

Abstract
INTRODUCTION
Evidence suggests microglial activation precedes regional tau and neurodegeneration in Alzheimer's disease (AD). We characterized microglia with translocator protein (TSPO) positron emission tomography (PET) within an AD progression model where global amyloid beta (Aβ) precedes local tau and neurodegeneration, resulting in cognitive impairment.
METHODS
Florbetaben, PBR28, and MK‐6240 PET, T1 magnetic resonance imaging, and cognitive measures were performed in 19 cognitively unimpaired older adults and 22 patients with mild cognitive impairment or mild AD to examine associations among microglia activation, Aβ, tau, and cognition, adjusting for neurodegeneration. Mediation analyses evaluated the possible role of microglial activation along the AD progression model.
RESULTS
Higher PBR28 uptake was associated with higher Aβ, higher tau, and lower MMSE score, independent of neurodegeneration. PBR28 mediated associations between tau in early and middle Braak stages, between tau and neurodegeneration, and between neurodegeneration and cognition.
DISCUSSION
Microglia are associated with AD pathology and cognition and may mediate relationships between subsequent steps in AD progression.
Keywords: AD progression, Alzheimer's disease, neuroinflammation, TSPO PET
1. BACKGROUND
Diffuse amyloid β (Aβ) deposition throughout the neocortex 1 can occur as early as two decades before Alzheimer's disease (AD) symptom onset and is believed to facilitate the development of other AD pathologies, including the aggregation of hyperphosphorylated tau into neurofibrillary tangles. 2 , 3 The spread of tau pathology in AD has been well characterized into Braak stages at autopsy, 4 starting with early accumulation in the entorhinal cortex and hippocampal regions before spreading to other regions of the temporal, frontal, and parietal regions. Of note, the spatiotemporal trajectory of tau aggregation precedes that of neurodegeneration (eg, neuronal death, synaptic loss), which gives rise to AD symptoms. 5 This model of Aβ deposition to tau aggregation across Braak stages to neurodegeneration to cognitive impairment will be referred to as the AD progression model throughout this article.
Neuroinflammation is an immune response in which glial cells in the brain, such as microglia, are recruited to protect tissue from pathogens, respond to injury, and help with the upkeep of tissue maintenance. Microglia are heterogeneous in their developmental origins as well as their varied response to stimuli during surveillance, including the adoption of different cellular morphologies and differential transcriptomic expression through aging, injury, and disease, suggesting context‐dependent functionality over time within a single microglia. 6 Within the context of AD, microglial activity plays a complex role, where it may protect against disease pathology, promote the spread of disease pathology, or be a product of disease progression. Microglia may serve a protective function, such as clearing soluble Aβ or corralling larger Aβ deposits in the extracellular space, maintaining an innate immune memory; however, primed microglia can develop a complex neurotoxic phenotype with exaggerated immune responses leading to chronic inflammation or an attenuated immune response leading to unchecked pathology, depending on the stimuli, the microenvironment, and the cellular cross‐talk with neurons and other inflammatory cells. 7 , 8 These dysfunctional microglia are associated with increased Aβ and AD risk. 9 Further, apolipoprotein E (APOE) and triggering receptor expressed on myeloid cells 2 (TREM2) mutations in microglia may promote amyloidosis and tauopathies through their role in leading to or suppressing microglial activation. 10 Microglial activation may also promote disease pathology and progression via cytokine release 9 and the NOD‐, LRR‐, and pyrin domain‐containing protein 3 (NLRP3) inflammasome, 2 , 11 , 12 , 13 , 14 , 15 , 16 , 17 preceding 13 , 14 , 18 , 19 , 20 and seeding 21 , 22 , 23 tau pathology to surrounding unaffected areas as well as contributing to amyloid‐initiated, tau‐dependent synaptic loss. 9 , 24 Microglia, as a product of disease progression, can perform cellular functions related to the upkeep of the extracellular space (ie, removing cellular waste and debris). 25 The accumulation of insoluble myelin debris within surveilling microglia can ultimately lead to microglia dysfunction, brain aging, 26 and cognitive impairment. 27 The complex age‐ and disease stage‐dependent role of microglia likely contributes to the mixed results obtained in clinical trials of non‐steroidal anti‐inflammatory drugs (NSAIDs) to prevent or delay AD. 28 , 29 , 30 For a review of microglia heterogeneity and nomenclature, see Healy et al. 6 and Paolicelli et al. 8
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional sources (eg, PubMed) and cite these studies appropriately. The role of key proteins, including amyloid β (Aβ) and tau, has been well characterized in Alzheimer's disease (AD), while the mechanisms by which neuroinflammation is related to disease progression remain uncertain.
Interpretation: This study shows that neuroinflammation is not only related to measures of AD such as Aβ and tau burden but also may mediate important steps in the progression of disease pathology. These findings support the hypothesis that activated microglia contribute to the spread of AD pathology and, in turn, symptomology.
Future directions: Future studies should explore this in a longitudinal manner by repeating measures in the same patient to explore within‐subject AD progression. A better understanding of AD progression will contribute to improved treatments and cognitive outcomes for those with the disease.
Moving beyond study designs that only include canonical Aβ, tau, and neurodegeneration biomarkers, recent advancements have made it possible to incorporate the role of neuroinflammatory markers throughout AD progression. 31 The 18 kDa translocator protein (TSPO) can be imaged in vivo using positron emission tomography (PET) 32 ; an elevated signal from TSPO PET can indicate activated microglia and/or increased microglial density. Regional TSPO expression, induced by amyloid, propagates across Braak stages and may be a driver of tau aggregation and spread in patients with AD, 33 yet TSPO in activated microglia is also closely associated with neurodegeneration. 34 We sought to determine whether TSPO had both upstream (ie, preceding neurodegeneration) and downstream (ie, following neurodegeneration) associations with AD pathology. First, we examined the association of TSPO with measures of (1) global Aβ, (2) regional tau across different Braak stages, and (3) cognition, while accounting for neurodegeneration. Next, we considered the role of TSPO in AD based on a progression model that began with Aβ deposition and continues with tau aggregation and neurodegeneration in early, middle, and late Braak stages, followed by cognitive impairment.
2. METHODS
2.1. Participants
Forty‐one research participants underwent magnetic resonance imaging (MRI), multiple PET scans, and a cognitive assessment. These participants were retrospectively selected from and harmonized across studies (K23AG052633, R01AG026158, R56AG034189, P50AG008702, P01AG07232, R01AG037212, RF1AG054023), 35 , 36 based on the availability of imaging and cognitive measures. Of these participants, 19 were cognitively unimpaired, while 22 were cognitively impaired based on having a primary memory complaint and meeting clinical criteria for either amnestic mild cognitive impairment (MCI, single‐ or multiple‐domain) 37 or AD, 38 as described previously. 36 Participants were evaluated with the Mini‐Mental State Examination (MMSE 39 ), and domain‐specific tests including the Selective Reminding Test‐Delayed Recall (SRT‐DR 40 ), Trail Making Test Part B (Trails B 41 ), and categorical fluency (CF‐Animals 42 ). Domain‐specific cognitive test scores were transformed into z‐scores using age‐, sex‐, and education‐adjusted normative data derived from the National Alzheimer's Coordinating Center Uniform Dataset (NACC). 43 All participants (or their legally authorized representatives) provided informed consent according to the Declaration of Helsinki, and all study procedures were approved by the Columbia University Irving Medical Center Institutional Review Board.
2.2. TSPO genotyping
TSPO binding affinity was determined at screening, as previously described. 36 Briefly, genomic DNA from each subject was used to genotype the rs6971 polymorphism using a TaqMan assay. 44 Participants were high‐affinity binders (HH) or mixed‐affinity binders (HL), and the proportion did not differ between controls and patients. Low‐affinity binders (LL) were excluded.
2.3. APOE genotyping
APOE information was only available in a subset of 17 participants (10 controls, seven patients). All patients were APOE ε4 non‐carriers, which precluded robust interpretation of models when including APOE ε4 status. However, APOE ε4 carriers had significantly greater global 18F‐florbetaben (FBB) SUVR and MK6240 SUVR and lower global cognition, with slightly greater PBR28 SUVR and lower %GM (Table S1).
2.4. Magnetic resonance imaging
MRI scans were acquired as previously described. 35 , 36 In short, all participants underwent T1‐weighted MRI scanning on a 3T scanner. FreeSurfer 6.0 (Massachusetts General Hospital, Harvard Medical School; http://surfer.nmr.mgh.harvard.edu) was used to segment the MRI scans and to determine gray matter (GM) volume. A global neurodegeneration measure (%GM) was calculated as the sum of the GM volume from composite AD‐related regions (hippocampus, inferior frontal gyrus, middle‐inferior and superior temporal cortex, medial temporal cortex, inferior and superior parietal cortex, precuneus, prefrontal cortex, and posterior cingulate) normalized by the total intracranial volume. Neurodegeneration measures were also calculated for early (I+II), middle (III+IV), and late (V+VI) Braak stages (Table S2). Divisions of Braak staging have been described elsewhere. 35 , 45
2.5. PET
2.5.1. Image acquisition, processing, and quantification
2.5.1.1. 18F‐Florbetaben (FBB)
FBB scans were acquired to evaluate Aβ as described previously. 36 In summary, image data from 50 to 70 min after FBB injection were aligned with the MRI, and regions of interest (ROIs) were defined using the Hammers‐N30R83‐1 MM atlas in the PNEURO module of PMOD 3.9 (PMOD Technologies 46 ). Amyloid positivity was defined as previously described. 36 Partial volume correction (PVC) was completed using the region‐based voxel‐wise method (PMOD Technologies 47 ). Standardized uptake value ratios (SUVRs) were calculated by normalizing the radioactivity in the ROIs to the activity in the GM of the cerebellum. A composite SUVR measure was calculated based on a volume‐weighted average of representative AD‐related regions consisting of the same regions as mentioned earlier for the composite MRI measure.
11C‐PBR28 (PBR28)
PBR28 scans were acquired to evaluate TSPO expression, as described previously. 36 In summary, image data from 60 to 90 min after PBR28 injection were aligned with the MRI, and ROIs were defined using the Hammers‐N30R83‐1 MM atlas in the PNEURO module of PMOD 3.9. PVC was completed using the region‐based voxel‐wise method in PMOD. SUVR measures were calculated by normalizing the radioactivity in the ROIs to the activity in the GM of the cerebellum, which was used as a reference region. A composite SUVR measure was calculated based on a volume‐weighted average of representative AD‐related regions consisting of the same regions as mentioned above for the composite FBB and MRI measures.
18F‐MK‐6240 (MK‐6240)
MK‐6240 scans were acquired to evaluate the presence and extent of tau pathology, as described previously. 35 In summary, image data were collected from 90 to 110 min after MK‐6240 injection. ROIs were defined in FreeSurfer. Tau positivity thresholds for early, middle, and late Braak stages (individual regions outlined in Table S2) were 1.40, 1.50, and 1.53 SUVR, respectively, and were previously derived in a sample of 100 cognitively unimpaired adults from a community‐based study in Northern Manhattan. 35 PVC was implemented in MATLAB using the Muller‐Gartner method 47 as performed in the previous study. 35 SUVR measures were calculated using the inferior GM of the cerebellum as a reference region, which avoids spill‐in of radiotracer binding in the occipital lobe and off‐target binding in the falx cerebelli. Regional SUVR measures were calculated for early (I+II), middle (III+IV), and late (V+VI) Braak stages, as described above for the regional MRI measures.
2.6. Statistical analysis
Demographic characteristics between the control and patient groups were compared with t‐tests for continuous variables and chi‐squared tests for proportional variables for context related to AD progression, but data from both groups were combined for the association and mediation analyses. To test whether TSPO expression was associated with AD biomarkers and cognition independently of neurodegeneration, we used a series of linear regressions with PBR28 SUVR as the independent variable and AD biomarkers (FBB SUVR, MK‐6240 SUVR) or cognitive performance (MMSE, SRT‐DR, Trails B, CF‐Animals) as the dependent variable controlling for age, education, race, sex, and TSPO genotypes, with and without %GM in an AD composite region (Figure S1). To evaluate whether TSPO expression mediated pathways along the AD progression model, we performed a series of structural equation models (lavaan package, R version 4.1.2 48 ; standard errors computed using Delta method 49 ), adjusting each path for age, education, race, sex, and TSPO genotypes. For the AD progression model, regional %GM within early, middle, and late Braak stages was used rather than %GM in an AD composite region.
The main analyses were performed using PVC PET data to account for spill‐out of PET signal due to neurodegeneration within a voxel; PVC methods may add noise to data, 50 and analyses using uncorrected PET data are shown in Figures S2 and S3 and Table S3. Some PET measures were skewed. While very high values in patients relative to controls are biologically plausible and meaningful, we repeated the analysis after log transformation. Reported results were not biased by the skewness of PET data. Corrections for multiple comparisons were not performed. All analyses were performed using R version 4.1.2.
3. RESULTS
3.1. Demographic characteristics
Age did not differ between controls and patients, and there were more men, more White participants, and greater years of education in the patient group (Table 1). As expected, the patient group had greater FBB SUVR, greater PBR28 SUVR, greater MK‐6240 SUVR in early, middle, and late Braak stages, and lower MMSE, SRT‐DR, Trails B, and CF‐Animals scores. Notably, the patient group had the greatest MK‐6240 SUVR in middle Braak regions and the lowest SRT‐DR scores, as expected, but the greatest impairment (ie, lowest individual‐level z‐scores) in Trails B.
TABLE 1.
Descriptive statistics of demographic characteristics and primary outcomes.
| Control (N = 19) | Patient (N = 22) | All (N = 41) | p value | ||
|---|---|---|---|---|---|
| Age | Mean (SD) | 70 .0 (4.3) | 68.1 (8.7) | 69.0 (7.0) | .401 |
| Range | 60 to 76 | 53 to 83 | 53 to 83 | ||
| Sex | Female | 10 (53%) | 5 (22%) | 15 (37%) | .047 |
| Male | 9 (47%) | 17 (77%) | 26 (63%) | ||
| Race | Black/African‐American | 5 (26%) | 0 (0%) | 5 (12%) | .010 |
| White | 14 (74%) | 22 (100%) | 36 (88%) | ||
| Education | Mean (SD) | 15.4 (2.7) | 17.1 (2.4) | 16.3 (2.7) | .033 |
| Range | 10 to 20 | 12 to 20 | 10 to 20 | ||
| TSPO genotype | HH | 13 (68%) | 13 (59%) | 26 (63%) | .536 |
| HL | 6 (32%) | 9 (41%) | 15 (37%) | ||
| APOE ε4 status | Non‐carriers | 8 (42%) | 2 (9%) | 10 (24%) | <.001 |
| Carriers | 7 (37%) | 0 (0%) | 7 (17%) | ||
| Missing | 4 (21%) | 20 (91%) | 24 (59%) | ||
| Amyloid status | Negative | 12 (63%) | 8 (36%) | 20 (49%) | .087 |
| Positive | 7 (37%) | 14 (64%) | 21 (51%) | ||
| Early tau status | Negative | 10 (53%) | 2 (9%) | 12 (29%) | .002 |
| Positive | 9 (47%) | 20 (91%) | 29 (71%) | ||
| Middle tau status | Negative | 13 (68%) | 4 (18%) | 17 (41%) | .001 |
| Positive | 6 (32%) | 18 (82%) | 24 (59%) | ||
| Late tau status | Negative | 14 (74%) | 6 (27%) | 20 (49%) | .003 |
| Positive | 5 (26%) | 16 (73%) | 21 (51%) | ||
| Composite 18F‐FBB PVC SUVR | Mean (SD) | 1.47 (0.33) | 1.88 (0.46) | 1.69 (0.45) | .003 |
| Range | 1.12 to 2.17 | 1.11 to 2.81 | 1.11 to 2.81 | ||
| Composite 11C‐PBR28 PVC SUVR | Mean (SD) | 1.10 (0.11) | 1.21 (0.10) | 1.16 (0.12) | .001 |
| Range | 0.77 to 1.22 | 1.06 to 1.45 | 0.77 to 1.45 | ||
| Early Braak 18F‐MK‐6240 PVC SUVR | Mean (SD) | 1.50 (0.51) | 2.46 (0.91) | 2.02 (0.886) | <.001 |
| Range | 0.97 to 2.91 | 1.00 to 3.82 | 0.97 to 3.82 | ||
| Middle Braak 18F‐MK‐6240 PVC SUVR | Mean (SD) | 1.41 (0.21) | 2.85 (1.54) | 2.18 (1.34) | <.001 |
| Range | 1.11 to 1.90 | 1.02 to 5.79 | 1.02 to 5.79 | ||
| Late Braak 18F‐MK‐6240 PVC SUVR | Mean (SD) | 1.39 (0.19) | 2.55 (1.41) | 2.01 (1.18) | <.001 |
| Range | 1.09 to 1.75 | 0.98 to 5.99 | 0.98 to 5.99 | ||
| %GM volume | Mean (SD) | 0.44 (0.01) | 0.43 (0.01) | 0.44 (0.01) | .017 |
| Range | 0.43 to 0.46 | 0.39 to 0.45 | 0.39 to 0.46 | ||
| MMSE | Mean (SD) | 29.2 (1.31) | 25.6 (3.20) | 27.3 (3.07) | <.001 |
| Range | 26 to 30 | 18 to 30 | 18 to 30 | ||
| SRT‐DR z‐score | Mean (SD) | 0.53 (1.09) | −3.11 (0.66) | −1.42 (2.04) | <.001 |
| Range | −1.60 to 2.27 | −3.87 to 1.53 | −3.87 to 2.27 | ||
| Trails B z‐score | Mean (SD) | 0.15 (0.64) | −2.79 (2.57) | −1.43 (2.42) | <.001 |
| Range | −1.23 to 1.02 | −6.28 to 0.42 | −6.26 to 1.02 | ||
| CF‐Animals z‐score | Mean (SD) | −0.24 (0.88) | −1.65 (1.17) | −0.98 (1.25) | <.001 |
| Range | −2.06 to 1.26 | −3.31 to 2.38 | −3.31 to 2.38 |
Abbreviations: CF, categorical fluency; MMSE, Mini‐Mental State Examination; SRT‐DR, Selective Reminding Test‐Delayed Recall; SUVR, standardized uptake value ratio; TSPO, translocator protein.
3.2. TSPO correlates with AD PET and cognitive measures, independent of neurodegeneration
When correcting for %GM, greater PBR28 SUVR was associated with greater FBB SUVR (Figure 1; β = 0.46, p = .002). Greater PBR28 SUVR was also associated with greater MK‐6240 SUVR in middle (β = 0.45, p = .004) and late (β = 0.50, p = .002) Braak regions, with a non‐significant positive association in early Braak regions. Greater PBR28 SUVR was associated with lower MMSE (β = −0.53, p = .006), SRT‐DR (β = −0.39, p = .037), and Trails B scores (β = −0.50, p = .004), but not categorical fluency scores. When not correcting for %GM, greater PBR28 SUVR was associated with greater MK‐6240 SUVR in early Braak regions and lower CF‐Animals score (Figure S1).
FIGURE 1.
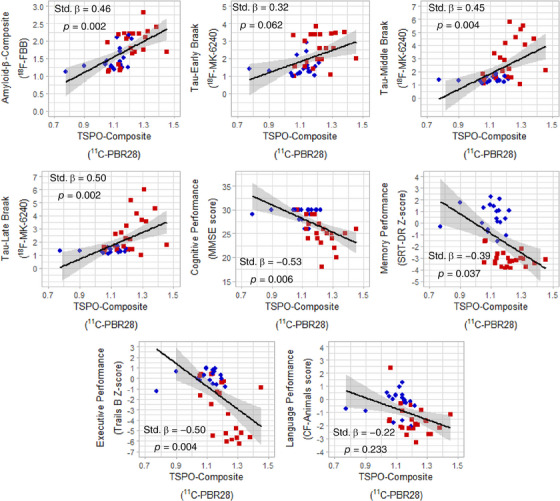
TSPO expression by 11C‐PBR28 uptake (SUVR) correlated with AD‐related PET and cognitive measures across controls (blue circles) and patients (red squares). All associations were corrected for age, sex, race, education, TSPO genotype, and neurodegeneration (%GM in AD composite region). PET, positron emission tomography; SUVR, standardized uptake value ratio; TSPO, translocator protein.
3.3. TSPO significantly mediates pathways along AD progression model
In a series of mediation analyses (Figure 2, Table 2), greater PBR28 SUVR mediated the association between greater MK‐6240 SUVR in early Braak regions and greater MK‐6240 SUVR in middle Braak regions (indirect effect = 0.118, z‐score = 2.169, p = .030) and the association between greater MK‐6240 SUVR in middle Braak regions and greater neurodegeneration in middle Braak regions (indirect effect = −0.225, z‐score = −2.214, p = .027). Greater PBR28 SUVR mediated the association between greater neurodegeneration in both early (indirect effect = 0.233, z‐score = 2.307, p = .021) and middle Braak regions (indirect effect = 0.204, z‐score = 2.185, p = .029) and lower MMSE score. Greater PBR28 SUVR also mediated the association between greater neurodegeneration in both early (indirect effect = 0.283, z‐score = 2.579, p = .009) and middle Braak regions (indirect effect = 0.228, z‐score = 2.379, p = .017) and lower Trails B score.
FIGURE 2.
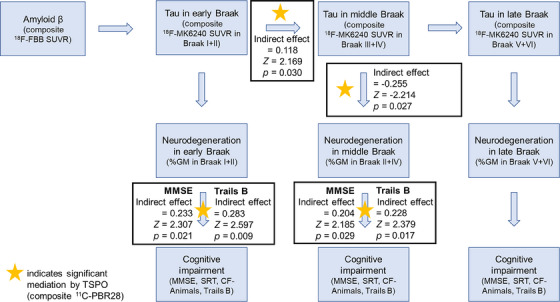
TSPO expression by 11C‐PBR28 uptake (SUVR) significantly mediated pathways along AD progression model. SUVR, standardized uptake value ratios; TSPO, translocator protein.
TABLE 2.
Results of mediation analyses for composite 11C‐PBR28 SUVR in AD progression model.
| X | Mediator | Y | Indirect effect (std. β, z, p) | Direct effect (std. β, z, p) | Total effect (std. β, z, p) |
|---|---|---|---|---|---|
| Global Aβ burden | Global TSPO expression | Tau in early Braak | 0.017, 0.195, .845 | 0.748, 5.259, .000 | 0.765, 6.895, 0.000 |
| Tau in early Braak | Neurodegeneration in early Braak | −0.157, −1.917, .055 | −0.229, −1.591, .112 | −0.386, −2.870, 0.004 | |
| Neurodegeneration in early Braak | Cognitive impairment – MMSE | 0.233, 2.307, .021 | 0.207, 1.310, .190 | 0.440, 2.821, 0.005 | |
| Cognitive impairment – SRT‐DR | 0.117, 1.536, .125 | 0.492, 3.368, .001 | 0.609, 4.553, 0.000 | ||
| Cognitive impairment – CF‐Animals | 0.076, 0.982, .326 | 0.391, 2.456, .014 | 0.468, 3.276, 0.001 | ||
| Cognitive impairment – Trails B | 0.283, 2.597, .009 | −0.057, −0.384, .701 | 0.226, 1.443, .149 | ||
| Tau in early Braak | Tau in middle Braak | 0.118, 2.169, .030 | 0.634, 7.078, .000 | 0.752, 8.750, .000 | |
| Tau in middle Braak | Neurodegeneration in middle Braak | −0.255, −2.214. .027 | −0.087, −0.506, .613 | −0.342, −2.295, .022 | |
| Neurodegeneration in middle Braak | Cognitive impairment – MMSE | 0.204, 2.185, .029 | 0.347, 2.277, .023 | 0.552, 3.768, .000 | |
| Cognitive impairment – SRT‐DR | 0.158, 1.794, .073 | 0.310, 1.959, .050 | 0.469, 3.198, .001 | ||
| Cognitive impairment – CF‐Animals | 0.061, 0.804, .422 | 0.454, 2.909, .004 | 0.515, 3.727, .000 | ||
| Cognitive impairment – Trails B | 0.228, 2.379, .017 | 0.211, 1.439, .150 | 0.439, 3.037, .002 | ||
| Tau in middle Braak | Tau in late Braak | 0.029, 0.956, .335 | 0.917, 18.22, .000 | 0.947, 23.03, .000 | |
| Tau in late Braak | Neurodegeneration in late Braak | −0.168, −1.382, .167 | 0.015, 0.077, .938 | −0.153, −0.966, .334 | |
| Neurodegeneration in late Braak | Cognitive impairment – MMSE | 0.0146, 1.588, .112 | 0.157, 1.074, .283 | 0.303, 1.850, .064 | |
| Cognitive impairment – SRT‐DR | 0.114, 1.497, .134 | 0.191, 1.283, .200 | 0.305, 1.933, .053 | ||
| Cognitive impairment – CF‐animals | 0.071, 1.251, .211 | 0.297, 1.983, .047 | 0.368, 2.450, .014 | ||
| Cognitive impairment – Trails B | 0.144, 1.603, .109 | 0.136, 1.003, .316 | 0.280, 1.180, .070 |
Note: Rows of significant mediations are shown in bold.
Abbreviations: CF, categorical fluency; MMSE, Mini‐Mental State Examination; SRT‐DR, Selective Reminding Test‐Delayed Recall; std, standardized; SUVR, standardized uptake value ratio; TSPO, translocator protein.
4. DISCUSSION
We examined the associations of microglial recruitment and/or density, measured by TSPO expression, and key AD biomarkers and cognitive measures with two goals – to disentangle components of neuroinflammation that drive pathology from those that respond to neurodegenerative processes and to elucidate the role of neuroinflammation along an AD progression model. Our findings add to the growing body of evidence that neuroinflammation tracks in severity with Aβ and tau pathology and cognition, independently of the microglial response to neurodegenerative processes. The findings additionally demonstrate that neuroinflammation may be the mechanistic link by which tau spreads across Braak regions and between tau burden, neurodegeneration, and cognitive impairment. 51 Neuroinflammation should be incorporated into large‐scale, multimodal studies of AD to understand the role of activated microglia in the links between Aβ and tau, tau burden across Braak stage regions, and tau and downstream neurodegeneration and cognitive impairment such that specific inflammatory processes can be targeted at specific points along the AD continuum.
We found that greater TSPO expression was associated with AD severity in terms of neuropsychological testing results, GM volumes, and both Aβ and tau burden, aligning with previous studies. 33 , 52 , 53 , 54 However, previous studies did not correct for neurodegeneration, so whether microglia were driving AD pathogenesis or responding to neurodegeneration had not been previously established. The current study suggests that the relationships between TSPO expression, AD biomarkers, and cognition are present regardless of the role of microglia in removing cellular debris and other neurodegeneration‐related products. While we did not have the statistical power to test associations within controls and patients separately, it should be noted that a range of TSPO expression was present at low levels of tau in controls (Figure 1), potentially suggesting that neuroinflammation precedes tau accumulation. Within patients, greater tau burden was observed in the presence of greater TSPO expression across Braak stage regions, and the association across controls and patients was independent of neurodegenerative processes in middle and late Braak stage regions. The association between greater TSPO expression and greater tau burden in early Braak stage regions did not survive adjustment for neurodegeneration. As tau spreads from across Braak stage regions, tau continues to increase in early Braak stage regions; tau burden in early Braak stage regions across controls and patients may reflect disease progression in a similar way to neurodegeneration. Therefore, there may be a mechanistic link between TSPO and tau in early Braak stage regions, but it may be too collinear with neurodegeneration across the clinical AD continuum to be identified using cross‐sectional data. Further work investigating regional TSPO, AD biomarkers, neurodegeneration, and cognition at distinct stages of the AD continuum is needed (eg, using amyloid‐positive controls to test associations in early Braak stage regions).
We found that TSPO expression mediated the association between tau in early and middle Braak stage regions, between tau and neurodegeneration in middle Braak stages, and between neurodegeneration and cognitive impairment in early and middle Braak stages. To our knowledge, this is the first study to examine this type of TSPO mediation in an AD progression model from Aβ through tau, neurodegeneration, and cognition. Our results do not show a mediation effect of TSPO on the relationships between Aβ and tau in early Braak stages, between tau in early Braak stages and neurodegeneration, or throughout any steps in the late Braak pathway. Patients had the greatest tau burden in middle Braak stage regions and the greatest range of impairment in Trails B; it may be the case that the current sample did not have advanced enough tau pathology in late Braak stage regions to elucidate the roles of tau and neurodegeneration in late Braak stage regions but advanced enough tau pathology for measures in early Braak stages to have plateaued. Regardless, these analyses are an important step toward characterizing neuroinflammation in AD progression.
The findings of this study are in agreement with the extant literature and current research frameworks of AD, for example, AT(N) 2 , 3 and ATX(N) 31 , which were the basis of the longitudinal AD progression model investigated here. Our well‐characterized sample with and without clinical impairment leveraged multiple studies and centers to perform broad neuroimaging characterization of TSPO PET in AD progression. Given that the results represented here are a secondary analysis of previously published data, PET methods (ROI definitions and PVC methods) for tau were defined differently than those of Aβ and TSPO; however, consistency within radioligands is more important than consistency across radioligands for each participant, particularly as each method has been shown to reliably quantify the underlying pathology. 55 , 56 , 57 In the current sample, approximately 35% of patients were amyloid‐negative. This aligns with previous reports of the mismatch between clinical AD and post mortem histopathological confirmation. 58 Participants who were amyloid‐negative but tau‐positive were similar to amyloid‐positive and tau‐positive participants in terms of age, cortical thickness, memory‐biomarker associations, and more. 59
The study's limitations include its small sample size, lack of APOE information, and TSPO PET as a general marker. The inclusion of only a few participants in the early stages of AD pathogenesis, that is, cognitively normal but amyloid‐positive and tau‐negative, may limit our ability to detect early effects (eg, TSPO‐mediating amyloid to tau associations). Therefore, our results may be capturing a microglial pathway specific to tau (eg, TSPO‐mediated tau spreading and downstream neurodegeneration and cognitive impairment) that is simply apparent in this range of the AD continuum. Alternatively, there may be a specific microglial response to tau that differs from that to amyloid, 60 and the pathway from amyloid to tau may depend on other aspects of neuroinflammation, including astrocyte reactivity, in cognitively normal, amyloid‐positive individuals. 61 APOE ε4 plays a modulatory role in neuroinflammation, such that APOE ε4 attenuates 62 or exaggerates 63 microglial activation, but the missingness pattern of APOE information precludes its inclusion in models. Simple main effects demonstrated higher PBR28 SUVR in APOE ε4 carriers, in addition to its known effects on amyloid, tau, neurodegeneration, and cognition. While TSPO is upregulated in microglial activation in rodent models, TSPO is not upregulated in humans due to a species‐specific promoter region on the TSPO gene. 64 Further, TSPO is expressed mostly in microglia 65 but to a lesser extent in astrocytes and endothelial cells. 64 , 66 , 67 However, changes in TSPO PET signal have been reported across many different neurodegenerative diseases, 68 and the majority of TSPO PET signals 69 , 70 likely reflect microglial recruitment and/or density rather than an explicit microglial activation process. Whether microglia are maintaining their homeostatic state, responding properly to a given stimulus in a given circumstance, or responding improperly to a given stimulus in a given circumstance is unknown with TSPO PET. Regardless, we have identified tau spreading and tau‐related neurodegeneration and cognitive impairment as a critical time point to further investigate with cell type‐ and function‐specific methods such as glycogen synthase kinase 3 (GSK‐3), cyclooxygenase‐1 (COX‐1), or COX‐2 71 in larger longitudinal studies.
Neuroinflammatory processes have been implicated in late‐onset AD, 72 , 73 early‐onset AD, 74 , 75 autosomal‐dominant AD, 76 and AD in adults with Down syndrome. 77 In our work, neuroinflammation may play a critical role in tau spreading across Braak stages as well as downstream neurodegenerative and cognitive consequences of tau burden, independently of neuroinflammatory processes that clear neurodegeneration‐related products. Other studies have also suggested that elevated TSPO signal precedes tau deposition throughout Braak staging. 33 Specific pathways in the AD progression model (eg, tau burden in early and middle Braak stages) may be more or less relevant depending on disease stage and severity of the study sample, and further work should elucidate the specific neuroinflammatory processes and microglial activation states on which to intervene. 10 , 78 Disease‐modifying drugs, particularly the recent U.S. Food and Drug Administration‐approved Aβ‐targeting antibodies, aducanumab 79 , 80 and lecanemab, 81 , 82 need to be better characterized in relation to the inflammatory responses that are induced to clear Aβ oligomers and protofibrils. Monitoring and controlling the inflammatory response may lead to a reduction in amyloid‐related imaging abnormalities (ARIA). 83 As other therapeutics are developed in clinical trials, treatment response stratification will be critical. One potential way to perform such stratification might be based on patterns of neuroinflammation that may precede distinct subtypes of tau burden 84 , 85 and neurodegeneration, 86 , 87 which have differential rates of cognitive decline. 88 Overall, neuroinflammation may be the mechanism by which tau spreads across Braak stage regions and leads to downstream neurodegeneration and cognitive impairments. TSPO PET using either 11C‐PBR28 or third‐generation TSPO radioligands, such as 11C‐ER17671, offers a suitable method of identifying key regionally and temporally specific neuroinflammatory processes in AD and related dementias.
CONFLICT OF INTEREST STATEMENT
WCK has a consulting agreement with Cerveau Technologies. However, Cerveau was not involved in the study design or interpretation of these results. No authors have conflicts of interest to report. Author disclosures are available in the Supporting Information.
CONSENT STATEMENT
All human subjects provided informed consent.
Supporting information
Supplemental Information.
Supplemental Information.
ACKNOWLEDGMENTS
This study utilized data acquired from research supported by National Institutes of Health (NIH) grants: K23AG052633, R01AG026158, R56AG034189. Research reported in this publication was supported by the National Institute on Aging of the NIH under Award P30AG066462 (Columbia University Alzheimer's Disease Research Center). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. This study was additionally supported by the Washington Heights‐Inwood Columbia Aging Project (WHICAP, P01AG07232, R01AG037212, RF1AG054023), and R00AG065506. 18F‐Florbetaben was supplied by Life Molecular Imaging. 18F‐MK‐6240 was supplied by Cerveau Technologies. The authors acknowledge Dr. Regina Santella, who performed the TSPO genotyping work.
Rossano SM, Johnson AS, Smith A, et al. Microglia measured by TSPO PET are associated with Alzheimer's disease pathology and mediate key steps in a disease progression model. Alzheimer's Dement. 2024;20:2397–2407. 10.1002/alz.13699
REFERENCES
- 1. Thal DR, Rub U, Orantes M, Braak H. Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791‐1800. [DOI] [PubMed] [Google Scholar]
- 2. Jack CR Jr., Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jack CR Jr., Bennett DA, Blennow K, et al. NIA‐AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Braak H, Braak E. Evolution of the neuropathology of Alzheimer's disease. Acta Neurol Scand Suppl. 1996;165:3‐12. [DOI] [PubMed] [Google Scholar]
- 5. Bejanin A, Schonhaut DR, La Joie R, et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer's disease. Brain. 2017;140(12):3286‐3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Healy LM, Zia S, Plemel JRJCB. Towards a definition of microglia heterogeneity. Commun Biol. 2022;5:1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li X, Li Y, Jin Y, et al. Transcriptional and epigenetic decoding of the microglial aging process. Nature Aging. 2023:1‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Paolicelli RC, Sierra A, Stevens B, et al. Microglia states and nomenclature: a field at its crossroads. Neuron. 2022;110:3458‐3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hansen DV, Hanson JE, Sheng Morgan. Microglia in Alzheimer's disease. J Cell Biol. 2018;217:459‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krasemann S, Madore C, Cialic R, et al. The TREM2‐APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47:566‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dani M, Wood M, Mizoguchi R, et al. Microglial activation correlates in vivo with both tau and amyloid in Alzheimer's disease. Brain. 2018;141:2740‐2754. [DOI] [PubMed] [Google Scholar]
- 12. Hanslik KL, Ulland TK. The role of microglia and the Nlrp3 inflammasome in Alzheimer's disease. Front Neurol. 2020;11:570711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hopp SC, Lin Y, Oakley D, et al. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer's disease. J Neuroinflammation. 2018;15:269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ising C, Venegas C, Zhang S, et al. NLRP3 inflammasome activation drives tau pathology. Nature. 2019;575:669‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee M, McGeer E, McGeer PL. Activated human microglia stimulate neuroblastoma cells to upregulate production of beta amyloid protein and tau: implications for Alzheimer's disease pathogenesis. Neurobiol Aging. 2015;36:42‐52. [DOI] [PubMed] [Google Scholar]
- 16. Stancu I‐C, Cremers N, Vanrusselt H, et al. Aggregated Tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non‐exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019;137:599‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Venegas C, Kumar S, Franklin BS, et al. Microglia‐derived ASC specks cross‐seed amyloid‐β in Alzheimer's disease. Nature. 2017;552:355‐361. [DOI] [PubMed] [Google Scholar]
- 18. Eikelenboom P, Van Exel E, Hoozemans JJ, Veerhuis R, Rozemuller AJ, Van Gool WA. Neuroinflammation–an early event in both the history and pathogenesis of Alzheimer's disease. Neurodegener Dis. 2010;7:38‐41. [DOI] [PubMed] [Google Scholar]
- 19. Serrano‐Pozo A, Mielke ML, Gómez‐Isla T, et al. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer's disease. Am J Pathol. 2011;179:1373‐1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sheffield LG, Marquis JG, Berman NE. Regional distribution of cortical microglia parallels that of neurofibrillary tangles in Alzheimer's disease. Neurosci Lett. 2000;285:165‐168. [DOI] [PubMed] [Google Scholar]
- 21. DeVos SL, Miller RL, Schoch KM, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9:eaag0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takeda S, Commins C, DeVos SL, et al. Seed‐competent high‐molecular‐weight tau species accumulates in the cerebrospinal fluid of Alzheimer's disease mouse model and human patients. Ann Neurol. 2016;80:355‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Walsh DM, Selkoe DJ. A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat Rev Neurosci. 2016;17:251‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rajendran L, Paolicelli RC. Microglia‐mediated synapse loss in Alzheimer's disease. J Neurosci. 2018;38:2911‐2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461‐553. [DOI] [PubMed] [Google Scholar]
- 26. Santos EN, Fields RD Regulation of myelination by microglia. Sci Adv. 2021;7:eabk1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang F, Ren S‐Y, Chen JF, et al. Myelin degeneration and diminished myelin renewal contribute to age‐related deficits in memory. Nat Neurosci. 2020;23:481‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hoozemans JJM, Veerhuis R, Rozemuller JM, Eikelenboom PJC, Targets ND‐D. Soothing the inflamed brain: effect of non‐steroidal anti‐inflammatory drugs on Alzheimer's disease pathology. CNS Neurol Disord Drug Targets. 2011;10:57‐67. [DOI] [PubMed] [Google Scholar]
- 29. Arvanitakis Z, Grodstein F, Bienias J, et al. Relation of NSAIDs to incident AD, change in cognitive function, and AD pathology. Neurology. 2008;70:2219‐2225. [DOI] [PubMed] [Google Scholar]
- 30. In'T Veld BA, Ruitenberg A, Hofman A, et al. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N Engl J Med. 2001;345:1515‐1521. [DOI] [PubMed] [Google Scholar]
- 31. Hampel H, Cummings J, Blennow K, Gao P, Jack CR Jr., Vergallo A. Developing the ATX(N) classification for use across the Alzheimer disease continuum. Nat Rev Neurol. 2021;17(9):580‐589. [DOI] [PubMed] [Google Scholar]
- 32. Kreisl WC, Fujita M, Fujimura Y, et al. Comparison of [11C]‐(R)‐PK 11195 and [11C] PBR28, two radioligands for translocator protein (18 kDa) in human and monkey: implications for positron emission tomographic imaging of this inflammation biomarker. Neuroimage. 2010;49:2924‐2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pascoal TA, Benedet AL, Ashton NJ, et al. Microglial activation and tau propagate jointly across Braak stages. Nat Med. 2021;27:1592‐1599. [DOI] [PubMed] [Google Scholar]
- 34. Kreisl WC, Lyoo CH, Liow JS, et al. Distinct patterns of increased translocator protein in posterior cortical atrophy and amnestic Alzheimer's disease. Neurobiol Aging. 2017;51:132‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kreisl WC, Lao PJ, Johnson A, et al. Patterns of tau pathology identified with 18F‐MK‐6240 PET imaging. Alzheimers Dement. 2022;18(2):272‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zou J, Tao S, Johnson A, et al. Microglial activation, but not tau pathology, is independently associated with amyloid positivity and memory impairment. Neurobiol Aging. 2020;85:11‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:270‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Folstein MF, Robins LN, Helzer JE. The mini‐mental state examination. Arch Gen Psychiatry. 1983;40:812. [DOI] [PubMed] [Google Scholar]
- 40. Ruff RM, Light RH, Quayhagen MJ. Selective reminding tests: a normative study of verbal learning in adults. J Clin Exp Neuropsychol. 1989;11:539‐550. [DOI] [PubMed] [Google Scholar]
- 41. Tombaugh TN. Trail making test A and B: normative data stratified by age and education. Neuropsychol. 2004;19:203‐214. [DOI] [PubMed] [Google Scholar]
- 42. Rosen WG. Verbal fluency in aging and dementia. J Clin Neurophysiol. 1980;2:135‐146. [Google Scholar]
- 43. Shirk SD, Mitchell MB, Shaughnessy LW, et al. A web‐based normative calculator for the uniform data set (UDS) neuropsychological test battery. Alzheimers Res Ther. 2011;3:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Owen DR, Yeo AJ, Gunn RN, et al. An 18‐kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab. 2012;32:1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pascoal TA, Benedet AL, Ashton NJ, et al. Publisher correction: microglial activation and tau propagate jointly across Braak stages. Nat Med. 2021;27:2048‐2049. [DOI] [PubMed] [Google Scholar]
- 46. Hammers A, Allom R, Koepp MJ, et al. Three‐dimensional maximum probability atlas of the human brain, with particular reference to the temporal lobe. Hum Brain Mapp. 2003;19:224‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thomas BA, Erlandsson K, Modat M, et al. The importance of appropriate partial volume correction for PET quantification in Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2011;38:1104‐1119. [DOI] [PubMed] [Google Scholar]
- 48. Rosseel YJ. Lavaan: an R package for structural equation modeling. J Stat Softw. 2012;48:1‐36. [Google Scholar]
- 49. Sobel ME. Asymptotic confidence intervals for indirect effects in structural equation models. Sociol Methodol. 1982;13:290‐312. [Google Scholar]
- 50. Yang J, Hu C, Guo N, Dutta J, et al. Partial volume correction for PET quantification and its impact on brain network in Alzheimer's disease. Sci Rep. 2017;7:13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17:157‐172. [DOI] [PubMed] [Google Scholar]
- 52. Kreisl WC, Lyoo CH, McGwier M, et al. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer's disease. Brain. 2013;136:2228‐2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Klein J, Yan X, Johnson A, et al. Olfactory impairment is related to tau pathology and neuroinflammation in Alzheimer's disease. J Alzheimers Dis. 2021;80:1051‐1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bradburn S, Murgatroyd C, Ray NJ. Neuroinflammation in mild cognitive impairment and Alzheimer's disease: a meta‐analysis. Ageing Res Rev. 2019;50:1‐8. [DOI] [PubMed] [Google Scholar]
- 55. Barthel H, Gertz H‐J, Dresel S, et al. Cerebral amyloid‐β PET with florbetaben (18F) in patients with Alzheimer's disease and healthy controls: a multicentre phase 2 diagnostic study. Lancet Neurol. 2011;10:424‐435. [DOI] [PubMed] [Google Scholar]
- 56. Betthauser TJ, Cody KA, Zammit MD, et al. In vivo characterization and quantification of neurofibrillary tau PET radioligand 18F‐MK‐6240 in humans from Alzheimer disease dementia to young controls. J Nucl Med. 2019;60:93‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lyoo CH, Ikawa M, Liow J‐S, et al. Cerebellum can serve as a pseudo‐reference region in Alzheimer disease to detect neuroinflammation measured with PET radioligand binding to translocator protein. J Nucl Med. 2015;56:701‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005‐2010. J Neuropathol Exp Neurol. 2012;71:266‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Weigand AJ, Edwards LE, Thomas KR, Bangen KJ, Bondi MW. Comprehensive characterization of elevated tau PET signal in the absence of amyloid‐beta. Brain Commun. 2022;4:fcac272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Navarro V, Sanchez‐Mejias E, Jimenez S, et al. Microglia in Alzheimer's disease: activated, dysfunctional or degenerative. Front Aging Neurosci. 2018;10:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bellaver B, Povala G, Ferreira PC, et al. Astrocyte reactivity influences amyloid‐β effects on tau pathology in preclinical Alzheimer's disease. Nat Med. 2023;29(7):1775‐1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yin Z, Rosenzweig N, Kleemann KL, et al. APOE4 impairs the microglial response in Alzheimer's disease by inducing TGFβ‐mediated checkpoints. Nat Immunol. 2023;24(11):1839‐1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ferrari‐Souza JP, Lussier FZ, Leffa DT, et al. APOE ε4 associates with microglial activation independently of Aβ plaques and tau tangles. Sci Adv. 2023;9:eade1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nutma E, Fancy N, Weinert M, et al. Translocator protein is a marker of activated microglia in rodent models but not human neurodegenerative diseases. Nat Commun. 2023;14:5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Venneti S, Wang G, Nguyen J, Wiley CA, Neurology E. The positron emission tomography ligand DAA1106 binds with high affinity to activated microglia in human neurological disorders. J Neuropathol Exp Neurol. 2008;67:1001‐1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Masdeu JC, Pascual B, Fujita MJ. Imaging neuroinflammation in neurodegenerative disorders. J Nucl Med. 2022;63:45S‐52S. [DOI] [PubMed] [Google Scholar]
- 67. Nutma E, Gebro E, Marzin MC, et al. Activated microglia do not increase 18 kDa translocator protein (TSPO) expression in the multiple sclerosis brain. Glia. 2021;69:2447‐2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gouilly D, Saint‐Aubert L, Ribeiro MJ, et al. Neuroinflammation PET imaging of the translocator protein (TSPO) in Alzheimer's disease: an update. Eur J Neurosci. 2022;55:1322‐1343. [DOI] [PubMed] [Google Scholar]
- 69. Zhou R, Ji B, Kong Y, et al. PET imaging of neuroinflammation in Alzheimer's disease. Front Immunol. 2021;12:739130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wimberley C, Lavisse S, Hillmer A, Hinz R, Turkheimer F, Zanotti‐Fregonara P. Kinetic modeling and parameter estimation of TSPO PET imaging in the human brain. Eur J Nucl Med Mol Imaging. 2021;49(1):246‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Narayanaswami V, Dahl K, Bernard‐Gauthier V, Josephson L, Cumming P, Vasdev NJ. Emerging PET radiotracers and targets for imaging of neuroinflammation in neurodegenerative diseases: outlook beyond TSPO. Mol Imaging. 2018;17:1536012118792317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Walker KA, Ficek BN, Westbrook RJ. Understanding the role of systemic inflammation in Alzheimer's disease. ACS Chem Neurosci. 2019;10(8):3340‐3342. [DOI] [PubMed] [Google Scholar]
- 73. Xie J, Van Hoecke L, Vandenbroucke RE. The impact of systemic inflammation on Alzheimer's disease pathology. Front Immunol. 2022;12:796867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shepherd CE, Grace EM, Mann DMA, Halliday GM. Relationship between neuronal loss and ‘inflammatory plaques’ in early onset Alzheimer's disease. Neuropathol Appl Neurobiol. 2007;33:328‐333. [DOI] [PubMed] [Google Scholar]
- 75. Elahi FM, Casaletto KB, La Joie R, et al. Plasma biomarkers of astrocytic and neuronal dysfunction in early‐and late‐onset Alzheimer's disease. Alzheimers Dement. 2020;16(4):681‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Arboleda‐Velasquez JF, Lopera F, O'Hare M, et al. Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019;25:1680‐1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Petersen ME, Zhang F, Schupf N, et al. Proteomic profiles for Alzheimer's disease and mild cognitive impairment among adults with Down syndrome spanning serum and plasma: an Alzheimer's Biomarker Consortium–Down Syndrome (ABC–DS) study. Alzheimers Dement (Amst). 2020;12:e12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hamelin L, Lagarde J, Dorothée G, et al. Distinct dynamic profiles of microglial activation are associated with progression of Alzheimer's disease. Brain. 2018;141:1855‐1870. [DOI] [PubMed] [Google Scholar]
- 79. Budd Haeberlein S, Aisen P, Barkhof F, et al. Two randomized phase 3 studies of aducanumab in early Alzheimer's disease. J Prev Alzheimers Dis. 2022;9:197‐210. [DOI] [PubMed] [Google Scholar]
- 80. Haddad HW, Malone GW, Comardelle NJ, Degueure AE, Kaye AM, Kaye ADJHPR. Aducanumab, a novel anti‐amyloid monoclonal antibody, for the treatment of Alzheimer's disease: a comprehensive review. Health Psychol Res. 2022;10:31925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, et al. Lecanemab in early Alzheimer's disease. N Engl J Med. 2023;388:9‐21. [DOI] [PubMed] [Google Scholar]
- 82. McDade E, Cummings JL, Dhadda S, et al. Lecanemab in patients with early Alzheimer's disease: detailed results on biomarker, cognitive, and clinical effects from the randomized and open‐label extension of the phase 2 proof‐of‐concept study. Alzheimers Res Ther. 2022;14:1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sperling RA, Jack , Black SE, et al. Amyloid‐related imaging abnormalities in amyloid‐modifying therapeutic trials: recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7:367‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Charil A, Shcherbinin S, Southekal S, et al. Tau subtypes of Alzheimer's disease determined in vivo using flortaucipir PET imaging. J Alzheimers Dis. 2019;71:1037‐1048. [DOI] [PubMed] [Google Scholar]
- 85. Vogel JW, Young AL, Oxtoby NP, et al. Four distinct trajectories of tau deposition identified in Alzheimer's disease. Nat Med. 2021;27:871‐881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ferreira D, Nordberg A, Westman E. Biological subtypes of Alzheimer disease: a systematic review and meta‐analysis. Neurology. 2020;94:436‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wheatley S, Mohanty R, Ferreira D, et al. Neurodegenerative pathways in Alzheimer's disease subtypes. Alzheimer's Dement. 2022;18:e067749. [Google Scholar]
- 88. Ferreira D, Verhagen C, Hernández‐Cabrera JA, et al. Distinct subtypes of Alzheimer's disease based on patterns of brain atrophy: longitudinal trajectories and clinical applications. Sci Rep. 2017;7:46263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Information.
Supplemental Information.