fractions
Phosphorylated tau (p‐tau) and SV2A were analyzed in the frontal cortex P1 and P2 fractions of CN (n = 6; Pool 1 and 2, CN2, CN5–6, and CN10) and AD patients (n = 6; Pool 1 and 2, AD1–2, AD6, and AD9). BHs were also analyzed for p‐tau and SV2A, but only in the CN and AD Pool 1 and 2 (n = 2). Synaptic proteins synaptophysin (SYP) and synaptotagmin I/II (SYT I/II) were analyzed in the frontal cortex BHs, P1, and P2 fractions of CN (n = 2; Pool 1 and 2) and AD patients (n = 2; Pool 1 and 2). Samples were mixed with NuPAGE LDS sample buffer (Invitrogen, #NP0007), incubated for 5 min at 65°C, and briefly spun down before loading the gel. For AT8 immunoblotting, samples were incubated for 5 min at 95°C. Samples (10 μg) were run in NuPAGE 4%–12% Bis‐Tris Gel (Invitrogen, #NP0322BOX) at 60–150 V in NuPAGE MES SDS Running buffer (20×) (Invitrogen, #NP0002). Nitrocellulose membranes were equilibrated in transfer buffer for 5 min before assembling the blot sandwich. The transfer was performed for 1 h at constant 200 mA at room temperature. Then, membranes were washed using MilliQ water, stained using Revert 700 Total Protein Stain (P/N: 926–11,011), and total protein signal was imaged using LICOR Odyssey CLx imaging system. After de‐staining using the manufacturer's instructions, the membranes were blocked for 1–2 h in 5% (w/v) fat‐free milk in TBS‐T (0.1% tween‐20 in TBS at pH 7.4) at room temperature. Membranes were then incubated with AT8 (1:250; Invitrogen #MN1020), anti‐SV2A (1:500; Santa Cruz biotechnology #sc‐376234), anti‐synaptophysin (1:1000, Santa Cruz Biotechnology #sc‐17750), or anti‐synaptotagmin I/II antibody (1:500, Santa Cruz Biotechnology #sc‐393392), either at room temperature for 2 h or overnight at 4°C. Afterwards the membranes were incubated with anti‐mouse secondary antibodies (1:10,000 for LI‐COR antibodies) for 1 h at room temperature. The proteins were visualized with LICOR Odyssey CLx imaging system using the appropriate lasers. Band intensity was analyzed using Empiria software (LI‐COR) on the raw images and normalized for the total protein stain. Protein‐expression levels for all targets, unless stated, are then displayed as means ± standard error of the mean (SEM) of either percentage (%) of protein expression of respective CN or fold‐over total protein.
| Experiment | CN cases used | AD cases used |
|---|---|---|
| Saturation‐binding studies in BH, P1, and P2 fractions | Pools 1 and 2 | Pools 1 and 2 |
| Pre‐blocking saturation‐binding studies in P1 fractions | Pools 1 and 2 | |
| Competition binding studies in P1 and P2 fractions | Pool 1 | Pool 1 |
| Regional Binding studies in BH, P1, and P2 fractions | CN1, CN2, CN3, CN5, CN6, CN7, CN8, and CN10 | AD 1, AD2, AD6, and AD9 |
| Immunoblot analyses in P1 and P2 fractions | Pools 1 and 2, CN2, CN5, CN6, and CN10 | Pools 1 and 2, AD1, AD2, AD6, and AD9 |
Note: Regional binding studies were performed only on different brain regions from individual CN and AD cases. CN/AD Pool 1 and 2 samples were not used in regional binding studies. CN/AD Pool 1 and 2 samples were used for extensive saturation and competition binding studies, as they require much larger quantities of BHs, P1, and P2 fractions.
2.10. Statistical analysis
In regional binding studies, the mean differences across all CN and AD brain regions were determined using unpaired Welch's t‐test with no correction for multiple comparisons. For immunoassays, statistical and correlation analyses were performed using two‐way analysis of variance (ANOVA; Tukey's test) and Pearson correlation coefficient, respectively. A p < 0.05 was considered statistically significant. All the analyses were performed with GraphPad Prism 9.0 software.
3. RESULTS
3.1. 3H‐UCB‐J saturation‐binding studies in CN and AD BHs
The 3H‐UCB‐J saturation‐binding curves (total, NSP, and specific) and Scatchard plots for the frontal cortex CN and AD BHs are illustrated in Figure 1. 3H‐UCB‐J showed good saturation‐specific binding curves for both CN and AD BHs in the concentration range of 0–40 nM. In CN and AD BHs, 3H‐UCB‐J showed one binding site with almost comparable Kd values: CN Kd–2.1 nM and AD Kd–3.1 nM (Figure 1B,D and E). Surprisingly, the AD BH Bmax value of 0.18 pmol/mg was ≈1.12‐fold higher as compared to CN BH Bmax value (0.16 pmol/mg; Figure 1B,D and E) indicating relatively higher binding of 3H‐UCB‐J in AD BH as compared to CN BH. Moreover, we also observed relatively higher NSP binding in AD BH compared to CN BH (compare Figure 1A and C; blue curves). These observations were in complete opposition to the expected lower 3H‐UCB‐J binding in AD brains at the end stages and highlighted the potential interaction to other brain tissue component/proteins.
3.2. 3H‐UCB‐J saturation and competition binding studies in CN and AD subcellular P2 synaptosomal/membrane fraction
Because SV2A is a synaptic membrane protein, we performed subcellular fractionation on the frontal cortex of CN and AD brains as illustrated in Figure 2A to remove other unwanted brain components/proteins and obtain P2 synaptosomal/membrane fraction (P2 fraction) for 3H‐UCB‐J saturation and competition binding studies. The 3H‐UCB‐J total, NSP, and specific saturation‐binding curves and Scatchard plots for the frontal cortex CN and AD P2 fractions are presented in Figure 2B–F. In P2 fractions, 3H‐UCB‐J showed good saturation‐specific binding curves in the concentration range of 0–40 nM and extremely high specific binding in both CN and AD cases (Figure 2C,E and F). The specific binding in CN cases was ≈1.7‐fold higher than in the AD cases (CN Bmax–11.8 pmol/mg vs AD Bmax–6.9 pmol/mg) and showed ≈41% less 3H‐UCB‐J specific binding in AD cases (Figure 2F). The specific binding of 3H‐UCB‐J in P2 fractions were ≈38‐ to 73‐fold higher in AD and CN cases, respectively, as compared to BH saturation‐binding studies (compare Figure 1E with Figure 2F). In addition, the P2 fraction NSP binding was also low in both CN and AD cases and the saturation‐binding curve patterns were also different from BH studies (compare Figure 1A and C with Figure 2B and D). However, similar to BH studies, we still observed one‐binding site with nM affinity in P2 fractions of CN and AD cases (Figure 2C and E; inset). The Kd values were 4.8 nM and 5.1 nM in CN and AD cases, respectively. We further confirmed the presence of one nanomolar affinity binding site in P2 fractions of CN and AD cases with 3H‐UCB‐J versus UCB‐J competition binding studies (Figure 2G). P2 fraction saturation‐binding studies clearly demonstrated the expected loss of 3H‐UCB‐J binding (i.e., synaptic loss) in AD cases as compared to CN cases.
FIGURE 2.
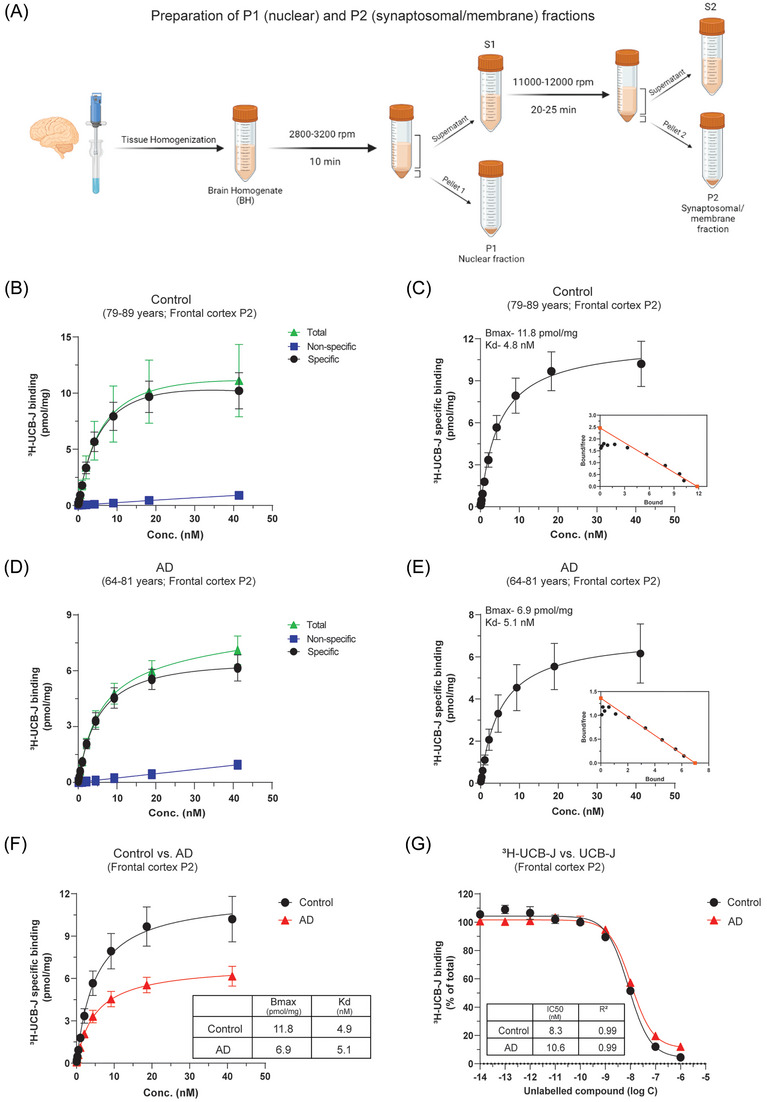
3H‐UCB‐J saturation and competition binding studies in CN and AD brain P2 synaptosomal/membrane fractions. 3H‐UCB‐J saturation‐binding studies were performed in the P2 synaptosomal/membrane fractions (P2) prepared from the pooled frontal cortex brain tissue homogenates from CN (Pools 1 and 2; 79–89 years) and AD patients (Pools 1 and 2; 64–81 years) using increasing concentrations of 3H‐UCB‐J (0–40 nM). (A) Illustration shows the subcellular fractionation protocol for the preparation of P1 nuclear, P2 synaptosomal/membrane, and S2 microsome/cytosolic proteins fractions from brain homogenates. (B–C) and (D–E) show the saturation‐binding curves for the CN and AD cases, respectively, together with the corresponding Scatchard plots (inset). (F) Shows the comparison of 3H‐UCB‐J specific binding in CN and AD cases. (G) 3H‐UCB‐J competition binding studies were performed in the P2 fractions prepared from the pooled frontal cortex brain tissue homogenates from CN (Pool 1; 79–84 years) and AD patients (Pool 1; 79–81 years) using a single concentration of 3H‐UCB‐J (5 nM) and increasing concentrations of unlabeled UCB‐J. Data are presented as means ± SEM. AD, Alzheimer's disease; CN, control; Bmax, density of binding sites; IC50, half‐maximal inhibitory concentration; Kd, dissociation constant.
3.3. 3H‐UCB‐J saturation and competition binding studies in CN and AD subcellular P1 nuclear fraction
Next, we performed similar studies in the frontal cortex subcellular P1 nuclear fraction (P1 fraction) of CN and AD cases (Figure 3). The saturation‐binding curves (total, NSP, and specific) and Scatchard plots showed similar patterns as P2 fraction saturation studies in both CN and AD cases. We observed unexpectedly high 3H‐UCB‐J specific binding in both CN and AD P1 fractions, and the binding in AD was ≈1.53‐fold higher than in CN cases (Figure 3B,D and E). The Bmax values were 16.9 pmol/mg and 25.9 pmol/mg in CN and AD cases, respectively. The specific binding of 3H‐UCB‐J in P1 fractions was ≈1.4‐ to 3.7‐fold higher in CN and AD cases, respectively, as compared to P2 fractions (compare Figure 2F with Figure 3E). In comparison to BH, the P1 fraction specific binding was ≈105‐ to 143‐fold higher in CN and AD cases, respectively (compare Figure 1E with Figure 3E). Despite these differences in specific binding values with regard to BH and P2 fractions, we again observed only one binding site with comparable nanomolar affinity in P1 fractions of CN and AD cases, demonstrated by Scatchard plots and 3H‐UCB‐J versus UCB‐J competition binding studies (Figure 3B,D and F). This higher specific binding in P1 fractions highlighted it as the potential source of off‐target interaction.
FIGURE 3.
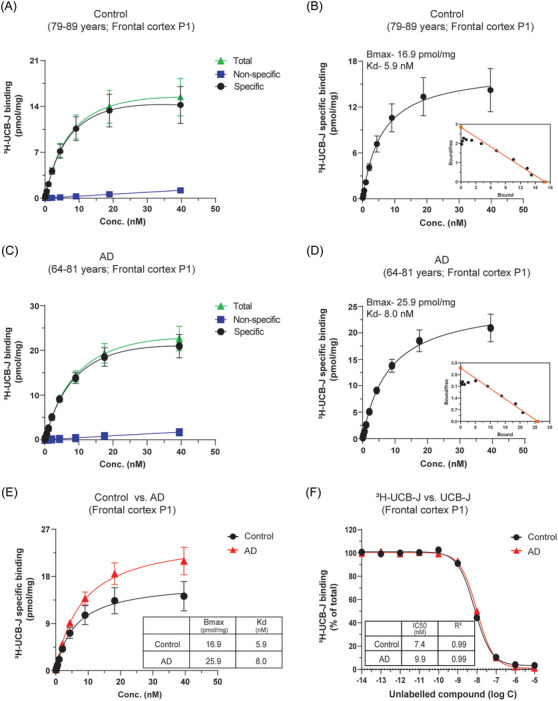
3H‐UCB‐J saturation and competition binding studies in CN and AD brain P1 nuclear fractions. 3H‐UCB‐J saturation‐binding studies were performed in the P1 nuclear fractions (P1) prepared from the pooled frontal cortex brain tissue homogenates from CN (Pools 1 and 2; 79–89 years) and AD patients (Pools 1 and 2; 64–81 years) using increasing concentrations of 3H‐UCB‐J (0–40 nM). (A–B) and (C–D) show the saturation‐binding curves for the CN and AD cases, respectively, together with the corresponding Scatchard plots (inset). (E) Shows the comparison of 3H‐UCB‐J‐specific binding in CN and AD cases. (F) 3H‐UCB‐J competition binding studies were performed in the P1 fractions prepared from the pooled frontal cortex brain tissue homogenates from CN (Pool 1; 79–84 years) and AD patients (Pool 1; 79–81 years) using a single concentration of 3H‐UCB‐J (5 nM) and increasing concentrations of unlabeled UCB‐J. Data are presented as means ± SEM. AD, Alzheimer's disease; CN, control; Bmax, density of binding sites; IC50, half‐maximal inhibitory concentration; Kd, dissociation constant.
3.4. 3H‐UCB‐J regional distribution binding studies in CN and AD BH, P1, and P2 fractions
To further confirm the observations of frontal cortex BH, P1, and P2 fraction studies, we performed 3H‐UCB‐J (5 nM) regional distribution binding studies in five brain regions (frontal cortex [FC], temporal cortex [TC], and parietal cortex [PC], hippocampus [Hipp], and cerebellum [Cb]) from CNs (mean age 75.6 ± 11.6 years) and patients with AD (mean age 71.7 ± 11.6 years) (Figure 4). The findings were complementary as we observed higher 3H‐UCB‐J specific binding in AD FC, PC, and Cb brain regions with BH as compared to CN brains (Figure 4A). Moreover, a significantly lower 3H‐UCB‐J‐specific binding in the BH of AD TC (p = 0.002) and Hipp (p = 0.0003) as compared to CN brains was observed. In the P2 fractions, we observed, as expected, a very evident and clear loss of 3H‐UCB‐J‐specific binding in all AD brain regions as compared to CN brains (Figure 4B). The reduction in 3H‐UCB‐J‐specific binding ranged from ≈26.4% to 70% in AD cortical and hippocampal regions as compared to CN. Again, the loss of 3H‐UCB‐J‐specific binding was significant in AD FC (≈55.9%; p = 0.010), TC (≈50.7%; p = 0.003), and Hipp (≈70.3%; p = 0.044) as compared to CN brains. The observation in P1 fractions was consistent and interesting at the same time; in AD FC brain regions, we again observed higher specific binding for 3H‐UCB‐J as compared to CN brains; however, in all the other AD brain regions the binding was lower as compared to CN brains except in Cb, where the binding was higher (Figure 4C). A significantly lower binding was observed in AD TC (p = 0.003) as compared to CN brains. 3H‐UCB‐J‐specific binding in the cortical regions P1 fractions is still relatively higher than BH and P2 fraction binding in CN and AD brains, further complementing the above saturation‐binding studies.
FIGURE 4.
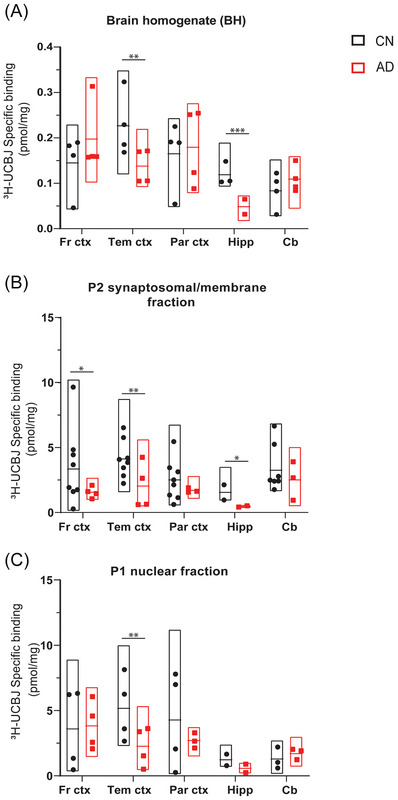
3H‐UCB‐J regional distribution binding studies in CN and AD BHs, P1, and P2 fractions. 3H‐UCB‐J regional distribution binding studies were performed in BHs (0.1 mg tissue), P1, and P2 fractions (0.01 mg protein) of frontal cortex, temporal cortex, parietal cortex, hippocampus, and cerebellum from CN (mean age 75.6 ± 11.6 years) and AD patients (mean age 71.7 ± 11.6 years) using a single concentration of 3H‐UCB‐J (5 nM) and unlabeled UCB‐J (1 μM). The graph shows the comparison of 3H‐UCB‐J‐specific binding (pmol/mg) in BH, P1, and P2 fractions prepared from different regions of CN and AD brains. Each data point represents one individual case. Data are presented as floating box plots (min to max) with means ± SEM for each region of CN and AD brains and analyzed using unpaired Welch's t‐test. * p < 0.05, ** p < 0.004, *** p < 0.0004. AD, Alzheimer's disease; CN, control; Fr ctx, frontal cortex; Tem ctx, temporal cortex; Par ctx, parietal cortex; Hipp, hippocampus; Cb, cerebellum.
3.5. 3H‐UCB‐J autoradiography regional binding studies in CN and sporadic AD (sAD) brain sections
To understand 3H‐UCB‐J regional binding behavior, we performed autoradiography studies in the cortical, subcortical, and hippocampal regions of CN and sAD brains. 3H‐UCB‐J regional binding autoradiograms of large frozen sections from one CN (56 years) and two sAD (57 and 79 years) brains are shown in Figure 5A–C. A visual/qualitative assessment demonstrated extremely specific 3H‐UCB‐J binding in the gray matter along with similar regional distribution pattern in CN and sAD cases (Figure 5A–C). The relative total binding in all regions of sAD cases appeared much higher as compared to CN brain (compare Figure 5A with B and C). To confirm the higher binding in sAD brains as compared to the CN brain, we performed 3H‐UCB‐J binding semiquantitative assessment in the gray matter of ROI such as the frontal lobe, temporal lobe, insula, and hippocampus. We calculated specific, NSP, and total binding in the gray matter of ROI, and the comparative results for each case are presented in Figure 5D. We observed ≈1.4‐ to 2.2‐fold higher total binding in sAD brain ROIs as compared to the CN brain (Figure 5D). The 3H‐UCB‐J NSP binding was determined by co‐incubation of CN and sAD brain sections with 1 μM unlabeled UCB‐J (Figure 5A–C; inset). We observed ≈11%–16% and ≈4%–11% NSP binding in CN and sAD brains ROI, respectively (Figure 5D). The specific binding of 3H‐UCB‐J in ROI was ≈84%–96% demonstrating high specificity of 3H‐UCB‐J in CN and sAD brains (Figure 5D). OF interest, similar to total binding, specific binding in sAD (57 years) was also ≈1.4‐ to 2.3‐fold higher as compared to CN brain in different ROI (compare CN and sAD specific binding values in Figure 5D). As expected, 3H‐UCB‐J binding was also displaceable by 10 μM levetiracetam but to a lower extent as compared to 1 μM unlabeled UCB‐J (Figure 5E and F). These findings further complemented our BH and P1 fraction data and demonstrate an overall increase of 3H‐UCB‐J binding in AD brains as compared to the CN brain.
FIGURE 5.
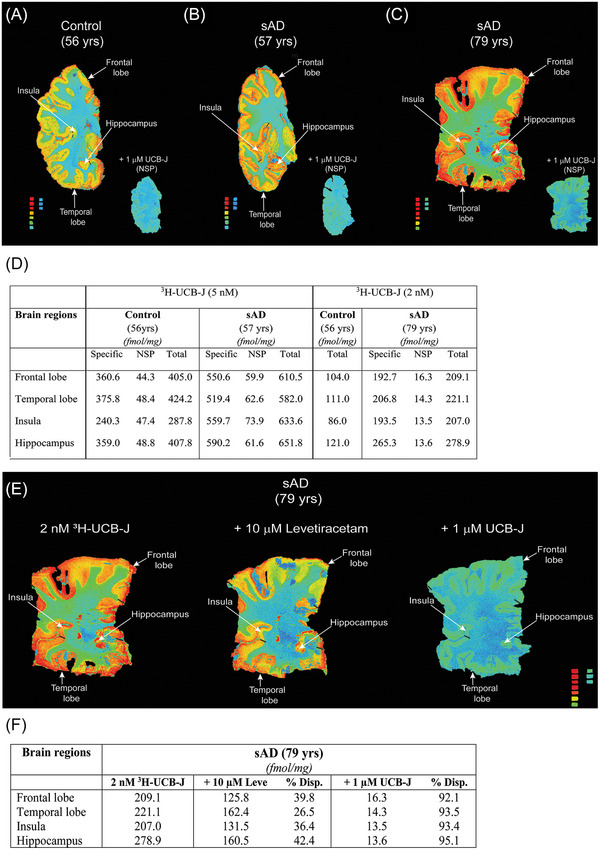
Head‐to‐head comparison of 3H‐UCB‐J autoradiography regional binding studies on large frozen post‐mortem brain sections from one CN and two sAD cases. (A–C) figure demonstrates the total binding of 2–5 nM 3H‐UCB‐J together with NSP binding in the presence of 1 μM unlabeled UCB‐J in different brain regions of CN and sAD cases, respectively. For comparison, 5 nM 3H‐UCB‐J autoradiography images of (A) CN (56 years) and (B) sAD (57 years) were set on the same color/brightness threshold levels of 53713 from the raw images (16 bits: 0–65,535 (color scale). The color scale standard from red to blue represents: 3562 fmol/mg to 7.5 fmol/mg, respectively. (C) sAD (79 years) 3H‐UCB‐J autoradiogram was set on the different color/brightness threshold levels (47545) as compared to (A–B) from the raw images (16 bits: 0–65,535 (color scale)) due to difference in the 3H‐UCB‐J concentration (2 nM). The color scale standard from red to green/blue represents: 3119–3647 fmol/mg to 6.6–7.7 fmol/mg, respectively. (D) 3H‐UCB‐J binding semi‐quantitative analyses in the gray matter of different regions of interest. Regions of interest as presented in the figure were manually drawn to calculate the specific, non‐specific, and total binding values in fmol/mg. (E–G) Autoradiograms and semi‐quantitative analyses showing the displacement of 2 nM 3H‐UCB‐J binding in the presence of 10 μM Levetiracetam and 1 μM in different regions of sAD (79 years) brain, respectively. All autoradiograms in (E) were set at a similar color/brightness threshold level of 47545 for comparison. The color scale standard from red to green/blue represents: 3119–3647 fmol/mg to 6.6–7.7 fmol/mg, respectively. Frontal and temporal lobe regions are highlighted with dark black bars. NSP, non‐specific binding; sAD, sporadic Alzheimer's disease; % Disp., percent displaced.
3.6. Immunoblot analyses of CN and AD BH, P1, and P2 fractions
To gain more mechanistic insight into 3H‐UCB‐J binding in CN and AD frontal cortex BH, P1, and P2 fractions and to identify the source of potential off‐target interaction, we performed immunoblot analyses with SV2A, other synaptic proteins (SYP and SYT I/II), and tau (AT8) antibodies. We observed the presence of SV2A protein in both CN and AD P1 fractions (Figure 6A). The amount of SV2A protein in AD P1 fraction was higher than the CN P1 and AD P2 fractions (p = 0.047; Figure 6B). Of interest, a relatively high AT8 immunoreactivity in AD P1 fractions as compared to CN P1 and AD P2 fractions was also observed (Figure 6E) along with some high‐molecular‐weight species (most likely tangles) in the well, which were not able to enter the gel (Figure 6A; single arrow). Moreover, our initial analysis with Aβ‐specific antibody 4G8, which targets the Aβ fibril core sequence (17–24 amino acid), showed low Aβ fibril immunoreactivity in AD P1 fraction (Figure S1). A significant positive association between SV2A and AT8 immunoreactivity was observed in AD P1 fractions (r = 0.94 and p = 0.004; Figure 6F). On the contrary, no such correlation was observed in AD P2 fractions (Figure 6G). SYP and STY I/II proteins showed a similar pattern as SV2A, with STY I/II showing significantly higher amounts in AD P1 fractions as compared to CN P1 (p = 0.034) and AD P2 (p = 0.017) fractions (Figure 6C and D). Moreover, we observed either very faint or no bands for all three proteins in the BH fractions of CN and AD cases despite loading the same amounts as P1 and P2 fractions (Figure 6A; BH lanes). This complemented the lower specific binding observed in BH binding studies.
FIGURE 6.
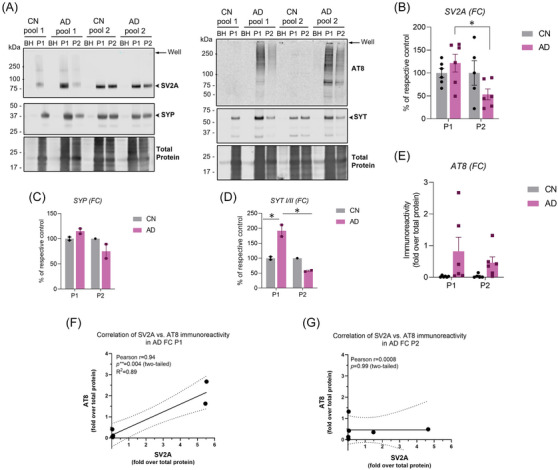
Immunoblot analyses in CN and AD brain BHs, P1, and P2 fractions. Immunoblot analyses for synaptic proteins SV2A, SYP, STY I/II and p‐tau (AT8) were performed in the BH, P1, and P2 fractions prepared from the frontal cortex brain tissue homogenates from CN and AD patients. (A) Representative immunoblots probed for synaptic proteins SV2A (93 kDa), SYP (38–48 kDa), SYT (65–67 kDa), and p‐tau in BH, P1, and P2 fractions prepared from CN and AD brains. Single arrows indicate the gel well. (B–E) Quantification of SV2A, SYP, STY I/II, and p‐tau (AT8) band intensity in CN and AD P1 and P2 fractions. Band intensity was quantified using LI‐COR Empiria software and signal was normalized to total protein stain (LICOR). Protein expression levels are displayed as means ± SEM of either percentage (%) of respective CN or fold‐over total protein and analyzed using two‐way ANOVA (Tukey's test). * p < 0.05. (F–G) Association between SV2A and AT8 immunoreactivity in AD P1 and P2 fractions were analyzed using Pearson correlation coefficient. The solid line represents the linear regression curve along with dotted line showing the 95% confidence interval. AD, Alzheimer's disease; CN, control; FC; frontal cortex; SV2A, synaptic vesicle protein 2A; SYP, synaptophysin; SYT I/II, synaptotagmin I/II.
3.7. 3H‐UCB‐J pre‐blocking saturation and competition binding studies in CN and AD P1 fractions with tau PET ligands
Following the immunoblot analyses findings, we investigated the possibility of 3H‐UCB‐J interaction with p‐tau species (specifically in the nuclear fraction). We first pre‐blocked the frontal cortex AD P1 fraction with 10 μM tau PET ligands AV‐1451 and MK6420, and then we performed similar saturation‐binding studies as above. With AV‐1451 and MK6240, we observed ≈22% reduction in 3H‐UCB‐J binding as compared to unblocked AD P1 fractions with almost similar Kd values in the nanomolar range (Figure 7A). 3H‐UCB‐J versus MK6240 competition binding in CN and AD P1 fraction showed ≈38% displacement of 3H‐UCB‐J binding but at a relatively higher concentration (100 μM; Figure 7B). However, with reverse 3H‐MK6240 versus UCB‐J competition binding in CN and AD P1 fractions, no displacement of 3H‐MK6240 binding was observed, even at 10 μM concentration (Figure 7C).
FIGURE 7.
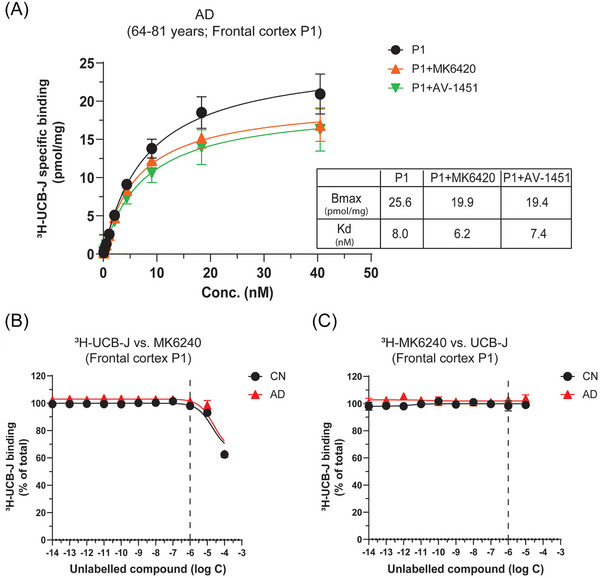
3H‐UCB‐J pre‐blocking saturation and competition binding studies in P1 fractions of CN and AD brains with tau PET ligands. (A) 3H‐UCB‐J pre‐blocked saturation‐binding studies with tau PET ligands MK6240 and AV‐1451 were performed in the P1 nuclear fractions (P1) prepared from the pooled frontal cortex brain tissue homogenates from AD patients using increasing concentrations of 3H‐UCB‐J (0–40 nM). The graph shows 3H‐UCB‐J‐specific binding curve in AD P1 fraction alone or in the presence of 10 μM of unlabeled MK640 or AV‐1451. The AD P1 fraction alone shows the combined data from experiments performed on Pools 1 and 2. The AV‐1451 blocking experiment was performed on AD Pool 1 only, whereas MK‐6240 blocking experiment was performed on both AD Pool 1 and 2 P1 fractions. Data are presented as means ± SEM of three to four experiments in duplicate. (B) 3H‐UCB‐J competition binding studies were performed in the P1 fractions prepared from the pooled frontal cortex brain tissue homogenates from CN (Pool 1; 79–84 years) and AD patients (Pool 1; 79–81 years) using a single concentration of 3H‐UCB‐J (5 nM) and increasing concentrations of unlabeled MK6240. (C) 3H‐MK6240 competition binding studies were performed in the P1 fractions prepared from the pooled frontal cortex brain tissue homogenates from CN (Pool 1; 79–84 years) and AD patients (Pool 1; 79–81 years) using a single concentration of 3H‐MK6240 (0.5 nM) and increasing concentrations of unlabeled UCB‐J. Data are presented as means ± SEM of 3–5 experiments in duplicate. AD, Alzheimer's disease; CN, control; Bmax, density of binding sites; Kd, dissociation constant.
4. DISCUSSION
Synaptic density markers such as SV2A could have immense clinical potential since synaptic loss is one of the early key pathological hallmarks of AD correlating with cognitive impairment. 11 , 12 , 13 , 32 In this study, we reported the in‐depth preclinical validation of SV2A PET‐tracer UCB‐J in post‐mortem AD and CN brains to understand its binding mechanism and explore its suitability as a novel synaptic‐PET tracer for AD.
Our initial 3H‐UCB‐J saturation‐binding studies in the frontal cortex BH demonstrated one binding site with comparable nanomolar affinity in both CN and AD brains but ≈1.12‐fold higher specific binding in AD brains as compared to CN (Bmax = 0.18 pmol/mg compared to Bmax = 0.16 pmol/mg, respectively). This observation was unexpected considering the significant loss of the number of synapses in the AD frontal cortex as compared to CN, as reported by an extensive meta‐analysis study of synapses and synaptic loss markers in post‐mortem AD brains. 33 Moreover, this also contrasted with recent UCB‐J post‐mortem studies by Patel et al., 25 where 57% loss of UCB‐J binding was reported in the AD cortex (undefined brain region) saturation‐binding analysis as compared to CN. In addition, we also observed relatively higher NSP binding in AD brains as compared to CN, which clearly suggested potential interaction with other brain components/proteins. Large frozen section autoradiography regional binding studies on one CN (56 years) and two sAD (57 and 79 years) brains corroborated these findings and once again demonstrated ≈1.4‐ to 2.4‐fold higher 3H‐UCB‐J binding in sAD brain ROI (frontal lobe, temporal lobe, insula, and hippocampus) as compared to the CN brain. The binding was well defined and specific to gray matter as well as displaceable by 10 μM levetiracetam (≈26%–42%) and 1 μM UCB‐J (≈92%–95%). We used only 10 μM levetiracetam in our studies to avoid any potential interference with 3H‐UCB‐J binding by very high concentrations of levetiracetam. Previous post‐mortem studies have used concentrations as high as 500 μM, which we believe is too high and might affect the tracer binding. 24 In fact, we tested different concentrations of levetiracetam and observed the same displacement as 1 μM UCB‐J at 1 mM concentration (Figure S2). Regardless of this, our findings were in agreement with a previous study by Metaxas et al., where no difference in 3H‐UCB‐J binding was observed between AD and CN cases in the middle frontal gyrus small brain section autoradiograms 24 and in contrast to a recent study by Mikkelsen et al., where they showed 19% reduction in the AD middle frontal cortex as compared to CN cases along with large inter‐individual variations. 34 Of interest, Mikkelsen et al. did not observe any changes in UCB‐J binding between AD and CN cases in the other examined cortical regions, further complementing our autoradiography findings.
There could be several possible explanations for increased UCB‐J binding in AD ROI as compared to CN: (1) a possible off‐target interaction with other brain tissue components/proteins as AD brains contain abundant plaques and tangles in ROI; (2) compensatory mechanisms at the end stages of the disease such as enlarged synaptic contact size in response to synaptic loss affecting tracer binding 12 , 35 ; (3) presynaptic proteins are dynamic in nature and it is possible that all proteins are not equally affected in the same/different brain regions and synaptic pathology could up‐/downregulate expressions of different synaptic proteins depending on disease stages and brain regions. 36
Considering the scope of this study, we next focused on detecting the expected synaptic loss in AD brains and exploring the potential off‐target interactor of UCB‐J. We performed subcellular fractionation to remove undesirable brain components/proteins and prepared synaptosomal/membrane P2 fractions from the frontal cortex of both CN and AD brains. Saturation‐binding studies in P2 fractions showed extremely high specific binding and relatively low NSP binding for 3H‐UCB‐J in CN and AD brains as compared to BH studies. Most importantly, P2 fractions demonstrated the expected loss of 3H‐UCB‐J binding in AD brains compared to CN (CN Bmax–11.8 pmol/mg vs AD Bmax–6.9 pmol/mg; ≈41% less). 3H‐UCB‐J regional binding studies in BH and P2 fractions of FC, TC, PC, Hipp, and Cb further confirmed these findings and showed the superiority of P2 fractions in highlighting synaptic loss (i.e., low UCB‐J specific binding) in all AD brain ROI compared to CN brains. The reduction in 3H‐UCB‐J‐specific binding showed the following order: Hipp > FC > TC > PC > Cb. Moreover, all the AD cases used for 3H‐UCB‐J regional binding studies had short symptom duration (5–7 years) and high disease severity (Braak Staging 4–6). Recent in vivo imaging studies have also reported similar findings and showed significant loss of 11C‐UCB‐J binding in the hippocampal and cortical brain regions of mild cognitive impairment (MCI) and mild dementia AD cases compared to CN. 20 , 21
As P2 fractions showed a loss of 3H‐UCB‐J binding in AD brains, we hypothesized that the potential off‐target interaction may be coming from other subcellular fractions such as nuclear P1 fractions. To test this theory, we performed similar saturation studies in the frontal cortex of CN and AD P1 fractions. The results were remarkable, as we observed unexpectedly high 3H‐UCB‐J‐specific binding in both CN and AD brains. Overall, the Bmax value pattern in both CN and AD followed the same trend: P1 Bmax > P2 Bmax > BH Bmax. The brain regional binding studies in BH, P2, and P1 fractions evidently reflected the case‐by‐case and ROI variability in CN and AD brains and highlighted the complexity and dynamic nature of synaptic proteins as discussed above.
To identify the source of P1 fractions high 3H‐UCB‐J‐specific binding, we probed the CN and AD brains frontal cortex BH, P1, and P2 fractions with SV2A, SYP, STY I/II, and p‐tau antibodies. The immunoblot analyses revealed some very interesting findings: (1) We observed significantly increased amounts of SV2A and other synaptic proteins in the AD P1 fractions as compared to P2 fractions, and (2) most importantly, increased AT8 immunoreactivity with larger p‐tau species (most likely tangles) in the wells and a significant positive correlation with SV2A in the AD P1 fractions as compared to P2 fractions. These important observations indicate the potential association between p‐tau species (specifically in the nuclear fraction) and SV2A, thereby fueling the speculations that UCB‐J is either interacting directly with p‐tau species or indirectly via SV2A bound to p‐tau species (p‐tau‐SV2A complex). This is highly possible, since tau pathologies have been heavily involved in the synaptic degeneration 6 , 7 , 9 and, interestingly, pathologic tau could also bind to synaptic vesicles in a “doughnut‐like” pattern, indicating potential interactions with membrane synaptic proteins, 37 with more recent studies showing high AT8 reactivity on synaptic vesicles isolated from the brains of AD patients. 38 A recent in vivo PET study by Coomans et al. 39 also showed a positive association between [18F]‐Flortaucipir and [11C]‐UCB‐J uptake in AD subjects with low neocortical tau load. However, two other in vivo PET studies with tau tracers and [11C]‐UCB‐J suggested negative association between p‐tau load and synaptic density in AD patients. 40 , 41 Due to the difference between these limited in vivo studies with MCI, amnestic MCI, and probable AD patients and our postmortem studies at the cellular/subcellular (synaptosomal) levels and end stage of the disease, we believe a direct comparison is not possible and advise against it.
In addition, in our blocking studies with 10 μM tau ligands AV‐1451 and MK6240 we were able to displace AD P1 fractions 3H‐UCB‐J specific binding by ≈22% as compared to unblocked AD P1 fractions. Moreover, MK6240 was able to displace ≈38% 3H‐UCB‐J binding in AD P1 fractions at 100 μM in competition studies. However, in the reverse competition studies (3H‐MK6240 vs UCB‐J), no displacement of MK620 binding was observed with UCB‐J until 10 μM final concentration in the assay. This observation is not surprising if you consider the complexity of tau folds and multiple/cryptic binding sites for tau ligands on AD tau fibrils. 27 , 28 , 42 , 43 , 44 , 45
The differences in 3H‐UCB‐J‐specific binding in P1 and P2 fractions could be attributed to the extent of tau phosphorylation in these fractions as highlighted by transgenic mouse model study of Sahara et al. 46 Similar to our findings, this study also showed the presence of p‐tau in P1 and P2 fractions and further suggested that the amount, phosphorylation, and types of tau species can vary between these fractions. The presence of SV2A and other presynaptic proteins (SYP and SYT) in the AD P1 fractions provides key mechanistic insight into synaptic pathology/plasticity and suggests increased co‐localization of synaptic proteins in the nucleus during pathological conditions via the nuclear import signaling mechanism (please refer to review by Ch'ng et al. 47 for further reading). All these results clearly indicate the potential interaction of UCB‐J to p‐tau species (especially the nuclear form), warranting further investigation to answer several questions critical for UCB‐J in vivo imaging studies such as: (1) What is the in vivo cell permeability of UCB‐J? (2) Could UCB‐J bind to different p‐tau species (ranging from pretangles to ghost tangles) in the same/different brain regions with similar selectivity? (3) Could the degree and extent of phosphorylation affect UCB‐J interaction with p‐tau? (4) Is UCB‐J targeting pathologic p‐tau (neurofibrillary tangles) spewed out by dying neurons?
4.1. Limitations
Our studies have a few limitations: (1) The autoradiography of large frozen brain sections used in this study originated from different anatomic levels; hence, a direct comparison between the cases should be carefully interpreted. It is important to keep in mind that large brain sections are rare and very hard to acquire. (2) 3H‐UCB‐J regional binding studies were performed on a small number of AD brains and have limited statistical power. However, the mean differences across all CN and AD regions were evident and consistent with the findings in literature. (3) Correlation analyses between SV2A and AT8 immunoreactivity should be interpreted with caution due to a limited number of cases. Despite these limitations, the findings were conclusive and supported by extensive multiple experimental approaches not presented in a handful of previous post‐mortem studies. 24 , 25
5. CONCLUSION
Overall, in this study we reported, for the first time, detailed mechanistic insight into UCB‐J binding behavior in different regions of AD and CN brains with post‐mortem brain imaging techniques and saturation, competition, and regional binding studies in BHs, P1, and P2 fractions. We demonstrated the high specificity of UCB‐J for SV2A and the superiority of P2 fractions in tracing synaptic loss in post‐mortem AD brains. Of interest, our studies showed a potential interaction of UCB‐J at the subcellular level with nuclear p‐tau species in AD brains, which could have subsequent clinical implications for in vivo imaging studies. Whether this interaction is specific to AD or not still needs to be investigated. Further in vivo longitudinal studies in larger sample sizes along with post‐mortem studies focused on UCB‐J binding at cellular and subcellular levels are needed to establish the reliability of UCB‐J as a definite marker of synaptic density/loss. This will contribute significantly to understanding the role of synaptic loss in AD and related dementias and reinforce the existing and future diagnostic and therapeutic strategies.
AUTHOR CONTRIBUTIONS
Amit Kumar and Agneta Nordberg conceptualized the study. Amit Kumar designed the study. Amit Kumar performed the saturation and competition binding assays and autoradiography studies. Amit Kumar and Miriam Scarpa performed regional distribution experiments. Miriam Scarpa performed the immunoblot experiment and analyses. Amit Kumar analyzed the data. Amit Kumar wrote the first draft of the manuscript. All authors have provided critical input and feedback during the writing of the manuscript. All authors read and approved the final version of the manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare no competing financial interests.
CONSENT STATEMENT
Informed consent was obtained for all cases investigated.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
We would like to thank the Brain Bank at Karolinska Institutet for providing large frozen whole hemisphere Alzheimer’s disease (AD) brain tissue. We would also like to thank Prof. Bernardino Ghetti, Department of Pathology and Laboratory Medicine, Indiana University School of Medicine, Indianapolis, USA, for providing the large frozen whole hemisphere control (CN) and AD brain tissues. We would like to thank the Netherlands Brain Bank for providing the CN and AD human brain tissues used in the binding assays. We would like to thank Filipa Rocha for her help with the subcellular fractionation illustration. This study was financially supported by the Swedish Foundation for Strategic Research (SSF; RB13‐0192), the Swedish Research Council (projects 2017‐02965, 2017‐06086, 2020‐01990), the Stockholm County Council‐Karolinska Institute regional agreement on medical training and clinical research (ALF grant), the Swedish Brain Foundation (Hjärnfonden), the Swedish Alzheimer Foundation (Alzheimerfonden), the Foundation for Old Servants (Gamla Tjänarinnor), Gun and Bertil Stohne's Foundation, Magnus Bergvall's Foundation, the Swedish Dementia Foundation (Demensfonden), Stiftelsen Sigurd och Elsa Goljes Minne, the Center for Innovative Medicine (CIMED) Region Stockholm, Åhlén Foundation, the Alzheimer's Association, USA (AARF −21‐848395), Loo and Hans Osterman Foundation for Medical Research, Karolinska geriatrics foundation, Tore Nilsons Stiftelse för Medicinsk Forskning, the Recherche sur Alzheimer Foundation (Paris, France), and the private initiative “Innovative ways to fight Alzheimer's disease—Leif Lundblad Family and others.” Prof. Bernardino Ghetti would like to acknowledge the funding from Public Health Service (PHS; P30 AG010133).
Kumar A, Scarpa M, Nordberg A. Tracing synaptic loss in Alzheimer's brain with SV2A PET‐tracer UCB‐J. Alzheimer's Dement. 2024;20:2589–2605. 10.1002/alz.13720
REFERENCES
- 1. Burns ME, Augustine GJ. Synaptic structure and function: dynamic organization yields architectural precision. Cell. 1995;83:187‐194. [DOI] [PubMed] [Google Scholar]
- 2. Lepeta K, Lourenco MV, Schweitzer BC, et al. Synaptopathies: synaptic dysfunction in neurological disorders—A review from students to students. J Neurochem. 2016;138:785‐805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Heurling K, Ashton NJ, Leuzy A, et al. Synaptic vesicle protein 2A as a potential biomarker in synaptopathies. Mol Cell Neurosci. 2019;97:34‐42. [DOI] [PubMed] [Google Scholar]
- 4. Klein WL. Synaptotoxic amyloid‐β oligomers: a molecular basis for the cause, diagnosis, and treatment of Alzheimer's disease? J Alzheimers Dis. 2013;33:S49‐S65. [DOI] [PubMed] [Google Scholar]
- 5. Forner S, Baglietto‐Vargas D, Martini AC, Trujillo‐Estrada L, LaFerla FM. Synaptic Impairment in Alzheimer's Disease: a Dysregulated Symphony. Trends Neurosci. 2017;40:347‐357. [DOI] [PubMed] [Google Scholar]
- 6. Shankar GM, Li S, Mehta TH, et al. Amyloid‐β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837‐842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Z, Jackson RJ, Hong W, et al. Human brain‐derived Aβ oligomers bind to synapses and disrupt synaptic activity in a manner that requires APP. J Neurosci. 2017;37:11947‐11966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spires‐Jones TL, Hyman BT. The intersection of amyloid beta and tau at synapses in Alzheimer's disease. Neuron. 2014;82:756‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Spires‐Jones Tara L, Hyman BT. The intersection of amyloid beta and tau at synapses in Alzheimer's disease. Neuron. 2014;82:756‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Colom‐Cadena M, Spires‐Jones T, Zetterberg H, et al. The clinical promise of biomarkers of synapse damage or loss in Alzheimer's disease. Alzheimer's. Res Ther. 2020;12:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tzioras M, McGeachan RI, Durrant CS, Spires‐Jones TL. Synaptic degeneration in Alzheimer disease. Nat Rev Neurol. 2023;19:19‐38. [DOI] [PubMed] [Google Scholar]
- 12. DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27:457‐464. [DOI] [PubMed] [Google Scholar]
- 13. Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572‐580. [DOI] [PubMed] [Google Scholar]
- 14. Marcus C, Mena E, Subramaniam RM. Brain PET in the diagnosis of Alzheimer's disease. Clin Nucl Med. 2014;39:e413‐22; quiz e23‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zimmer ER, Parent MJ, Souza DG, et al. [18F]FDG PET signal is driven by astroglial glutamate transport. Nat Neurosci. 2017;20:393‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jack CR Jr, Bennett DA, Blennow K, et al. NIA‐AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nabulsi NB, Mercier J, Holden D, et al. Synthesis and preclinical evaluation of 11C‐UCB‐J as a PET tracer for imaging the synaptic vesicle glycoprotein 2A in the brain. J Nucl Med. 2016;57:777‐784. [DOI] [PubMed] [Google Scholar]
- 18. Finnema SJ, Nabulsi NB, Eid T, et al. Imaging synaptic density in the living human brain. Sci Transl Med. 2016;8:348ra96. [DOI] [PubMed] [Google Scholar]
- 19. Bajjalieh S, Frantz G, Weimann J, McConnell S, Scheller R. Differential expression of synaptic vesicle protein 2 (SV2) isoforms. J Neurosci. 1994;14:5223‐5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen MK, Mecca AP, Naganawa M, et al. Assessing synaptic density in Alzheimer disease with synaptic vesicle glycoprotein 2A positron emission tomographic imaging. JAMA Neurol. 2018;75:1215‐1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mecca AP, Chen MK, O'Dell RS, et al. In vivo measurement of widespread synaptic loss in Alzheimer's disease with SV2A PET. Alzheimers Dement. 2020;16:974‐982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Holland N, Jones PS, Savulich G, et al. Synaptic loss in primary tauopathies revealed by [11 C]UCB‐J positron emission tomography. Mov Disord. 2020;35:1834‐1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Holland N, Jones PS, Savulich G, et al. Longitudinal synaptic loss in primary tauopathies: an in vivo [11 C]UCB‐J positron emission tomography study. Mov Disord. 2023;38:1316‐1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Metaxas A, Thygesen C, Briting SRR, Landau AM, Darvesh S, Finsen B. Increased inflammation and unchanged density of synaptic vesicle glycoprotein 2A (SV2A) in the postmortem frontal cortex of Alzheimer's disease patients. Front Cell Neurosci. 2019;13:538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Patel S, Knight A, Krause S, et al. Preclinical in vitro and in vivo characterization of synaptic vesicle 2A‐targeting compounds amenable to F‐18 labeling as potential PET radioligands for imaging of synapse integrity. Mol Imaging Biol. 2020;22:832‐841. [DOI] [PubMed] [Google Scholar]
- 26. Kumar A, Koistinen NA, Malarte M‐L, et al. Astroglial tracer BU99008 detects multiple binding sites in Alzheimer's disease brain. Mol Psychiatry. 2021;26:5833‐5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malarte ML, Gillberg PG, Kumar A, Bogdanovic N, Lemoine L, Nordberg A. Discriminative binding of tau PET tracers PI2620, MK6240 and RO948 in Alzheimer's disease, corticobasal degeneration and progressive supranuclear palsy brains. Mol Psychiatry. 2022;28:1272‐1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Malarte ML, Nordberg A, Lemoine L. Characterization of MK6240, a tau PET tracer, in autopsy brain tissue from Alzheimer's disease cases. Eur J Nucl Med Mol Imaging. 2021;48:1093‐1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. GraphPad for Windows Version 9.0 (GraphPad software Inc, La Jolla, CA, USA).
- 30. Fontana IC, Kumar A, Okamura N, Nordberg A. Multitracer approach to understanding the complexity of reactive astrogliosis in Alzheimer's brains. ACS Chem Neurosci. 2023;15(2):328‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lemoine L, Saint‐Aubert L, Nennesmo I, Gillberg PG, Nordberg A. Cortical laminar tau deposits and activated astrocytes in Alzheimer's disease visualised by 3H‐THK5117 and 3H‐deprenyl autoradiography. Sci Rep. 2017;7:45496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scheff SW, Neltner JH, Nelson PT. Is synaptic loss a unique hallmark of Alzheimer's disease? Biochem Pharmacol. 2014;88:517‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. de Wilde MC, Overk CR, Sijben JW, Masliah E. Meta‐analysis of synaptic pathology in Alzheimer's disease reveals selective molecular vesicular machinery vulnerability. Alzheimers Dement. 2016;12:633‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mikkelsen JD, Kaad S, Aripaka SS, Finsen B. Synaptic vesicle glycoprotein 2A (SV2A) levels in the cerebral cortex in patients with Alzheimer's disease: a radioligand binding study in postmortem brains. Neurobiol Aging. 2023;129:50‐57. [DOI] [PubMed] [Google Scholar]
- 35. Scheff SW, Price DA. Alzheimer's disease‐related alterations in synaptic density: neocortex and hippocampus. J Alzheimer's Dis. 2006;9:101‐115. [DOI] [PubMed] [Google Scholar]
- 36. Honer WG. Pathology of presynaptic proteins in Alzheimer's disease: more than simple loss of terminals. Neurobiol Aging. 2003;24:1047‐1062. [DOI] [PubMed] [Google Scholar]
- 37. Zhou L, McInnes J, Wierda K, et al. Tau association with synaptic vesicles causes presynaptic dysfunction. Nat Commun. 2017;8:15295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McInnes J, Wierda K, Snellinx A, et al. Synaptogyrin‐3 mediates presynaptic dysfunction induced by tau. Neuron. 2018;97:823‐835.e8. [DOI] [PubMed] [Google Scholar]
- 39. Coomans EM, Schoonhoven DN, Tuncel H, et al. In vivo tau pathology is associated with synaptic loss and altered synaptic function. Alzheimers Res Ther. 2021;13:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mecca AP, Chen MK, O'Dell RS, et al. Association of entorhinal cortical tau deposition and hippocampal synaptic density in older individuals with normal cognition and early Alzheimer's disease. Neurobiol Aging. 2022;111:44‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vanderlinden G, Ceccarini J, Vande Casteele T, et al. Spatial decrease of synaptic density in amnestic mild cognitive impairment follows the tau build‐up pattern. Mol Psychiatry. 2022;27:4244‐4251. [DOI] [PubMed] [Google Scholar]
- 42. Murugan NA, Nordberg A, Ågren H. Cryptic sites in tau fibrils explain the preferential binding of the AV‐1451 PET tracer toward Alzheimer's tauopathy. ACS Chem Neurosci. 2021;12:2437‐2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shi Y, Zhang W, Yang Y, et al. Structure‐based classification of tauopathies. Nature. 2021;598:359‐363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou Y, Li J, Nordberg A, Ågren H. Dissecting the binding profile of PET tracers to corticobasal degeneration tau fibrils. ACS Chem Neurosci. 2021;12:3487‐3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li J, Kumar A, Långström B, Nordberg A, Ågren H. Insight into the binding of first‐ and second‐generation PET tracers to 4R and 3R/4R tau protofibrils. ACS Chem Neurosci. 2023;14:3528‐3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sahara N, Murayama M, Higuchi M, Suhara T, Takashima A. Biochemical distribution of tau protein in synaptosomal fraction of transgenic mice expressing human P301L tau. Front Neurol. 2014;5:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ch'ng TH, Martin KC. Synapse‐to‐nucleus signaling. Curr Opin Neurobiol. 2011;21:345‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information