

Abstract
INTRODUCTION
Late‐onset Alzheimer's disease (LOAD) has a strong genetic component. Participants in Long‐Life Family Study (LLFS) exhibit delayed onset of dementia, offering a unique opportunity to investigate LOAD genetics.
METHODS
We conducted a whole genome sequence analysis of 3475 LLFS members. Genetic associations were examined in six independent studies (N = 14,260) with a wide range of LOAD risk. Association analysis in a sub‐sample of the LLFS cohort (N = 1739) evaluated the association of LOAD variants with beta amyloid (Aβ) levels.
RESULTS
We identified several single nucleotide polymorphisms (SNPs) in tight linkage disequilibrium within the MTUS2 gene associated with LOAD (rs73154407, p = 7.6 × 10−9). Association of MTUS2 variants with LOAD was observed in the five independent studies and was significantly stronger within high levels of Aβ42/40 ratio compared to lower amyloid.
DISCUSSION
MTUS2 encodes a microtubule associated protein implicated in the development and function of the nervous system, making it a plausible candidate to investigate LOAD biology.
Highlights
Long‐Life Family Study (LLFS) families may harbor late onset Alzheimer's dementia (LOAD) variants.
LLFS whole genome sequence analysis identified MTUS2 gene variants associated with LOAD.
The observed LLFS variants generalized to cohorts with wide range of LOAD risk.
The association of MTUS2 with LOAD was stronger within high levels of beta amyloid.
Our results provide evidence for MTUS2 gene as a novel LOAD candidate locus.
Keywords: genetic risk, late‐onset Alzheimer's disease, microtubule protein, MTUS2 gene, whole genome sequence
1. BACKGROUND
Alzheimer's disease (AD) affects an estimated 50 million people worldwide. Approximately 90% of cases are sporadic and occur after 65 years of age, that is, late‐onset AD (LOAD). 1 LOAD is complex and multifactorial with genetic and environmental factors 2 contributing to the brain accumulation of beta amyloid (Aβ) and tau proteins. 3 Twin studies have demonstrated the high heritability of LOAD, with overall estimates ranging from 58% to 79%. 4 Genome‐wide association studies (GWAS) revealed that LOAD risk is driven by multiple loci. These GWAS genes confer only weak to modest AD risk when compared with a 4‐ to 15‐fold increase in AD risk due to apolipoprotein E (APOE)‐ε4 inheritance. The identified susceptibility loci explain a small proportion of the LOAD indicating that undiscovered loci remain. 5 , 6
The largest GWAS meta‐analysis to date 7 identified 75 variants using genetic data from 880,000 subjects. Instead of identifying LOAD risk factors using large samples of the general population, we focused our discovery efforts on the Long‐Life Family Study (LLFS). LLFS aims to examine the genetic and non‐genetic factors associated with exceptional longevity. The LLFS cohort appears to be healthier than random cohorts of the same age/sex. 8 Previous studies examining the health of LLFS participants 9 have shown that offspring of long‐lived family members exhibit lower rates of diabetes mellitus, pulmonary disease, and peripheral arterial disease. Consistent with previously reports, 10 rates of dementia and memory decline among offspring of parents with exceptional longevity 10 are lower, and cognitive tests scores in the LLFS offspring generation are higher on average when compared to spouse controls. 11 LLFS results demonstrated that individuals who are part of exceptionally long‐lived families are protected against cognitive impairment characteristic of LOAD. 12 The genetic advantage is likely due to variants that decrease risk for aging‐related diseases. The exceptionally healthy LLFS population provides a unique opportunity to investigate the genetic contributions to LOAD. Our previous research has also shown that reduction of the frequency of APOE‐ε4 allele and the increase in the frequency of the APOE‐ε2 allele in the LLFS offspring generation contribute to longevity. 13
The majority of the genetic studies in LOAD have consisted of GWAS based on genotyping single nucleotide polymorphism (SNP) arrays or a combination of microarray‐based genotyping with high dense imputation efforts. 6 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 However, decreasing costs and advances in analytic methods have made high‐throughput sequencing a feasible alternative. 24 Whole genome sequence (WGS) GWAS (Seq‐GWAS) offers a significant advantage with the potential to detect all variants present in a sample, not only those present on an array or imputation reference panel. However, to date, those few Seq‐GWAS studies of LOAD have either exclusively focused on rare variants 25 or specific populations. 26
The present study aims to: (1) examine the association between WGS variants and LOAD in the LLFS cohort, and (2) validating the findings of LLFS in additional cohorts with different risk of dementia: high risk populations (i.e., familial LOAD and Down's syndrome [DS]), AD referral‐base cohort, and population‐based cohorts.
The selection of the DS datasets as validation cohorts is based on its strong association with AD. 27 This association has a genetic basis through a gene‐dose effect of the triplication of amyloid precursor protein (APP) gene in this population. Ninety percent of individuals with DS will have developed AD by age 70 (vs. 11% of people over age 65 in the general population). Because of these similarities, DS has been conceptualized as genetically determined AD, just like the autosomal dominant forms.
In the case of the familial LOAD, the most significant LOAD risk factor after advanced age is family history. The observed incidence rates for LOAD are estimated to be three to five times higher in multiplex families. 28 Individuals with a first‐degree relative affected by AD, are at 4‐ to 10‐fold higher risk for developing LOAD. 29 , 30 , 31 , 32
2. METHODS
2.1. Study cohorts
In addition to LLFS, the present study examined the findings in six external datasets. These cohorts include varying LOAD risk, ranging from those with high risk (The National Institute on Aging Alzheimer's Disease Family Based Study [NIA‐LOAD FBS], The Alzheimer Biomarkers Consortium—Down Syndrome [ABC‐DS], and The Multiomic Studies of Alzheimer's Disease in Adults with Down Syndrome Study [omicsADDS]), as well as those from the general population (the Religious Orders Study/Memory and Aging Project [ROSMAP], the Alzheimer's Disease Genetics Consortium [ADGC], and the Washington Heights/Inwood Columbia Aging Project [WHICAP]).
RESEARCH IN CONTEXT
Systematic review: Late‐onset Alzheimer's disease (LOAD) susceptibility loci identified explain a small proportion of its heritability, indicating that undiscovered loci remain. We focused on the Long‐Life Family Study (LLFS), a cohort designed to assess genetic and environmental factors associated with exceptional longevity. The exceptionally healthy LLFS population provides a unique opportunity to investigate LOAD genetics. Our study identified MTUS2 gene's variants associated with LOAD. The MTUS2 association with LOAD was observed in six independent studies with a wide range of LOAD risk and was significantly stronger within high plasma levels of beta amyloid.
Interpretation: MTUS2 gene encodes a microtubule associated protein implicated in nervous system development and function, making it a plausible candidate for LOAD.
Future directions: Future studies may seek to conduct functional validation analyses. Additionally, future studies may aim to consider the contribution of socioeconomic status, mental/behavioral health, and environmental factors to LOAD risk.
2.1.1. The Long‐Life Family Study (LLFS)
The LLFS is a longitudinal, family‐based study designed to assess genetic and environmental risk factors associated with exceptional longevity. Study design details can be found elsewhere. 8 AD status was determined by a dementia review committee. Briefly, LLFS participants were selected for dementia review based on the presence of any of the following: (i) Clinical Dementia Rating 33 score greater than zero, (ii) cognitive impairment consistent with dementia using a previously published diagnostic algorithm, 12 and (iii) if previous data were missing, the informant report of a cognitive problem was used. Participants included in the current analyses included those diagnosed with probable LOAD with and without stroke. Participants with mild cognitive impairment (MCI) and other types of dementia were not included in the analyses. Additionally, Danish participants were excluded due to the lack of clinical consensus dementia diagnosis.
Whole genome sequencing was carried out by the McDonnell Genome Institute (MGI) at Washington University by Illumina Sequencers. MGI subsequently quality control procedures included: (i) alignment to build GRCh38 with BWA‐MEM; (ii) marking duplicates with Picard; (iii) base quality score recalibration with GATK; (iv) lossless conversion to CRAM format with SAMtools. The quality of the derived CRAM files was reviewed using to filter out samples with high level of contamination and haploid coverage. Final analysis files excluded SNP variants by coverage, lack of informativeness (monomorphic or missing), and failing Hardy‐Weinberg test because of excess heterozygosity.
Plasma was collected in a random sample of 1739 LLFS participants, demographically comparable to the entire sample, and their corresponding Aβ levels were processed after 24–48 h. Centrifuged plasma aliquoted in polypropylene tubes and stored at −80°C was used to measure Aβ42 and Aβ40 using Luminex technology.
2.1.2. The National Institute on Aging Alzheimer's Disease Family Based Study (NIA‐LOAD FBS)
The NIA‐LOAD FBS is the largest collection of multiplex LOAD families recruited and longitudinally assessed worldwide. A detailed description of the study can be found elsewhere. 28 The present study used data from 4079 study participants. For analysis purposes, LOAD cases were defined using the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and related disorders association (NINCDS‐ADRDA) criteria. 34 Non‐demented subjects were defined as any individual with no evidence of LOAD, and subjects with MCI were excluded from analyses. Genome‐wide microarray data 35 were imputed using the HRC panel through the Michigan Imputation Server. 36
2.1.3. The Alzheimer Biomarkers Consortium‐Down Syndrome (ABC‐DS) and The Multiomic Studies of Alzheimer's Disease in Adults with Down Syndrome Study (omicsADDS)
The ABC‐DS is a multisite longitudinal study examining biomarkers of AD in a large cohort of adults with DS. 37 The omicsADDS study aims to identify multiomic profiles for successful cognitive aging in adults with DS. The recruitment and clinical assessment have been previously detailed. 38 , 39 , 40 For both cohorts, the determination of diagnostic status was made during case consensus conferences. As previously described, 38 diagnostic status considered for the present analyses were (1) cognitively stable, (2) MCI‐DS, (3) AD‐dementia. To maximize our statistical power, subjects with MCI were grouped with cognitively stable.
Genome‐wide genotyping in both cohorts was performed using the Illumina Infinium Global Screening Array‐24 v2.0 at the Center for Applied Genomics at Children's Hospital of Pennsylvania. Imputation was performed using the TOPMed imputation server. 36 For the present analysis, the ABC‐DS sample consisted of 366 participants and the omicsADDS consisted of 244 participants.
2.1.4. The Religious Orders Study/Memory and Aging Project (ROSMAP)
Study participants were drawn from two different population‐based cohorts: (i) The Religious Orders Study (ROS) study, which includes older Catholic nuns, priests, and brothers from groups across the United States and (ii) The Rush Memory and Aging Project (MAP), which includes older individuals from the metropolitan Chicago area. Detailed AD dementia are based on criteria of the joint working group of the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association (NINCDS/ADRDA). 34 Participants with MCI were excluded for the present analysis. As previously described, a subset of the ROSMAP samples (n = 1200) underwent whole genome sequencing. 42 To minimize population structure bias, analyses were restricted to those with White ancestry, a total of 912 samples.
2.1.5. The Alzheimer's Disease Genetics Consortium (ADGC)
The samples genotyped by the ADGC are distributed by the National Alzheimer's Coordinating Center (NACC). The NACC was established by the National Institute on Aging in 1999 to facilitate collaborative research by using data collected from the approximately 30 NIA‐funded Alzheimer's Disease Centers across the United States. Clinical diagnosis of Alzheimer's dementia is based on NINCDS‐ADRDA criteria. 34 Participants classified as mild cognitive impaired were excluded for this current analysis. The genotyping of NACC samples was performed on a variety of genotyping platforms as previously described. 22 Genetic imputation was carried out using IMPUTE v2 software 43 using the 1,000 Genomes Project as a reference. 44
2.1.6. The Washington Heights/Inwood Columbia Aging Project (WHICAP)
Participants were drawn from a community‐based multiethnic cohort via random sampling of Medicare beneficiaries living in northern Manhattan, New York. 45 Based on information obtained at the baseline and follow‐up visits, dementia diagnosis was made by consensus during diagnostic conferences. The dementia diagnosis was based on Diagnostic and Statistical Manual of Mental Disorders 46 as well NINCDS‐ADRDA criteria. 34 Mild cognitive impaired participants were not included in the present analysis. As previously described, 47 genotypic data underwent imputation using IMPUTE2 36 with the HRC r1.1.2016 reference panel.
2.2. Statistical analyses
SNP‐based association analyses in LLFS and in NIA‐LOAD FBS were conducted using generalized logistic mixed models implemented in GMMAT software. 48 To account for relatedness among participants in both cohorts, standardized relatedness matrix files were computed using GEMMA software. 49 Diagnosis of LOAD was used as outcome variable and sex, age, education, site of recruitment, kinship matrix, and first three principal components were modeled as covariates. In the DS cohorts, association analysis with MTUS2 variants were performed using PLINK software. 50 Because of their intellectual disability, educational levels are not available for DS individuals. However, based on previous findings, 51 all analyses have been adjusted for the severity of their intellectual disability. All models were adjusted for sex, age, level of functioning, and principal components. In ROSMAP, WHICAP, and ADGC cohorts, logistic models implemented in EPACTS software 52 were used to investigate the association of MTUS2 variants with LOAD. All analyses included sex, age, education, and principal components as covariates. In the LLFS cohort, SNPs were retained based on their minimum allele frequency (MAF ≥ 1%), Hardy‐Weinberg p‐value ≥ 5.7 × 10−7, and genotype missing proportion ≥5%.
In LLFS, gene‐based test of association for MTUS2 gene were conducted using EPACTS 52 software. Sex, age, education, and principal components adjusted analyses were conducted using the sequence kernel association test SKAT and incorporating the efficient Mixed‐Model Association eXpedited (EMMAX) algorithm. 49 , 53
Plasma Aβ levels (Aβ40 and Aβ42) were collected at visit two in a sub‐sample of 1739 LLFS participants. The socio‐demographic characteristics of the sample were comparable to the whole LLFS cohort (data not shown). Based on previous findings that have consistently reported that a lower Aβ42/40 ratio in plasma is associated with higher risk of AD, 54 , 55 , 56 , 57 we investigated whether the association of MTUS2 with LOAD was influenced by Aβ42/40 levels. Because of the relatively limited sample size, we speculated that association effects will be more evident using quartiles of Aβ42/40 ratio. The Aβ42/40 ratio quartiles were categorized as a dichotomous variable: quartiles 1 and 2 were considered as lowest quartiles; quartiles 3 and 4 were considered the highest quartiles. Association analyses were performed using generalized estimating equations in SPSS software to adjust for relatedness among LLFS's participants. We investigated the association of Aβ42/40 ratio levels with the MTUS2 variants identified in the GWAS analysis (11 SNPs in tight linkage disequilibrium [LD]), adjusting for sex, age, and LOAD diagnosis.
3. RESULTS
Characteristics of the study's participants are summarized in Table 1. The proportion of women ranged from 46% in ABC‐DS to 66% in ROSMAP. The average age of participants from the proband LLFS generation and ROSMAP was significantly older (89 ± 7 years and 89 ± 6, respectively) when compared to the participants from the other cohorts. The highest prevalence of LOAD (69%) was observed for ROSMAP participants, followed by the NIA‐LOAD FBS (60%). The highest educational level was observed for ROSMAP and ADGC with an average of 16 years of education.
TABLE 1.
Characteristics of study cohorts.
| LLFS | Replication datasets | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| All | Proband | Offspring | NIA‐FBS | ABC‐DS | omicsADDS | ROSMAP | ADGC | WHICAP | |
| N | 3475 | 1303 | 2172 | 4079 | 336 | 244 | 912 | 6977 | 851 |
| Percentage of women | 55 | 53 | 56 | 61 | 46 | 65 | 66 | 57 | 60 |
| Age (mean ± SD) | 71 ± 16 | 89 ± 7 | 61 ± 8 | 74 ± 11 | 46 ± 10 | 56 ± 7 | 89 ± 6 | 79 ± 8 | 81 ± 7 |
| Education (mean ± SD) | 14 ± 3 | 13 ± 2 | 16 ± 3 | 14 ± 3 | – | – | 16 ± 4 | 16 ± 3 | 14 ± 4 |
| Prevalence of LOAD (%) | 7 | 15 | 2 | 60 | 21 | 28 | 69 | 48 | 21 |
Abbreviations: ABC‐DS, Alzheimer's Biomarkers Consortium‐Down Syndrome; ADGC, Alzheimer's Disease Genetic Consortium; LLFS, Long‐Life Family Study; LOAD, late‐onset Alzheimer's disease; NIA‐FBS, The National Institute on Aging Alzheimer's Disease Family Based Study; omicsADDS, The Multiomic Studies of Alzheimer's Disease in Adults with Down Syndrome Study; ROSMAP, the Religious Orders Study/Memory and Aging Project; WHICAP, the Washington Heights/Inwood Columbia Aging Project.
As described in the manuscript, educational levels are not available for both Down Syndrome cohorts.
In LLFS, association results from the SNP‐based analysis are shown in Figures 1 and 2. Several SNPs in chromosome 1 reached genome‐wide significance levels (p ≤ 5 × 10−8); however, they cover a large genomic area of more than 500Kb, which encompassed at least four different genes, none of which reached significance at the gene level. Tables S1–S, 5 list the single‐SNP association results for those chromosomal regions in which at least one SNP reached the genome‐wide significance threshold (chromosomes 1, 6, 13, 20, and 21). In addition, Figures S1–S, 5 show the corresponding chromosomal regional association plots using Locuszoom. 58 , 59 On chromosome 13, a tight LD block of 11 SNPs spanning 1.6Kb (Table 2) was mapped to Microtubule Associated Scaffold Protein 2 gene, MTUS2. The strongest association was observed at MTUS2 intronic variant rs73154407 (p = 7.6 × 10−9). Gene‐based analyses also reached statistical significance, nominating MTUS2 gene as a LOAD risk locus (p = 2.6 × 10−8). In secondary analyses considering adjustment for APOE‐ε4 allele, MTUS2 associations remained statistically significant (data not shown). Likewise, we did not observe either a significant association of APOE locus with AD, consistent with our previous reports 13 on the lower likelihood of carrying an APOE‐ε4 allele as well as an elevated allele frequency of APOE‐ε2 60 among LLFS family members compared to similar‐aged spouses’ controls.
FIGURE 1.
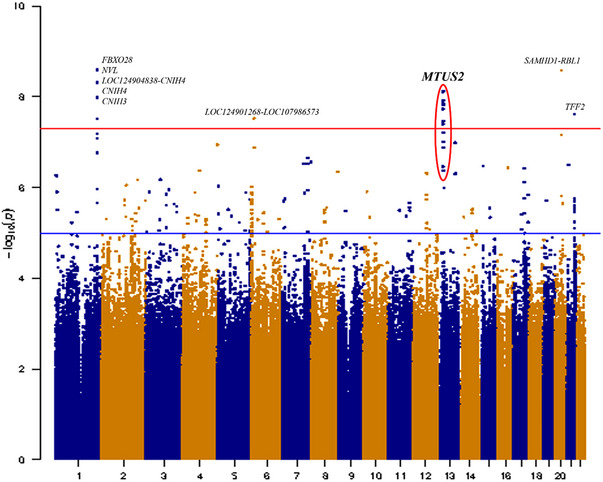
Manhattan plot of Seq‐GWAS results in LLFS cohort. The X‐axis represents the genomic position for each of the SNPs analyzed; Y‐axis represents the ‐log10 transformed p‐values. The red line indicates the genome‐wide significance threshold (p < 5 × 10−8). GWAS, genome‐wide association studies; LLFS, Long‐Life Family Study; SNP, single nucleotide polymorphism.
FIGURE 2.
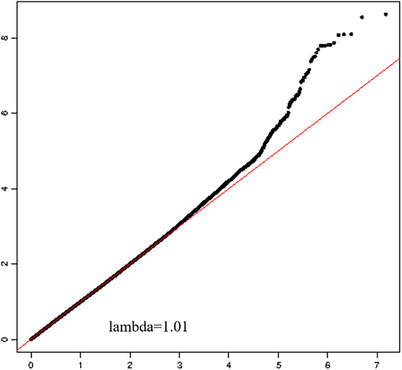
Q‐Q plot of Seq‐GWAS results in LLFS cohort. The X‐axis represents the expected ‐log10 transformed p‐values for each SNP association; the Y‐axis represents the observed ‐log10 transformed p‐values. GWAS, genome‐wide association studies; LLFS, Long‐Life Family Study; SNP, single nucleotide polymorphism.
TABLE 2.
GWAS top significant MTUS2 variants associated with late‐onset Alzheimer's disease.
| Chr | SNP | hg38 | A1 | A2 | N | MAF | B | SE | p‐Value |
|---|---|---|---|---|---|---|---|---|---|
| 13 | rs7330699 | 29,483,430 | G | A | 3,200 | 0.010 | 2.89 | 0.51 | 1.4 × 10−8 |
| 13 | rs73154407 | 29,483,753 | G | T | 3,184 | 0.010 | 2.97 | 0.51 | 7.6 × 10−9 |
| 13 | rs11842476 | 29,483,758 | G | A | 3,184 | 0.013 | 2.47 | 0.46 | 6.1 × 10−8 |
| 13 | rs73154410 | 29,483,772 | A | T | 3,187 | 0.010 | 2.96 | 0.51 | 7.8 × 10−9 |
| 13 | rs1350113738 | 29,483,860 | T | C | 3,184 | 0.010 | 2.88 | 0.51 | 1.5 × 10−8 |
| 13 | rs17073514 | 29,484,261 | G | T | 3,202 | 0.013 | 2.41 | 0.45 | 9.8 × 10−8 |
| 13 | rs116813765 | 29,484,326 | C | T | 3,204 | 0.010 | 2.89 | 0.51 | 1.4 × 10−8 |
| 13 | rs73154413 | 29,484,390 | C | T | 3,207 | 0.011 | 2.85 | 0.51 | 1.8 × 10−8 |
| 13 | rs73154414 | 29,484,568 | A | G | 3,201 | 0.010 | 2.91 | 0.51 | 1.2 × 10−8 |
| 13 | rs73154415 | 29,484,701 | A | G | 3,206 | 0.010 | 2.77 | 0.50 | 3.5 × 10−8 |
| 13 | rs41291229 | 29,484,987 | A | G | 3,186 | 0.010 | 2.75 | 0.50 | 4.0 × 10−8 |
Note: Physical positions in base pair (bp) correspond to the hg38 genome build.
Abbreviations: GWAS, genome‐wide association studies; MAF, minimum allele frequency; SNP, single nucleotide polymorphism.
The association of MTUS2 with LOAD observed in LLFS was validated using six different independent cohorts (Table 3). Due to the different genetic coverage of WGS and GWAS data, MTUS2 associated SNPs differ across the cohorts, and therefore meta‐analyses cannot be performed. We performed LD calculations using the NIH Web‐based application LDlink (LD matrix module; https://ldlink.nci.nih.gov/ = home). When assessing the LD between the top SNP in LLFS and SNPs in the replication cohorts, our results showed high LD measured as D’, but moderate‐low values as measured by r 2 (r 2 < 0.5).
TABLE 3.
Results of MTUS2 association with LOAD in independent cohorts.
| Cohort | SNP | bp (hg37) | N | B | SE | p | MAF | D' |
|---|---|---|---|---|---|---|---|---|
| NIA‐LOAD | rs775323299 | 28,795,404 | 4079 | 3.92 | 0.50 | 8.4 × 10−5 | 1.5 × 10−4 | NA |
| ABC‐DS | rs73164590 | 29,064,021 | 366 | 2.01 | 0.35 | 0.002 | 0.01 | 1.00 |
| omicsADDS | rs55646198 | 29,486,171 | 244 | 1.08 | 0.24 | 0.009 | 0.06 | 1.00 |
| ROSMAP | rs118034475 | 29,426,891 | 625 | 1.15 | 0.40 | 0.004 | 0.01 | 1.00 |
| WHICAP_NHW | rs68054178 | 29,128,208 | 839 | 4.12 | 0.62 | 3.7 × 10−5 | 0.07 | 1.00 |
| ADGC | rs581625 | 28,956,466 | 6977 | 3.15 | 0.60 | 0.002 | 0.17 | 0.42 |
Abbreviations: ABC‐DS, Alzheimer's Biomarkers Consortium‐Down Syndrome; ADGC, Alzheimer's Disease Genetic Consortium; LOAD, late‐onset Alzheimer's disease; NIA‐LOAD, The National Institute on Aging Alzheimer's Disease‐Late‐Onset Alzheimer's disease; omicsADDS, The Multiomic Studies of Alzheimer's Disease in Adults with Down Syndrome Study; ROSMAP, the Religious Orders Study/Memory and Aging Project; SNP, single nucleotide polymorphism; WHICAP, the Washington Heights/Inwood Columbia Aging Project.
The strongest association in the NIA‐LOAD FBS was observed at SNP variant rs775323299 (p = 8.4 × 10−5). Next, we analyzed two independent samples of DS. SNP variants rs4769747 and rs55646198 yielded the most significant association signal (ABC‐DS p = 0.009 and omicsADDS p = 0.009, respectively). In ROSMAP, WHICAP, and ADGC, the strongest associations were achieved at SNP rs9551636 (p = 4 × 10−4), rs68054178 (p = 3.7 × 10−5), and rs581625 (p = 0.002), respectively.
Results from LLFS (Table S6, Figure S6) revealed that MTUS2 variant rs73154415 was strongly associated with plasma Aβ42/40 ratio (B = 1.06, SE = 0.01, p = 1.9 × 10−5). Secondary analyses (Figure S1) demonstrated that the association of variant rs73154415 with LOAD become significantly stronger within high Ab42/40 ratio compared to lower amyloid ratio (B = 3.46, SE = 0.74, p = 3 × 10−6 vs. B = 1.66, SE = 0.67, p = 0.005, respectively).
4. DISCUSSION
Genome‐wide analysis of WGS data from healthy aging families identified MTUS2 as a novel LOAD genetic risk factor. The association of MTUS2 variants with LOAD was also observed in the six independent cohorts, including populations at high risk of LOAD. The association became stronger among subjects with high levels of plasma Aβ42/40 ratio.
MTUS2 is a plus end tracking protein involved in elongation of microtubules, 61 expressed in brain during development and adulthood. 62 Few studies reported variants in MTUS2 associated with LOAD. In a genome‐wide interaction study of cerebrospinal fluid T‐tau/Aβ42 ratio using the Alzheimer's Disease Neuroimaging Initiative cohort, MTUS2 variants significantly interacted with RLBP1L1 to explain a relatively high‐level variance of this biomarker, demonstrating their potential association with LOAD pathology. 63 Variants in MTUS2 were reported associated with an inflammatory LOAD subtype after having integrated post mortem brain gene co‐expression data from LOAD samples. 64 Chen and colleagues, 65 nominated MTUS2 as likely associated with AD using an epistasis detection method that completes a spatial search through a genetic algorithm. Results from the Accelerating Medicines Partnership for Alzheimer's disease (AMP‐AD) consortium RNA‐seq analysis of post mortem brains of more than 1100 individuals (https://agora.adknowledgeportal.org/), showed that MTUS2 was significantly expressed across different brain regions (Figure S7). Moreover, three of the brain regions (cerebellum, parahippocampal gyrus, and temporal cortex) showed significant change in MTUS2 gene expression when comparing LOAD cases and cognitively healthy samples (Figure S8).
Our results suggest that MTUS2 may play a role in Aβ metabolism. Aβ and tau protein deposition are the two principal pathological hallmarks of AD. The imbalance between Aβ production and clearance leads to the aggregation of amyloid plaques, while hyperphosphorylated tau protein transform into neurofibrillary tangles. 66 , 67 However, the relationship between amyloid and tau in the pathophysiology of LOAD remains unclear. It is known that tangles deposition is associated with cognitive decline, 68 , 69 while mutations in the APP cause familial AD. 70 The current literature suggests that plaques and tangles may act synergistically as both are necessary for cognitive decline and glucose hypometabolism. Evidence in cellular and mouse models also shows a synergistic effect, with the presence of amyloid peptides increasing the formation of certain species of hyperphosphorylated tau as well as of its aggregates. 71 , 72 The role of tau in amyloid plaque formation is less clear, although in at least one experiment, the presence of mutant tau increased the number of plaques in an APP/PSEN1 mouse model. 73 In another study using tau‐null animals, synapses were protected from Aβ toxicity, highlighting the potential synergy between the two proteins. 74
The integrity of microtubules is essential for neurons to maintain their morphology and to transport components between cell body and synaptic terminals. Aβ peptides can disrupt this transport through cytoskeleton reorganization which increases APP retention and its vesicular cleavage. 75 The mechanism through which Aβ affects microtubule stability involves detyrosination that causes hyperstabilization of the microtubules, which is a process dependent on APP expression. Although no direct link has been observed between MTUS2 and tau phosphorylation or microtubule hyperstabilization, the fact that it is involved in microtubule dynamics could explain the association of genetic variations of this protein and LOAD. Indeed, the fact that our variant is associated with plasma Aβ ratio supports the idea that microtubule stability is involved in APP metabolism.
Our study presents some limitations. First, differences in methodological approaches across the studies may introduce heterogeneity through biases variably, consequently affecting the results. Nonetheless, we have consistently shown an association between MTUS2 gene and AD in all the studies, suggesting that our findings are robust and reliable. Second, we did not consider the contribution of additional socioeconomic status, mental or behavioral health, and clinical comorbid conditions that may be associated with LOAD risk. Third, additional genetic factors in conjunction with lifestyle‐behavior‐environmental factors and their interactions may be needed for developing the disease. Fourth, functional analyses are needed to validate MTUS2 as candidate risk gene.
CONFLICT OF INTEREST STATEMENT
Bradley Christian receives PET precursor and compounds from Avid Radiopharmaceuticals Inc and equipment from Cerveau Technologies. Handen receives funding from National Institute of Child Health and Human Development, Autism Speaks, and Roche Pharmaceuticals. Mapstone is an inventor on patents related to biomarkers of neurodegenerative diseases owned by Georgetown University and the University of Rochester. The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All study participants provided written informed consent and the study procedures were approved by the institutional review boards within each institution. All study procedures were performed in accordance with the Declaration of Helsinki ethical principles.
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
We thank Wayne Silverman for his valuable comments and suggestions which helped us in improving the quality of the manuscript. The Long Family Study is supported by National Institute on Aging – National Institutes of Health grants (U01‐AG023746, U01‐AG023712, U01‐AG023749, U01‐AG023755, U01‐AG023744, and U19 AG063893). We would like to thank the research staff and LLFS participants for their substantial contribution to the study. Data collection and sharing for this project was supported by the Washington Heights‐Inwood Columbia Aging Project (WHICAP, R01 AG072474, RF1 AG066107) funded by the National Institute on Aging (NIA) and by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant Number UL1TR001873. The NIA‐LOAD study supported the collection of samples used in this study through National Institute on Aging (NIA) grants U24AG026395 and R01AG041797. The National Institutes of Health, National Institute on Aging (NIH‐NIA) supported this work through the following grants: ADGC, U01 AG032984, RC2 AG036528; Samples from the National Cell Repository for Alzheimer's Disease (NCRAD), which receives government support under a cooperative agreement grant (U24 AG21886) awarded by the National Institute on Aging (NIA), were used in this study. We thank contributors who collected samples used in this study, as well as patients and their families, whose help and participation made this work possible; Data for this study were prepared, archived, and distributed by the National Institute on Aging Alzheimer's Disease Data Storage Site (NIAGADS) at the University of Pennsylvania (U24‐AG041689); GCAD, U54 AG052427; NACC, U01 AG016976; NIA LOAD (Columbia University), U24 AG026395, U24 AG026390, R01AG041797; Columbia University, P50 AG008702, R37 AG015473, R01 AG037212, R01 AG028786; The NACC database is funded by NIA/NIH Grant U24 AG072122. NACC data are contributed by the NIA‐funded ADRCs: P30 AG062429 (PI James Brewer, MD, PhD), P30 AG066468 (PI Oscar Lopez, MD), P30 AG062421 (PI Bradley Hyman, MD, PhD), P30 AG066509 (PI Thomas Grabowski, MD), P30 AG066514 (PI Mary Sano, PhD), P30 AG066530 (PI Helena Chui, MD), P30 AG066507 (PI Marilyn Albert, PhD), P30 AG066444 (PI John Morris, MD), P30 AG066518 (PI Jeffrey Kaye, MD), P30 AG066512 (PI Thomas Wisniewski, MD), P30 AG066462 (PI Scott Small, MD), P30 AG072979 (PI David Wolk, MD), P30 AG072972 (PI Charles DeCarli, MD), P30 AG072976 (PI Andrew Saykin, PsyD), P30 AG072975 (PI David Bennett, MD), P30 AG072978 (PI Neil Kowall, MD), P30 AG072977 (PI Robert Vassar, PhD), P30 AG066519 (PI Frank LaFerla, PhD), P30 AG062677 (PI Ronald Petersen, MD, PhD), P30 AG079280 (PI Eric Reiman, MD), P30 AG062422 (PI Gil Rabinovici, MD), P30 AG066511 (PI Allan Levey, MD, PhD), P30 AG072946 (PI Linda Van Eldik, PhD), P30 AG062715 (PI Sanjay Asthana, MD, FRCP), P30 AG072973 (PI Russell Swerdlow, MD), P30 AG066506 (PI Todd Golde, MD, PhD), P30 AG066508 (PI Stephen Strittmatter, MD, PhD), P30 AG066515 (PI Victor Henderson, MD, MS), P30 AG072947 (PI Suzanne Craft, PhD), P30 AG072931 (PI Henry Paulson, MD, PhD), P30 AG066546 (PI Sudha Seshadri, MD), P20 AG068024 (PI Erik Roberson, MD, PhD), P20 AG068053 (PI Justin Miller, PhD), P20 AG068077 (PI Gary Rosenberg, MD), P20 AG068082 (PI Angela Jefferson, PhD), P30 AG072958 (PI Heather Whitson, MD), P30 AG072959 (PI James Leverenz, MD). The Religious Orders Study is funded by P30AG010161, R01AG015819, R01AG017917, RF1AG022018. The Alzheimer's Biomarkers Consortium–Down Syndrome (ABC‐DS) is funded by the National Institute on Aging and the National Institute for Child Health and Human Development (U01 AG051406, U01 AG051412, U19 AG068054). The work contained in this publication was also supported through the following National Institutes of Health Programs: The Alzheimer's Disease Research Centers Program (P50 AG008702, P30 AG062421, P50 AG16537, P50 AG005133, P50 AG005681, P30 AG062715, and P30 AG066519), the Eunice Kennedy Shriver Intellectual and Developmental Disabilities Research Centers Program (U54 HD090256, U54 HD087011, and P50 HD105353), the National Center for Advancing Translational Sciences (UL1 TR001873, UL1 TR002373, UL1 TR001414, UL1 TR001857, UL1 TR002345), the National Centralized Repository for Alzheimer Disease and Related Dementias (U24 AG21886), and DS‐Connect® (The Down Syndrome Registry) supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD). In Cambridge, UK this research was supported by the NIHR Cambridge Biomedical Research Centre and the Windsor Research Unit, CPFT, Fulbourn Hospital Cambridge, UK. The authors are grateful to the ABC‐DS study participants, their families and care providers, and the ABC‐DS research and support staff for their contributions to this study. This manuscript has been reviewed by ABC‐DS investigators for scientific content and consistency of data interpretation with previous ABC‐DS study publications. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH, the CPFT, the NIHR or the UK Department of Health and Social Care.
Xicota L, Cosentino S, Vardarajan B, et al. Whole genome‐wide sequence analysis of long‐lived families (Long‐Life Family Study) identifies MTUS2 gene associated with late‐onset Alzheimer's disease. Alzheimer's Dement. 2024;20:2670–2679. 10.1002/alz.13718
Contributor Information
Sandra Barral, Email: sandritabarral@gmail.com.
Alzheimer's Disease Genetic Consortium (ADGC):
Erin Abner, Perrie M. Adams, Alyssa Aguirre, Marilyn S. Albert, Roger L. Albin, Mariet Allen, Lisa Alvarez, Howard Andrews, Liana G. Apostolova, Steven E. Arnold, Sanjay Asthana, Craig S. Atwood, Gayle Ayres, Robert C. Barber, Lisa L. Barnes, Sandra Barral, Jackie Bartlett, Thomas G. Beach, James T. Becker, Gary W. Beecham, Penelope Benchek, David A. Bennett, John Bertelson, Sarah A. Biber, Thomas D. Bird, Deborah Blacker, Bradley F. Boeve, James D. Bowen, Adam Boxer, James B. Brewer, James R. Burke, Jeffrey M. Burns, William S. Bush, Joseph D. Buxbaum, Goldie Byrd, Laura B. Cantwell, Chuanhai Cao, Cynthia M. Carlsson, Minerva M. Carrasquillo, Kwun C. Chan, Scott Chasse, Yen‐Chi Chen, Marie‐Francoise Chesselet, Nathaniel A. Chin, Helena C. Chui, Jaeyoon Chung, Suzanne Craft, Paul K. Crane, Marissa Cranney, Carlos Cruchaga, Michael L. Cuccaro, Jessica Culhane, C. Munro Cullum, Eveleen Darby, Barbara Davis, Philip L. De Jager, Charles DeCarli, John C. DeToledo, Dennis W. Dickson, Nic Dobbins, Ranjan Duara, Nilufer Ertekin‐Taner, Denis A. Evans, Kelley M. Faber, Thomas J. Fairchild, Daniele Fallin, Kenneth B. Fallon, David W. Fardo, Martin R. Farlow, John Farrell, Lindsay A. Farrer, Victoria Fernandez‐Hernandez, Tatiana M. Foroud, Matthew P. Frosch, Douglas R. Galasko, Adriana Gamboa, Kathryn M. Gauthreaux, Tamar Gefen, Daniel H. Geschwind, Bernardino Ghetti, John R. Gilbert, Alison M. Goate, Thomas Grabowski, Neill R. Graff‐Radford, Anthony R. Griswold, Jonathan L. Haines, Hakon Hakonarson, Kathleen Hall, James R. Hall, Ronald L. Hamilton, Kara L. Hamilton‐Nelson, Xudong Han, Oscar Harari, John Hardy, Lindy E. Harrell, Elizabeth Head, Victor Henderson, Michelle Hernandez, Lawrence S. Honig, Ryan M. Huebinger, Matthew J. Huentelman, Christine M. Hulette, Bradley T. Hyman, Linda Hynan, Laura Ibanez, Gail P. Jarvik, Suman Jayadev, Lee‐Way Jin, Kimberly Johnson, Leigh Johnson, Bruce Jones, Gyungah Jun, M. Ilyas Kamboh, Moon Il Kang, Anna Karydas, Mindy J. Katz, John S. K. Kauwe, Jeffrey A. Kaye, C. Dirk Keene, Benjamin Keller, Aisha Khaleeq, Ronald Kim, Janice Knebl, Neil W. Kowall, Joel H. Kramer, Walter A. Kukull, Brian W. Kunkle, Amanda P. Kuzma, Frank M. LaFerla, James J. Lah, Eric B. Larson, Melissa Lerch, Alan J. Lerner, Yuk Ye Leung, James B. Leverenz, Allan I. Levey, Andrew P. Lieberman, Richard B. Lipton, Oscar L. Lopez, Kathryn L. Lunetta, Constantine G. Lyketsos, Douglas Mains, Jennifer Manly, Logue Mark, David Marquez, Daniel C. Marson, Eden R. Martin, Eliezer Masliah, Paul Massman, Arjun V. Masurkar, Richard Mayeux, Wayne C. McCormick, Susan M. McCurry, Stefan McDonough, Ann C. McKee, Marsel Mesulam, Jesse Mez, Bruce L. Miller, Carol A. Miller, Charles Mock, Abhay Moghekar, Thomas J. Montine, Edwin Monuki, Sean D. Mooney, John C. Morris, Shubhabrata Mukherjee, Amanda J. Myers, Adam C. Naj, Trung Nguyen, James Noble, Kelley Nudelman, Sid E. O'Bryant, Kyle Ormsby, Marcia Ory, Raymond Palmer, Joseph E. Parisi, Henry L. Paulson, Valory Pavlik, David Paydarfar, Victoria Perez, Margaret A. Pericak‐Vance, Ronald C. Petersen, Marsha Polk, Liming Qu, Mary Quiceno, Joseph F. Quinn, Ashok Raj, Farid Rajabli, Vijay Ramanan, Eric M. Reiman, Joan S. Reisch, Christiane Reitz, John M. Ringman, Erik D. Roberson, Monica Rodriguear, Ekaterina Rogaeva, Howard J. Rosen, Roger N. Rosenberg, Donald R. Royall, Mary Sano, Andrew J. Saykin, Gerard D. Schellenberg, Julie A. Schneider, Lon S. Schneider, William W. Seeley, Richard M. Sherva, Dean K. Shibata, Scott Small, Amanda G. Smith, Janet Smith, Yeunjoo Song, Salvatore Spina, Peter St George‐Hyslop, Robert A. Stern, Alan Stevens, Stephen Strittmatter, David Sultzer, Russell H. Swerdlow, Andrew Teich, Jeffrey Tilson, Giuseppe Tosto, John Q. Trojanowski, Juan C. Troncoso, Debby W. Tsuang, Otto Valladares, Vivianna M. Van Deerlin, Christopher Van Dyck, Linda J. Van Eldik, Jeffery M. Vance, Badri N. Vardarajan, Robert Vassar, Harry V. Vinters, Li‐San Wang, Sandra Weintraub, Kathleen A. Welsh‐Bohmer, Nick Wheeler, Ellen Wijsman, Kirk C. Wilhelmsen, Benjamin Williams, Jennifer Williamson, Henrick Wilms, Thomas S. Wingo, Thomas Wisniewski, Randall L. Woltjer, Martin Woon, Steven G. Younkin, Lei Yu, Yi Zhao, Xiongwei Zhou, and Congcong Zhu
Alzheimer's Biomarkers Consortium‐Down Syndrome (ABC‐DS):
Howard J. Aizenstein, Beau M. Ances, Howard F. Andrews, Karen Bell, Rasmus M. Birn, Adam M. Brickman, Peter Bulova, Amrita Cheema, Kewei Chen, Bradley T. Christian, Isabel Clare, Ann D. Cohen, John N. Constantino, Eric W. Doran, Anne Fagan, Eleanor Feingold, Tatiana M. Foroud, Benjamin L. Handen, Jordan Harp, Sigan L. Hartley, Elizabeth Head, Rachel Henson, Christy Hom, Lawrence Honig, Milos D. Ikonomovic, Sterling C Johnson, Courtney Jordan, M. Ilyas Kamboh, David Keator, William E. Klunk, Julia K. Kofler, Sharon J. Krinsky‐McHale, Florence Lai, Patrick Lao, Charles Laymon, Joseph Lee, Ira T. Lott, Victoria Lupson, Mark Mapstone, Chester A. Mathis, Davneet Singh Minhas, Neelesh Nadkarni, Sid O'Bryant, Melissa Parisi, Deborah Pang, Melissa Petersen, Julie C. Price, Margaret Pulsifer, Michael S. Rafii, Eric Reiman, Batool Rizvi, Herminia Diana Rosas, Laurie Ryan, Frederick Schmitt, Nicole Schupf, Wayne P. Silverman, Dana L. Tudorascu, Rameshwari Tumuluru, Badri Varadarajan, Desiree A. White, Michael A. Yassa, Shahid Zaman, and Fan Zhang
REFERENCES
- 1. Harman D. Alzheimer's disease pathogenesis: role of aging. Ann N Y Acad Sci. 2006;1067:454‐460. [DOI] [PubMed] [Google Scholar]
- 2. Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168‐174. [DOI] [PubMed] [Google Scholar]
- 3. Ibanez L, Cruchaga C, Fernandez MV. Advances in genetic and molecular understanding of Alzheimer's disease. Genes (Basel). 2021;12(8):1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Karlsson IK, Escott‐Price V, Gatz M, et al. Measuring heritable contributions to Alzheimer's disease: polygenic risk score analysis with twins. Brain Commun. 2022;4:fcab308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(10):a006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jansen IE, Savage JE, Watanabe K, et al. Genome‐wide meta‐analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019;51:404‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bellenguez C, Kucukali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat Genet. 2022;54:412‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wojczynski MK, Jiuan Lin S, Sebastiani P, et al. NIA long life family study: objectives, design, and heritability of cross‐sectional and longitudinal phenotypes. J Gerontol A Biol Sci Med Sci. 2022;77:717‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Newman AB, Glynn NW, Taylor CA, et al. Health and function of participants in the Long Life Family Study: a comparison with other cohorts. Aging (Albany NY). 2011;3:63‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lipton RB, Hirsch J, Katz MJ, et al. Exceptional parental longevity associated with lower risk of Alzheimer's disease and memory decline. J Am Geriatr Soc. 2010;58:1043‐1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barral S, Cosentino S, Costa R, et al. Cognitive function in families with exceptional survival. Neurobiol Aging. 2012;33:619 e1‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cosentino S, Schupf N, Christensen K, Andersen SL, Newman A, Mayeux R. Reduced prevalence of cognitive impairment in families with exceptional longevity. JAMA Neurol. 2013;70:867‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schupf N, Barral S, Perls T, et al. Apolipoprotein E and familial longevity. Neurobiol Aging. 2013;34:1287‐1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Rojas I, Moreno‐Grau S, Tesi N, et al. Common variants in Alzheimer's disease and risk stratification by polygenic risk scores. Nat Commun. 2021;12:3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kunkle BW, Grenier‐Boley B, Sims R, et al. Genetic meta‐analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414‐430. Erratum in: Nat Genet. 2019;51(9):1423-1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lambert JC, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marioni RE, Harris SE, Zhang Q, et al. GWAS on family history of Alzheimer's disease. Transl Psychiatry. 2018;8:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schwartzentruber J, Cooper S, Liu JZ, et al. Genome‐wide meta‐analysis, fine‐mapping and integrative prioritization implicate new Alzheimer's disease risk genes. Nat Genet. 2021;53:392‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wightman DP, Jansen IE, Savage JE, et al. A genome‐wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer's disease. Nat Genet. 2021;53:1276‐1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harold D, Abraham R, Hollingworth P, et al. Genome‐wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41:1088‐1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moreno‐Grau S, de Rojas I, Hernandez I, et al. Genome‐wide association analysis of dementia and its clinical endophenotypes reveal novel loci associated with Alzheimer's disease and three causality networks: the GR@ACE project. Alzheimers Dement. 2019;15:1333‐1347. [DOI] [PubMed] [Google Scholar]
- 22. Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late‐onset Alzheimer's disease. Nat Genet. 2011;43:436‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seshadri S, Fitzpatrick AL, Ikram MA, et al. Genome‐wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303:1832‐1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McMahon A, Lewis E, Buniello A, et al. Sequencing‐based genome‐wide association studies reporting standards. Cell Genom. 2021;1:100005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Prokopenko D, Morgan SL, Mullin K, et al. Whole‐genome sequencing reveals new Alzheimer's disease‐associated rare variants in loci related to synaptic function and neuronal development. Alzheimers Dement. 2021;17:1509‐1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shigemizu D, Asanomi Y, Akiyama S, Mitsumori R, Niida S, Ozaki K. Whole‐genome sequencing reveals novel ethnicity‐specific rare variants associated with Alzheimer's disease. Mol Psychiatry. 2022;27:2554‐2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Iulita MF, Garzon Chavez D, Klitgaard Christensen M, et al. Association of Alzheimer disease with life expectancy in people with down syndrome. JAMA Netw Open. 2022;5:e2212910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reyes‐Dumeyer D, Faber K, Vardarajan B, et al. The National Institute on Aging Late‐Onset Alzheimer's Disease Family Based Study: a resource for genetic discovery. Alzheimers Dement. 2022;18:1889‐1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cupples LA, Farrer LA, Sadovnick AD, Relkin N, Whitehouse P, Green RC. Estimating risk curves for first‐degree relatives of patients with Alzheimer's disease: the REVEAL study. Genet Med. 2004;6:192‐196. [DOI] [PubMed] [Google Scholar]
- 30. Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta‐analysis. APOE and Alzheimer disease meta analysis consortium. JAMA. 1997;278:1349‐1356. [PubMed] [Google Scholar]
- 31. Green RC, Cupples LA, Go R, et al. Risk of dementia among white and African American relatives of patients with Alzheimer disease. JAMA. 2002;287:329‐336. [DOI] [PubMed] [Google Scholar]
- 32. Silverman JM, Ciresi G, Smith CJ, Marin DB, Schnaider‐Beeri M. Variability of familial risk of Alzheimer disease across the late life span. Arch Gen Psychiatry. 2005;62:565‐573. [DOI] [PubMed] [Google Scholar]
- 33. Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412‐2414. [DOI] [PubMed] [Google Scholar]
- 34. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wijsman EM, Pankratz ND, Choi Y, et al. Genome‐wide association of familial late‐onset Alzheimer's disease replicates BIN1 and CLU and nominates CUGBP2 in interaction with APOE. PLoS Genet. 2011;7:e1001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Das S, Forer L, Schonherr S, et al. Next‐generation genotype imputation service and methods. Nat Genet. 2016;48:1284‐1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Handen BL, Lott IT, Christian BT, et al. The Alzheimer's Biomarker Consortium‐Down Syndrome: rationale and methodology. Alzheimers Dement (Amst). 2020;12:e12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Krinsky‐McHale SJ, Zigman WB, Lee JH, et al. Promising outcome measures of early Alzheimer's dementia in adults with Down syndrome. Alzheimers Dement (Amst). 2020;12:e12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee JH, Lee AJ, Dang LH, et al. Candidate gene analysis for Alzheimer's disease in adults with Down syndrome. Neurobiol Aging. 2017;56:150‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schupf N, Lee A, Park N, et al. Candidate genes for Alzheimer's disease are associated with individual differences in plasma levels of beta amyloid peptides in adults with Down syndrome. Neurobiol Aging. 2015;36:2907 e1‐2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS. Overview and findings from the rush Memory and Aging Project. Curr Alzheimer Res. 2012;9:646‐663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. De Jager PL, Ma Y, McCabe C, et al. A multi‐omic atlas of the human frontal cortex for aging and Alzheimer's disease research. Sci Data. 2018;5:180142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome‐wide association studies by imputation of genotypes. Nat Genet. 2007;39:906‐913. [DOI] [PubMed] [Google Scholar]
- 44. Genomes Project C , Abecasis GR, Auton A, Brooks LD, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tang MX, Cross P, Andrews H, et al. Incidence of AD in African‐Americans, Caribbean Hispanics, and Caucasians in northern Manhattan. Neurology. 2001;56:49‐56. [DOI] [PubMed] [Google Scholar]
- 46. Guze SB. Diagnostic and Statistical Manual of Mental Disorders: DSM‐IV. 4th ed. American Psychiatric Association; 1994. [Google Scholar]
- 47. Lee AJ, Raghavan NS, Bhattarai P, et al. FMNL2 regulates gliovascular interactions and is associated with vascular risk factors and cerebrovascular pathology in Alzheimer's disease. Acta Neuropathol. 2022;144:59‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen H, Huffman JE, Brody JA, et al. Efficient variant set mixed model association tests for continuous and binary traits in large‐scale whole‐genome sequencing studies. Am J Hum Genet. 2019;104:260‐274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou X, Stephens M. Genome‐wide efficient mixed‐model analysis for association studies. Nat Genet. 2012;44:821‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Purcell S, Neale B, Todd‐Brown K, et al. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet. 2007;81:559‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Burt DB, Loveland KA, Chen YW, Chuang A, Lewis KR, Cherry L. Aging in adults with Down syndrome: report from a longitudinal study. Am J Ment Retard. 1995;100:262‐270. [PubMed] [Google Scholar]
- 52. EPACTS: Efficient and Parallelizable Association Container Toolbox. http://genome.sph.umich.edu/wiki/EPACTS
- 53. Kang HM, Sul JH, Service SK, et al. Variance component model to account for sample structure in genome‐wide association studies. Nat Genet. 2010;42:348‐354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Graff‐Radford NR, Crook JE, Lucas J, et al. Association of low plasma Abeta42/Abeta40 ratios with increased imminent risk for mild cognitive impairment and Alzheimer disease. Arch Neurol. 2007;64:354‐362. [DOI] [PubMed] [Google Scholar]
- 55. Lambert JC, Schraen‐Maschke S, Richard F, et al. Association of plasma amyloid beta with risk of dementia: the prospective Three‐City Study. Neurology. 2009;73:847‐853. [DOI] [PubMed] [Google Scholar]
- 56. Chouraki V, Beiser A, Younkin L, et al. Plasma amyloid‐beta and risk of Alzheimer's disease in the Framingham Heart Study. Alzheimers Dement. 2015;11:249‐257. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. van Oijen M, Hofman A, Soares HD, Koudstaal PJ, Breteler MM. Plasma Abeta(1‐40) and Abeta(1‐42) and the risk of dementia: a prospective case‐cohort study. Lancet Neurol. 2006;5:655‐660. [DOI] [PubMed] [Google Scholar]
- 58. Boughton AP, Welch RP, Flickinger M, et al. LocusZoom.js: interactive and embeddable visualization of genetic association study results. Bioinformatics. 2021;37:3017‐3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome‐wide association scan results. Bioinformatics. 2010;26:2336‐2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sweigart B, Andersen SL, Gurinovich A, et al. APOE E2/E2 is associated with slower rate of cognitive decline with age. J Alzheimers Dis. 2021;83:853‐860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jiang K, Wang J, Liu J, et al. TIP150 interacts with and targets MCAK at the microtubule plus ends. EMBO Rep. 2009;10:857‐865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Du Puy L, Beqqali A, Monshouwer‐Kloots J, Haagsman HP, Roelen BA, Passier R. CAZIP, a novel protein expressed in the developing heart and nervous system. Dev Dyn. 2009;238:2903‐2911. [DOI] [PubMed] [Google Scholar]
- 63. Li J, Zhang Q, Chen F, et al. Genome‐wide association and interaction studies of CSF T‐tau/Aβ42 ratio in ADNI cohort. Neurobiol Aging. 2017;57:247 e1‐247.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Milind N, Preuss C, Haber A, et al. Transcriptomic stratification of late‐onset Alzheimer's cases reveals novel genetic modifiers of disease pathology. PLoS Genet. 2020;16:e1008775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen Y, Xu F, Pian C, et al. EpiMOGA: an epistasis detection method based on a multi‐objective genetic algorithm. Genes (Basel). 2021;12:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353‐356. [DOI] [PubMed] [Google Scholar]
- 67. Xiao X, Jiao B, Liao X, et al. Association of genes involved in the metabolic pathways of amyloid‐β and tau proteins with sporadic late‐onset Alzheimer's disease in the southern Han Chinese population. Front Aging Neurosci. 2020;12:584801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Arriagada PV, Growdon JH, Hedley‐Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631‐639. [DOI] [PubMed] [Google Scholar]
- 69. Hanseeuw BJ, Betensky RA, Jacobs HIL, et al. Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol. 2019;76:915‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Blacker D, Tanzi RE. The genetics of Alzheimer disease: current status and future prospects. Arch Neurol. 1998;55:294‐296. [DOI] [PubMed] [Google Scholar]
- 71. Rajmohan R, Reddy PH. Amyloid‐beta and phosphorylated tau accumulations cause abnormalities at synapses of Alzheimer's disease neurons. J Alzheimers Dis. 2017;57:975‐999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zheng WH, Bastianetto S, Mennicken F, Ma W, Kar S. Amyloid beta peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience. 2002;115(1):201‐211. [DOI] [PubMed] [Google Scholar]
- 73. Guo Q, Li H, Cole AL, Hur JY, Li Y, Zheng H. Modeling Alzheimer's disease in mouse without mutant protein overexpression: cooperative and independent effects of Aβ and tau. PLoS One. 2013;8:e80706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Pickett EK, Herrmann AG, McQueen J, et al. Amyloid beta and tau cooperate to cause reversible behavioral and transcriptional deficits in a model of Alzheimer's disease. Cell Rep. 2019;29:3592‐3604.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Henriques AG, Vieira SI, da Cruz ESEF, da Cruz ESOA. Abeta promotes Alzheimer's disease‐like cytoskeleton abnormalities with consequences to APP processing in neurons. J Neurochem. 2010;113:761‐771. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information