

Abstract
INTRODUCTION
Although dementia‐related proteinopathy has a strong negative impact on public health, and is highly heritable, understanding of the related genetic architecture is incomplete.
METHODS
We applied multidimensional generalized partial credit modeling (GPCM) to test genetic associations with dementia‐related proteinopathies. Data were analyzed to identify candidate single nucleotide variants for the following proteinopathies: Aβ, tau, α‐synuclein, and TDP‐43.
RESULTS
Final included data comprised 966 participants with neuropathologic and WGS data. Three continuous latent outcomes were constructed, corresponding to TDP‐43‐, Aβ/Tau‐, and α‐synuclein‐related neuropathology endophenotype scores. This approach helped validate known genotype/phenotype associations: for example, TMEM106B and GRN were risk alleles for TDP‐43 pathology; and GBA for α‐synuclein/Lewy bodies. Novel suggestive proteinopathy‐linked alleles were also discovered, including several (SDHAF1, TMEM68, and ARHGEF28) with colocalization analyses and/or high degrees of biologic credibility.
DISCUSSION
A novel methodology using GPCM enabled insights into gene candidates for driving misfolded proteinopathies.
Highlights
Latent factor scores for proteinopathies were estimated using a generalized partial credit model.
The three latent continuous scores corresponded well with proteinopathy severity.
Novel genes associated with proteinopathies were identified.
Several genes had high degrees of biologic credibility for dementia risk factors.
Keywords: Alzheimer's Coordinating Center, Alzheimer's Disease Neuroimaging Initiative, Alzheimer's disease neuropathologic changes (ADNC), Alzheimer's Disease Sequencing Project, ARHGEF28, Item response theory, Lewy, neuropathology, Religious Orders Study, RGNEF, Rush Memory and Aging Project (MAP), SDHAF1, TMEM68
1. BACKGROUND
Many older persons’ brains harbor multiple comorbid misfolded protein aggregates, 1 , 2 , 3 , 4 termed “multi‐proteinopathy,” which is a complex spectrum of abnormally aggregated proteins. Indeed, neuropathology (NP) in the aged brain is rarely “pure” but instead tends to occur in combination. 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 We reported that at least three of four pathologically misfolded dementia‐related proteins (Aβ, tau, α‐synuclein, and TDP‐43) were observed in 50% of brains with any tau pathology in the University of Kentucky Alzheimer's Disease Research Center (UK‐ADRC) cohort. 12 To address the complicated challenges of “mixed” NP‐based genetic association studies, a new toolkit is required, which accomplishes at least two goals: (1) the use of proteinopathy (as opposed to less specific clinical features) as the endophenotype of genetic association studies, and (2) optimal classification criteria and statistical methods to systematically analyze the complex combinations of pathologies.
Dimensionality reduction techniques are often employed to facilitate the analysis of multiple phenotypes. Item response theory (IRT), one such technique, is widely used for estimating latent traits in educational testing and psychometrics. 18 IRT was initially developed for dichotomous item responses in the context of an exam with questions correctly or incorrectly answered. 19 Over the past decades, it has been extended from dichotomous to polytomous, nominal, or graded data; from parametric to non‐parametric models; and, from unidimensional to multidimensional models. The generalized partial credit model (GPCM) 20 is a two‐parameter model (discrimination and difficulty parameters) for two or more ordered item responses that are not necessarily spaced evenly and do not necessarily have the same number of response options among items. As such, GPCM is a potentially ideal approach to analyze multi‐proteinopathy data because: (1) neuropathological features are often measured as a mixture of dichotomous and ordered polytomous variables; (2) each specific NP subtype theoretically contributes differentially to overall disease severity; (3) stages of different neuropathologies do not progress in parallel, as a brain may have a severe burden of one NP subtype but a mild burden of another. Further, GPCM produces continuous unobserved latent traits, which can theoretically increase statistical power (over categorical outcome data) even in a small sample study. 21 , 22 , 23
In the present study, we investigated the potential of multidimensional GPCM to address the complexities of analyzing genetic associations with multi‐proteinopathies by creating latent, continuous, and aggregated proteinopathy measures, thus yielding an overall measure of both the presence and severity of individual brain proteinopathies. We used a combined set of resources that included detailed neuropathologic data from the National Alzheimer's Coordinating Center (NACC), the Alzheimer's Disease Neuroimaging Initiative (ADNI), and the Religious Orders Study (ROS) and the Rush Memory and Aging Project (MAP) [together referred to as ROSMAP], and genetic data from the Alzheimer's Disease Sequencing Project (ADSP).
2. METHODS
2.1. Study cohorts and participants
We obtained NP data from three different cohorts. The NACC NP data https://www.naccdata.org/ were derived from the September 2022 data freeze and measured via the NACC NP v10‐11 forms; this included data from 37 different National Institute on Aging‐funded Alzheimer's Disease Research Centers (ADRCs). Brain autopsies were performed on site at each of the contributory ADRCs. The second set of NP data comprised ADNI. Data used in the preparation of this article were obtained from the ADNI database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public‐private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early Alzheimer's disease (AD). The third source of NP data was a harmonized dataset from the ROSMAP study. 24 Since all NP data in ROSMAP came from those who died aged 65 years or older, we excluded participants who were younger than 65 years old at death in the NACC and ADNI NP data. We also excluded participants who were diagnosed with at least one of 19 rare brain diseases at autopsy (see Figure 1 and ref. 25 ) from the NACC and ADNI NP data. The excluded conditions included frontotemporal lobar degeneration (FTLD), chronic traumatic encephalopathy, multiple sclerosis, multiple system atrophy, amyotrophic lateral sclerosis (ALS), triplet repeat (e.g., Huntington's and other) diseases, and prion diseases. Similar exclusion criteria were not applied to ROSMAP due to a lack of data availability. Finally, we removed participants who had missing data in any NP variables described below. ADRCs obtained written informed consent from their participants and maintained their own separate IRB review and approval from their institution prior to submitting data to NACC.
FIGURE 1.
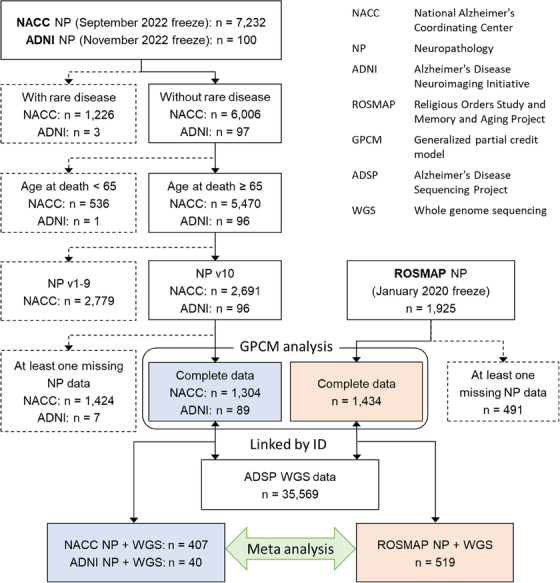
Work flow diagram of the present study.
2.2. NP data
The NP features included in this study are listed in Supplementary Table 1. AD neuropathologic changes (ADNC) include amyloid plaques, neocortical neuritic plaques, and tau neurofibrillary tangles (NFTs), all of which were specified as ordinal variables. Regional progression of amyloid plaques was represented by modified Thal Aβ phase ratings (A score: Thal Aβ phase A0–A3) 26 in NACC and ADNI. In ROSMAP, where Thal Aβ phase was not available, diffuse plaque burden across regions (plaq_d = 0, plaq_d ≤ 0.5, plaq_d ≤ 1, and plaq_d > 1), which was calculated as the average of scaled counts determined by microscopic examination from five brain regions (middle frontal gyrus cortex, middle temporal gyrus cortex, inferior parietal cortex, entorhinal cortex, and hippocampus), was included instead of Thal Aβ phase ratings. Regional progression of tau NFTs was operationalized by modified Braak NFT stage (B score: Stage 0 (B0), Stage I or II (B1), Stage III or IV (B2), and Stage V or VI (B3)), 27 and density of neocortical neuritic plaques by Consortium to Establish a Registry for Alzheimer's Disease (CERAD) ratings (C score: none (C0), sparse (C1), moderate (C2), and frequent (C3)). 28 , 29 TDP‐43 immunoreactive inclusions were specified as indicators for the presence of inclusions in three brain regions: amygdala, entorhinal/inferior temporal cortex and/or hippocampus, and neocortex. Lewy body pathology (LBP) data were categorized into three levels: 0 = none, 1 = present in non‐neocortical regions, and 2 = present in neocortical regions. Hippocampal sclerosis (HS) was determined by bilateral or unilateral HS of the CA1 region. Since NACC and ADNI NP data were collected using the “NACC NEUROPATHOLOGY DATA FORM” but ROSMAP NP data were not, we combined the NACC and ADNI NP data (hereafter referred as NACC/ADNI) and created two completed NP datasets (i.e., NACC/ADNI and ROSMAP) for the multidimensional GPCM analyses (Figure 1).
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using various sources including PubMed, meeting abstracts, and conference presentations. While the scientific problems regarding proteinopathy are increasingly recognized, we currently lack key instruments to augment efforts to understand the genetic architecture underlying dementia‐related proteinopathy. Relevant citations are appropriately cited.
Interpretation: Our approach discovered novel suggestive genes associated with limbic‐predominant age‐related TDP‐43 encephalopathy neuropathologic change, Alzheimer's disease neuropathologic changes, and Lewy body pathology.
Future directions: Future research will examine the functionalities of those genes using omics data analyses and elucidate the roles of biological pathways.
2.3. Whole genome sequencing data
ADSP whole genome sequencing (WGS) variant calling data (NG00067.v9) formatted with Variant Call Format (VCF) 30 , 31 were downloaded from DSS NIAGADS (https://dss.niagads.org/). The data consisted of biallelic/multiallelic single nucleotide variants (SNVs) and short insertions/deletions (INDELs) mapped to Genome Reference Consortium Human Build 38 (GRCh38). The ADSP WGS data were linked to the NACC/ADNI and ROSMAP NP data by ADSP sample IDs. Primary quality control (QC) was performed using bcftools version 1.10.2 32 based on the VCF's INFO field for each variant and the VCF's FORMAT for individual genotype calling. The filtering criteria and bcftools command lines are described in Supplementary Table 2. After the primary QC, the VCF format files were converted to PLINK format files pruned for each of the two NP datasets using PLINK v1.90a. 33 , 34 Hardy‐Weinberg equilibrium (HWE) was tested for AD cases and AD controls in the whole ADSP WGS data (the clinically diagnosed AD status data were available in DSS NIAGADS https://dss.niagads.org/ for NACC/ADNI and in SYNAPSE https://www.synapse.org/ for ROSMAP) using PLINK with the “‐‐hardy” option to evaluate excess homozygosity and heterozygosity. Variants with minor allele count < 5, or missing call rate > 5%, or p‐value < 5 × 10−8 in the HWE test for the AD control group were removed. To derive orthogonal principal components (PCs), which were used as covariates in the genetic association analyses, PC analyses (PCA) were performed using PLINK with the “‐‐pca” option using a linkage disequilibrium (LD) pruned subset of markers (pairwise r2 < 0.2) in NACC/ADNI and ROSMAP, separately. We determined the appropriate number of PCs for covariate adjustment based on the screen plots shown in Supplementary Figure 1.
2.4. Statistical analysis
The multidimensional GPCM analyses were conducted using R version 4.2.1. 35 The two‐parameter models were run by specifying a normal distribution (dentype = “Gaussian”) with z‐score constraints (mean = 0 and standard deviation (SD) = 1) and quasi‐Monte Carlo EM estimation (method = “QMCEM”) in the “mirt” function from the “mirt” R package version 1.37.1. 36 To determine the best fitting number of dimensions, or factors, we first performed leave‐one‐out cross‐validation for the responses to a set of the eight NPs and compared standardized root mean square residual (SRMSR) and comparative fit index (CFI) values among two to four dimensions models. Supplementary Figures 2A and 2B for NACC/ADNI show the evidence that three‐dimensional GPCMs were more appropriate than two‐ and four‐dimensional models. As shown in Supplementary Figures 2C and 2D, although four‐dimensional model had larger CFI values in ROSMAP, the model seemed unstable according to the SRMSR distribution (Supplementary Figures 2C). From this assessment, we specified model = 3 as a number of dimensions (i.e., three factors) in the “mirt” function. We then estimated three factor scores with 10,000 plausible value imputations (i.e., plausible.draws = 10000 in the “fscores” function from the “mirt” package) and then computed the means of the 10,000 plausible value imputations as individual factor score estimates.
For each of the three factor scores, we performed SNV association tests under an additive mode of inheritance. We ran two linear mixed effects models, which incorporated the kinship matrix: (1) model adjusted for age at death, sex, and the top three PCs; and (2) additionally adjusted for the other two factor score estimates. These analyses were implemented by GEMMA (https://github.com/genetics‐statistics/GEMMA) 37 in NACC/ADNI and ROSMAP, separately. Then we conducted meta‐analyses along with the heterogeneity tests using METAL (released 3/25/2011). 38 We included SNVs which were contained in both the NACC/ADNI and ROSMAP, and excluded SNVs with p < 1×10−5 in the heterogeneity tests. Because genetic effects may be attenuated for those with genetic risks but dying before a NP feature develops, we also focused on participants who died aged 80 years or older as a sensitivity analysis. We set genome‐wide significance at p < 5×10−8 and “suggestive” significance at p < 1×10−5. We explored annotation (coding, intron, splice site, promoter, 5′ or 3′ untranslated region [UTR], or intergenic region) of significant and suggestively significant SNVs using the “locateVariants” function in the VariantAnnotation Bioconductor R package v1.44.1 39 and the “TxDb.Hsapiens.UCSC.hg38.knownGene” annotation R package v3.16.0. 40 Region association plots were created by LocusZoom software. 41
To examine whether any candidate loci were possibly functional, we performed Bayesian colocalization analyses 42 , 43 developed by Giambartolomei et al., in which only summary statistics are required from two independent association studies. We obtained publicly available summary statistics on gene expression quantitative trait loci (eQTL) from the Genotype‐Tissue Expression Project (GTEx), 44 ROSMAP 45 downloaded from https://mostafavilab.stat.ubc.ca/xqtl/, 45 and the Trans‐Omics for Precision Medicine (TOPMed) Program (https://topmeddemo.wesdemo.com/). We ran the “coloc.abf” function in the “coloc” R package v5.1.0.1 42 with three prior probabilities of 10−4, 10−5, and 10−6 that a SNV is associated with both traits. We evaluated the loci and ± 500 kbp flanking regions for each of the SNVs that had significant or suggestively significant associations with proteinopathy in participants who died aged 65 years or older. We then identified eQTL based on posterior probability of 0.9 or more for each of the three prior probabilities.
3. RESULTS
Of 2,691 NACC participants, more than half of the participants (n = 1,387) were excluded due to at least one missing data element (Figure 1). The comparison between included and excluded participant groups is displayed in Supplementary Table 3. The excluded participants were older at death, less severe Aβ and neuritic plaques, less TDP‐43 pathology in amygdala and more TDP‐43 pathology in hippocampus and/or entorhinal/inferior temporal cortex, and less LBP.
The numbers of participants included in the multidimensional GPCM analyses were 1,304 in NACC, 89 in ADNI, and 1,413 in ROSMAP (Figure 1). The mean age at death was 83.0 (standard deviation (SD) = 9.1), the mean of years in education was 16.5 (SD = 8.5), and 50.8% were females in NACC; the mean age at death was 83.0 (SD = 6.6), the mean of years in education was 16.3 (SD = 2.7), and 25.8% were females in ADNI; and the mean age at death was 89.7 (SD = 6.5), the mean of years in education was 16.2 (SD = 3.6), and 68.2% were females in ROSMAP. The NACC and ADNI cohorts included more severe AD patients and fewer people with TDP‐43 pathology than ROSMAP (Table 1).
TABLE 1.
Characteristics in subjects of National Alzheimer's Coordinating Center (NACC), Alzheimer's Disease Neuroimaging Initiative (ADNI), and Religious Orders Study and Memory and Aging Project (ROSMAP).
| Characteristics |
NACC (n = 1,304) |
ADNI (n = 89) |
ROSMAP (n = 1,413) |
|---|---|---|---|
| Age at death (years), mean ± SD | 83.0 ± 9.1 | 83.0 ± 6.6 | 89.7 ± 6.5 |
| Years in education, mean ± SD | 16.5 ± 8.5 | 16.3 ± 2.7 | 16.2 ± 3.6 |
| Sex, n (%) | |||
| Male | 641 (49.2) | 66 (74.2) | 450 (31.8) |
| Female | 663 (50.8) | 23 (25.8) | 963 (68.2) |
| Thal phase/diffuse plaques a , n (%) | |||
| 0/0 | 102 (7.8) | 5 (5.6) | 253 (17.9) |
| 1‐2/≤0.5 | 115 (8.8) | 6 (6.7) | 440 (31.1) |
| 3/≤1 | 114 (8.7) | 10 (11.2) | 338 (23.9) |
| 4‐5/> 1 | 973 (74.6) | 68 (76.4) | 382 (27.1) |
| Braak NFT stage, n (%) | |||
| 0 | 25 (1.9) | 1 (1.1) | 14 (1.0) |
| I‐II | 177 (13.6) | 19 (21.3) | 221 (15.6) |
| III‐IV | 266 (20.4) | 7 (7.9) | 802 (56.8) |
| V‐VI | 836 (64.1) | 62 (69.7) | 376 (26.6) |
| Neuritic plaques, n (%) | |||
| No | 196 (15.0) | 18 (20.2) | 319 (22.6) |
| Sparse | 140 (10.7) | 9 (10.1) | 126 (8.9) |
| Moderate | 222 (17.0) | 9 (10.1) | 507 (35.9) |
| Frequent | 746 (57.2) | 53 (59.6) | 461 (32.6) |
| TDP‐43 in amygdala, n (%) | |||
| No | 860 (66.0) | 52 (58.4) | 681 (48.2) |
| Yes | 444 (34.0) | 37 (41.6) | 732 (51.8) |
| TDP‐43 in hippocampus and/or entorhinal/inferior temporal cortex, n (%) | |||
| No | 884 (67.8) | 51 (57.3) | 943 (66.7) |
| Yes | 420 (32.2) | 38 (42.7) | 470 (33.3) |
| TDP‐43 in neocortex, n (%) | |||
| No | 1240 (95.1) | 76 (85.4) | 1,085 (76.8) |
| Yes | 64 (4.9) | 13 (14.6) | 328 (23.2) |
| Lewy bodies, n (%) | |||
| No | 713 (54.7) | 44 (49.4) | 1078 (76.3) |
| Other regions | 384 (29.4) | 25 (28.1) | 142 (10.0) |
| Neocortex | 207 (15.9) | 20 (22.5) | 193 (13.7) |
| Hippocampal sclerosis, n (%) | |||
| No | 1109 (85.0) | 80 (89.9) | 1,281 (90.7) |
| Yes | 195 (15.0) | 9 (10.1) | 132 (9.3) |
Abbreviations: NFT, neurofibrillary tangle; SD, standard deviation.
Thal phase and diffuse plaques across regions were used in NACC and ROSMAP, respectively.
Figure 2 and Supplementary Table 4 show the rotated factor loadings (rotate = “oblimin”) representing a strength of the association between each of the eight NPs and each of the latent factor scores (F1 to F3) estimated by three‐dimensional GPCMs. The predominant NPs were TDP‐43 pathology and HS for the factor 1 (F1), ADNC‐related NPs for the factor 2 (F2), and LBP for the factor 3 (F3). The factor correlations were 0.225 between factors 1 and 2, 0.165 between factors 1 and 3, 0.204 between factors 2 and 3 in NACC/ADNI, and 0.303 between factors 1 and 2, 0.104 between factors 1 and 3, 0.115 between factors 2 and 3 in ROSMAP (Figure 2). Supplementary Figures 3‐10 display the density plots in each estimated factor score by the eight NP outcomes, indicating that the factor scores reflected well the NP presence and absence as well as the severities.
FIGURE 2.
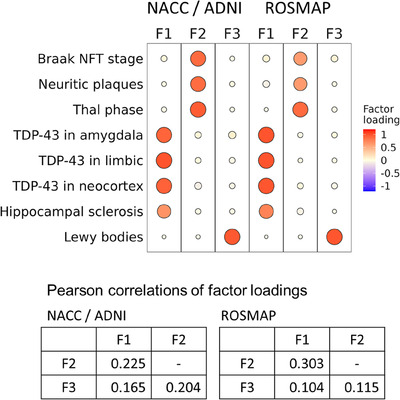
Rotated factor loadings with the oblimin rotation in NACC/ADNI and ROSMAP. ADNI = Alzheimer's Disease Neuroimaging Initiative; NFT, neurofibrillary tangle; NACC, National Alzheimer's Coordinating Center; ROSMAP, Religious Orders Study and Rush Memory and Aging Project.
After linking the NP data to ADSP genotype data, 447 (NACC/ADNI) and 519 (ROSMAP) participants were included in the subsequent SNV association tests (Figure 1). Most participants have predominantly European ancestry in both NACC/ADNI (Supplementary Figure 11A) and ROSMAP (Supplementary Figure 11B). We confirmed that known proteinopathy loci could be detected in our novel NP scoring: SNVs in TMEM106B and GRN, which are known as TDP‐43 pathology and HS related genes; 46 , 47 , 48 , 49 , 50 the SNVs in the BIN1 and APOE loci, which are strongly associated with ADNC; 51 , 52 and, the SNVs in GBA, which is a risk gene for dementia with LBP 53 , 54 (Table 2 and Supplementary Table 5 show the number of missing data for each SNV). Although the GBA and BIN1 did not reach the suggestive significance level, rs10950392 in TMEM106B and rs429358 in APOE were associated with the estimated factor 1 (TDP‐43 and HS related) score and factor 2 (ADNC related) score, respectively. Interestingly, the associations of rs429358 in APOE with each of the factor scores were attenuated after adjusting for the other factor scores (Supplementary Figures 12‐14).
TABLE 2.
Top single nucleotide variants in genes associated with proteinopathy.
| CHR | Gene | SNV | Position | Effect/reference | Model a | F1 b | F2 b | F3 b | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
Q‐value c |
|
Q‐value c |
|
Q‐value c | ||||||
| 1 | GBA | rs140335079 | 155237596 | A/T | 1 | 0.26 | 0.28 | −0.02 | 0.93 | −0.68 | 0.015 |
| 2 | 0.38 | 0.12 | 0.01 | 0.98 | −0.69 | 0.017 | |||||
| 2 | BIN1 | rs6733839 | 127135234 | T/C | 1 | 0.06 | 0.28 | 0.15 | 0.018 | 0.12 | 0.040 |
| 2 | 0.00 | 0.99 | 0.11 | 0.064 | 0.08 | 0.19 | |||||
| 7 | TMEM106B | rs10950392 | 12223912 | T/C | 1 | 0.18 | 0.0017 | −0.11 | 0.084 | −0.10 | 0.098 |
| 2 | 0.22 | 3.6×10−5 | −0.15 | 0.0097 | −0.10 | 0.14 | |||||
| 17 | GRN | rs5848 d | 44352876 | T/C | 1 | 0.19 | 0.0018 | −0.05 | 0.48 | −0.06 | 0.30 |
| 2 | 0.21 | 2.2×10−4 | −0.10 | 0.081 | −0.08 | 0.22 | |||||
| 19 | APOE | rs429358 | 44908684 | C/T | 1 | 0.36 | 5.1×10−8 | 0.64 | 1.1×10−23 | 0.22 | 0.0028 |
| 2 | 0.14 | 0.037 | 0.50 | 4.5×10−16 | 0.06 | 0.38 | |||||
Abbreviations: CHR, chromosome; SNV, single nucleotide variant.
Model 1 = adjusted for age at death, sex, and top three principal components; Model 2 = adjusted for age at death, sex, top three principal components, and other scores.
F1 = TDP‐43 and hippocampal sclerosis related score; F2 = Alzheimer's disease neuropathologic change related score; F3 = Lewy body pathology related score.
Q‐value indicates that false discovery rate (FDR) adjusted p‐value in each score and model.
The p‐value of Hardy‐Weinberg equilibrium test for rs5848 in AD controls was 1.2×10−61; however, we displayed the results because rs5848 is well‐known as TDP‐43 related single nucleotide variant.
Next, we performed the whole genome SNV analyses on each of the estimated factor scores without (Supplementary Figures 15 and 16) and with the adjustment of the other two scores (Supplementary Figures 17 and 18). The top significant and suggestive significant SNVs from the model additionally adjusted for the other factor scores for the three factor scores are shown in Supplementary Tables 6‐8 along with the individual quality values including RMS mapping quality (MQ), Phred‐scaled p‐value using Fisher's exact test to detect strand bias (FS), symmetric odds ratio of 2×2 contingency table to detect strand bias (SOR), variant confidence/quality by depth (QD), and p‐value from HWE test (Supplementary Tables 9‐11). We here highlighted some intriguing SNVs that reached to the significance (i.e., p < 5×10−8) or suggestive significance level (i.e., p < 1×10−5) in both people who died after 65 years or older (≥ 65 at death) and people who died after 80 years or older (≥ 80 at death) in the sensitivity analyses in Table 3 (Supplementary Table 5 displays the number of missing data for each SNV). The G allele of rs80190672 in ARHGEF28 on chromosome 5 was significantly associated with decreased factor 1 (TDP‐43 and HS related) score ( = −0.64 and p‐value = 4.7×10−8) in ≥ 65 at death, and the association remained in ≥ 80 at death at suggestive significance ( = −0.76 and p‐value = 5.8×10−8). rs141108370 in UNC13C on chromosome 15 was the second top SNV suggestively associated with the factor 1 score ( = −1.06 and p‐value = 5.7×10−7 in ≥ 65 at death and = −1.25 and p‐value = 2.8×10−7 in ≥ 80 at death). The T allele of rs72643142 in KAZN on chromosome 1 was also suggestively significant with the factor 1 score ( = −0.40 and p‐value = 7.1×10−7 in ≥ 65 at death and = −0.48 and p‐value = 5.6×10−7 in ≥ 80 at death). Other than APOE loci on chromosome 19, there were five loci that reached the suggestive significance level for the factor 2 (ADNC related) score and were confirmed in the sensitivity analysis for ≥ 80 at death. For the factor 3 (LBP related) score, we observed one locus that reached the suggestive significance level, and the associations retained even in ≥ 80 at death.
TABLE 3.
Significant and suggestive significant single nucleotide variants associated with each factor score both in subjects aged 65 years or older at death and in subjects aged 80 years or older at death.
| CHR | Gene | SNV | Position | Effect/reference | ≥ 65 at death | ≥ 80 at death | ||
|---|---|---|---|---|---|---|---|---|
|
|
p‐Value |
|
p‐Value | |||||
| Factor 1 (TDP‐43 pathology and hippocampal sclerosis) score | ||||||||
| 1 | KAZN | rs72643142 | 14148707 | T/C | −0.40 | 7.1×10−7 | −0.48 | 5.6×10−7 |
| 5 | ARHGEF28 | rs80190672 | 73973002 | G/A | −0.64 | 4.7×10 −8 | −0.76 | 5.8×10−8 |
| 15 | UNC13C | rs141108370 | 54631819 | G/A | −1.06 | 5.7×10−7 | −1.25 | 2.8×10−7 |
| Factor 2 (Alzheimer's disease neuropathologic change related) score | ||||||||
| 1 | C1orf185 | rs72692278 | 51129361 | A/G | −0.50 | 6.6×10−6 | −0.58 | 7.2×10−6 |
| 1 | ZNF281 | rs188482877 | 200431363 | T/A | −1.00 | 3.2×10−6 | −1.12 | 3.3×10−6 |
| 12 | GRIN2B | rs71457202 | 13748154 | A/G | 0.81 | 3.8×10−6 | 0.99 | 1.1×10−6 |
| 13 | LINC00559 | rs145442832 | 89883465 | T/C | −0.39 | 4.6×10−6 | −0.52 | 2.2×10−7 |
| 14 | TTLL5 | rs745536628 | 75760859 | A/AT | −1.04 | 5.4×10−6 | −1.21 | 6.3×10−6 |
| Factor 3 (Lewy body pathology related) score | ||||||||
| 6 | TNFRSF21 | rs78794444 | 47311493 | T/G | −0.71 | 3.0×10−6 | −0.87 | 1.5×10−6 |
Notes: Bolded result indicates statistical significance.
Abbreviations: CHR, chromosome; SNV, single nucleotide variant.
Some of the putative risk alleles also showed evidence of biological significance in eQTL (i.e., colocalization). Supplementary Tables 12‐14 show the full results of colocalization analyses with eQTL p‐value < 1×10−5. Figure 3 and Supplementary Figures 19‐22 highlighted the colocalization PPH4 > 0.9 for the prior probability of 10−5. The SNVs in TMEM68 on chromosome 8 (the top SNV rs28610182 was located in the promoter region shown in Supplementary Table 9) suggestively associated with the factor 1 (TDP‐43 and HS) score ( = −0.29 and p‐value = 1.1×10−6 for the A allele in ≥ 65 at death shown in Supplementary Table 6 and Figure 3A) colocalized with the TMEM68 expression in brain prefrontal cortex BA9 (Figure 3B) and brain hypothalamus (Figure 3C) in GTEx, and the colocalization of the TMEM68 expression in prefrontal cortex was replicated in ROSMAP (Figure 3D). This locus also colocalized with the TMEM68 expression in other tissues (Supplementary Table 12 and Supplementary Figure 20). We did not observe colocalization between the TMEM106B SNVs and gene expression in any brain region, but the TMEM106B locus colocalized with TMEM106B expression in several tissues (Supplementary Table 12 and Supplementary Figure 19). The SDHAF1 locus (the top SNV rs17706479 was located in the promoter region (Supplementary Table 9)) colocalized with SDHAF1 expression in lung and whole blood (Supplementary Table 12 and Supplementary Figure 21). The BMS1 locus suggestively associated with the factor 2 (ADNC related) score colocalized with the ENSG00000259869 in artery tibial and BMS1 expression in brain cerebellum (Supplementary Table 13 and Supplementary Figure 22). We did not observe a colocalization with PPH4 > 0.9 for the prior probability of 10−5 in the factor 3 (Lewy body pathology related) score (Supplementary Table 14).
FIGURE 3.
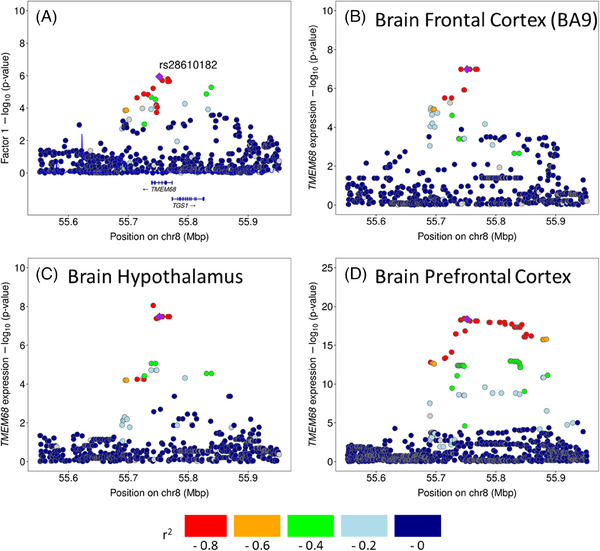
LocusZoom plots for the association of single nucleotide variants with the estimated factor 1 (TDP‐43 and hippocampal sclerosis) score in TMEM68 (A) and with the TMEM68 expressions in brain prefrontal cortex BA9 (B) and brain hypothalamus (C) from GTEx, and in brain prefrontal cortex (D) from ROSMAP. GTEx = Genotype‐tissue expression project; ROSMAP = Religious Orders Study and Rush Memory and Aging Project.
4. DISCUSSION
Multidimensional GPCM methods were used to generate new insights into dementia‐related proteinopathies. By applying this novel methodology, genetic analyses of WGS data were used to identify candidate risk‐associated SNVs for complex proteinopathies. Using data from multiple large autopsy studies with a meta‐analytic study design, our findings included the replication of prior studies’ results, as well as some novel findings. More specifically, our analyses helped validate known genotype/phenotype associations: APOE is a risk allele for ADNC; TMEM106B and GRN for LATE‐NC; and GBA for LBP. These findings also provided assurance about the validity of our approach (analogous to positive internal controls) and its potential to be applied to elucidate previously undiscovered genotype/phenotype associations. Indeed, novel risk allele candidates were identified and a subset of these are discussed below.
We demonstrated that NP phenotypes could be integrated via the IRT which provided three‐dimensional factor scores representing different severities on NP spectrums. The continuous factor scores allowed us to adjust the models for scores other than the outcome, that is, the adjustment removed the effect of other proteinopathies. For example, APOE is a well‐known risk gene of ADNC. Previous studies reported that APOE was also associated with TDP‐43 proteinopathy. 55 , 56 In our approach, the C allele of rs429358 was associated with increased burden of TDP‐43 without the adjustment of other scores; however, the association was greatly attenuated after adjustment for the ADNC and LBP related scores (Supplementary Figure 12). This implies that although TDP‐43 pathology commonly co‐exists with ADNC, APOE would primarily affect ADNC rather than directly promoting TDP‐43 pathology.
The three factors that we identified were predominantly associated with either LATE‐NC, ADNC, or LBP. As we expected, Braak NFT stage, neuritic plaques, and Thal Aβ phase/diffuse amyloid plaques had larger contributions to the same factor (i.e., factor 2 related to ADNC) which indicates that these measurements were involved in the same latent trait. We observed the similar structure in TDP‐43 pathology and HS (factor 1 related LATE‐NC). On the other hand, LBP was projected into the different latent space from ADNC, which was seen in both NACC/ADNI and ROSMAP. Previous studies reported that LBP was highly correlated and co‐existed with ADNC. 57 , 58 , 59 However, our study indicated that LBP had a different NP spectrum from ADNC and thus could be analyzed as a separate latent trait (termed factor 3).
At least several of the new potential risk alleles have compelling bases of biological credibility. Included are some novel and intriguing loci for ADNC (factor 2 in our study). However, because the genetic architecture of AD/ADNC has been exhaustively studied, we here focus on three putative novel genes that were linked in the current study to non‐ADNC dementia‐driving neuropathologies: ARHGEF28, TMEM68, and SDHAF1 as risk allele candidates for factor 1 (LATE‐NC).
The SNV rs80190672, which was significantly associated with TDP‐43 pathology and HS score (LATE‐NC) in participants with age at death ≥ 65 years, is located downstream of ARHGEF28 encoding rho guanine nucleotide exchange factor 28 (the cognate polypeptide is referred to as RGNEF). Of all the novel loci identified, this was the one with the lowest p‐value. ARHGEF28 has been reported as a putative ALS gene. 60 , 61 The large majority of ALS cases have TDP‐43 pathology in affected cells (i.e., motor neurons). 62 RGNEF has been found to localize to hallmark TDP‐43 immunoreactive inclusion bodies in ALS patient spinal cord motor neurons. 63 , 64 Although the SNVs located near ARHGEF28 are downstream of the transcript‐encoding gene sequences, they may be involved in pathogenesis through modulating the splicing or translation of the gene transcript.
Some of the genetic associations that were not statistically significant at the whole‐genome level were nonetheless intriguing, and follow‐up analyses indicated credible biological impacts. For example, rs28610182 in the TMEM68 gene had a nominal p‐value = 1.1×10−6 with TDP‐43 and HS pathologies (factor 1 score). The TMEM68 locus colocalized with the TMEM68 expression in brain frontal cortex in two independent datasets (i.e., GTEx and ROSMAP, PPH4 > 90%). TMEM68 encodes a protein named transmembrane protein 68, which is a putative “brain‐specific” acyltransferase involved in glycerolipid metabolism. 65 , 66 Recent studies showed that the deregulated metabolism of glycerolipid was associated with ALS risk. 67 , 68 Given that the A allele of rs28610182 had a protective effect (shown in Supplementary Table 6) and was associated with upregulated TMEM68 expression in human brain (shown in Supplementary Table 12), our findings may indicate a protective influence of this protein product and/or the glycerolipid metabolism pathway, in LATE‐NC.
Another SNV suggestively associated with Factor 1/LATE‐NC in the present study was rs17706479, which is in the SDHAF1 gene. SDHAF1 encodes a protein that serves as an assembly factor in mitochondrial complex II. 69 , 70 Mitochondrial dynamics have been implicated in numerous ways with TDP‐43 pathology. 71 , 72 We found colocalization between the strongest pathology‐associated allele and methylation at the SDHAF1 locus. Intriguingly, the SDHAF1 gene has previously been linked to neurological disease phenotypes, in addition to mitochondrial deficits with clinical impact. 69 , 73 Methylation of the SDHAF1 locus was specifically implicated in fetal alcohol syndrome, and mutations in the SDHAF1 gene were linked to the phenotype of white matter disease (specifically, pediatric leukoencephalopathy). 73 , 74 , 75 , 76 More work is required to ascertain if the association between SDHAF1 genetic variation and TDP‐43 pathology is robust in aging individuals.
There were limitations to the present study. Most importantly, most of the genotype‐NP phenotype associations identified (including the previously described ones, e.g., TMEM106B with LATE‐NC) did not reach the threshold for genome‐wide statistical significance, indicating that statistical power was only marginally capable of testing the null hypothesis. However, we were encouraged by the validation of prior pathology‐linked SNPs. Further, the pathologic data constitutes semiquantitative (ordinal, rather than quantitative) parameters according to consensus‐based rubrics, and these were generated in a manner that probably differs—even if subtly—from research center to research center. The NP phenotypes “pure” or “mixed” constitute the gold standard for disease presence and severity and the IRT methods we applied were meant to render these phenotypes testable with the quantitative genetic data. Nonetheless, the pathological phenotypes are still evolving as new clinical‐pathological relationships are elucidated. Although we showed that our NP scoring could detect known proteinopathy‐linked loci, some important SNVs that are associated with single proteinopathy may have been missed. For example, our scoring method aggregated Aβ and tau pathologies; thus, it may not be able to detect Aβ specific SNPs. We also note that the majority of the included research participants were people with European ancestry. In future research, we will need many sources to validate our findings, including different race/ethnicity sample groups and independent study cohorts of NACC, ADNI, and ROSMAP, and we will expand our scoring approach to multivariate outcomes representing more complicated patterns of mixed proteinopathies. In this study, we excluded participants who had at least one missing NP data element. The excluded participants had different NP characteristics, and thus our findings may be biased. In future studies, we will take into account the missing value issue using some techniques such as imputing missing data and handling missing data as a non‐answer (i.e., no contribution to any factor score) within an IRT framework.
Despite the significant caveats, we conclude that our novel application of multidimensional GPCM and human WGS data, combined with relatively sharp NP‐based endophenotype data from multiple high‐quality autopsy series, enabled novel insights into the genetic architecture of dementia‐associated brain pathologies. These findings require validation/corroboration from other data sets. However, they help underscore once again that, as with the clinical and pathological phenotypes involved, the underlying genetic factors that influence amnestic dementia are highly complex.
CONFLICT OF INTEREST STATEMENT
The authors report no competing interests. The authors have no conflicts of interest (see supporting information).
CONSENT STATEMENT
Alzheimer's Disease Research Centers obtained written informed consent from their participants and maintain their own separate IRB review and approval from their institution prior to submitting data to NACC.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
This work was supported by grants the National Institute on Aging RF1AG082339, R01AG082730, R01AG057187, U24AG07122, P01AG078116, the UK‐ADC P30AG072946, the National Institute of Neurological Disorders and Stroke F30NS124136, and the NACC New Investigator Award (https://naccdata.org/nacc‐productivity/new‐investigator‐awards) which are awarded each year to promising early‐career investigators from across the ADRC Program to support their career development and advance their research on Alzheimer's Disease Related Dementia (ADRD). The National Institutes of Health, National Institute on Aging (NIH‐NIA) supported this work through the following grants: ADGC, U01 AG032984, RC2 AG036528; Samples from the National Cell Repository for Alzheimer's Disease (NCRAD), which receives government support under a cooperative agreement grant (U24 AG21886) awarded by the National Institute on Aging (NIA), were used in this study.
We are extremely grateful to the research volunteers, clinicians, and staff who made this study possible.
Data for this study were prepared, archived, and distributed by the National Institute on Aging Alzheimer's Disease Data Storage Site (NIAGADS) at the University of Pennsylvania (U24‐AG041689‐01), funded by the National Institute on Aging.
The NACC database is funded by NIA/NIH Grant U24 AG072122. NACC data are contributed by the NIA‐funded ADRCs: P30 AG062429 (PI James Brewer, MD, PhD), P30 AG066468 (PI Oscar Lopez, MD), P30 AG062421 (PI Bradley Hyman, MD, PhD), P30 AG066509 (PI Thomas Grabowski, MD), P30 AG066514 (PI Mary Sano, PhD), P30AG066530 (PI Helena Chui, MD), P30 AG066507 (PI Marilyn Albert, PhD), P30 AG066444 (PI John Morris, MD), P30 AG066518 (PI Jeffrey Kaye, MD), P30 AG066512 (PI Thomas Wisniewski, MD), P30 AG066462 (PI Scott Small, MD), P30 AG072979 (PI David Wolk, MD), P30 AG072972 (PI Charles DeCarli, MD), P30 AG072976 (PI Andrew Saykin, PsyD), P30 AG072975 (PI David Bennett, MD), P30 AG072978 (PI Neil Kowall, MD), P30 AG072977 (PI Robert Vassar, PhD), P30 AG066519 (PI FrankLaFerla, PhD), P30 AG062677 (PI Ronald Petersen, MD, PhD), P30 AG079280 (PIEric Reiman, MD), P30 AG062422 (PI Gil Rabinovici, MD), P30 AG066511 (PI AllanLevey, MD, PhD), P30 AG072946 (PI Linda Van Eldik, PhD), P30 AG062715 (PI Sanjay Asthana, MD, FRCP), P30 AG072973 (PI Russell Swerdlow, MD), P30 AG066506 (PI Todd Golde, MD, PhD), P30 AG066508 (PI Stephen Strittmatter, MD, PhD), P30AG066515 (PI Victor Henderson, MD, MS), P30 AG072947 (PI Suzanne Craft, PhD), P30 AG072931 (PI Henry Paulson, MD, PhD), P30 AG066546 (PI Sudha Seshadri, MD), P20 AG068024 (PI Erik Roberson, MD, PhD), P20 AG068053 (PI Justin Miller, PhD), P20 AG068077 (PI Gary Rosenberg, MD), P20 AG068082 (PI Angela Jefferson, PhD), P30 AG072958 (PI Heather Whitson, MD), P30 AG072959 (PI James Leverenz, MD).
The Alzheimer's Disease Sequencing Project (ADSP) is comprised of two Alzheimer's Disease (AD) genetics consortia and three National Human Genome Research Institute (NHGRI) funded Large Scale Sequencing and Analysis Centers (LSAC).The two AD genetics consortia are the Alzheimer's Disease Genetics Consortium(ADGC) funded by NIA (U01 AG032984), and the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) funded by NIA (R01 AG033193), the National Heart, Lung, and Blood Institute (NHLBI), other National Institute of Health (NIH) institutes and other foreign governmental and non‐governmental organizations. The Discovery Phase analysis of sequence data is supported through UF1AG047133 (to Drs. Schellenberg, Farrer, Pericak‐Vance, Mayeux, and Haines); U01AG049505 to Dr. Seshadri; U01AG049506 to Dr. Boerwinkle; U01AG049507 to Dr. Wijsman; and U01AG049508 to Dr. Goate and the Discovery Extension Phase analysis is supported through U01AG052411 to Dr. Goate, U01AG052410 to Dr. Pericak‐Vance and U01 AG052409 to Drs. Seshadri and Fornage. Sequencing for the Follow Up Study (FUS) is supported through U01AG057659 (to Drs. PericakVance, Mayeux, and Vardarajan) and U01AG062943 (to Drs. Pericak‐Vance and Mayeux). Data generation and harmonization in the Follow‐up Phase is supported by U54AG052427 (to Drs. Schellenberg and Wang). The FUS Phase analysis of sequence data is supported through U01AG058589 (to Drs. Destefano, Boerwinkle, De Jager, Fornage, Seshadri, and Wijsman), U01AG058654 (to Drs. Haines, Bush, Farrer, Martin, and Pericak‐Vance), U01AG058635 (to Dr. Goate), RF1AG058066 (to Drs. Haines, Pericak‐Vance, and Scott), RF1AG057519 (to Drs. Farrer and Jun), R01AG048927 (to Dr. Farrer), and RF1AG054074 (to Drs. Pericak‐Vance and Beecham). The ADGCcohorts include: Adult Changes in Thought (ACT) (UO1 AG006781, UO1 HG004610, UO1 HG006375, U01 HG008657), the Alzheimer's Disease Centers (ADC) ( P30AG019610, P30 AG013846, P50 AG008702, P50 AG025688, P50 AG047266, P30 AG010133, P50 AG005146, P50 AG005134, P50 AG016574, P50 AG005138, P30 AG008051, P30AG013854, P30 AG008017, P30 AG010161, P50 AG047366, P30 AG010129, P50 AG016573, P50 AG016570, P50 AG005131, P50 AG023501, P30 AG035982, P30 AG028383, P30AG010124, P50 AG005133, P50 AG005142, P30 AG012300, P50 AG005136, P50 AG033514, P50 AG005681, and P50 AG047270), the Chicago Health and Aging Project (CHAP) (R01 AG11101, RC4 AG039085, K23 AG030944), Indianapolis Ibadan (R01 AG009956, P30 AG010133), the Memory and Aging Project (MAP) ( R01 AG17917), Mayo Clinic (MAYO) (R01 AG032990, U01 AG046139, R01 NS080820, RF1 AG051504, P50 AG016574), Mayo Parkinson's Disease controls (NS039764, NS071674, 5RC2HG005605), University of Miami (R01 AG027944, R01 AG028786, R01 AG019085, IIRG09133827, A2011048), the Multi‐Institutional Research in Alzheimer's Genetic Epidemiology Study (MIRAGE) (R01 AG09029, R01 AG025259), the National Cell Repository for Alzheimer's Disease (NCRAD) (U24 AG21886), the National Institute on Aging Late Onset Alzheimer's Disease Family Study (NIA‐ LOAD) (R01 AG041797), the Religious Orders Study (ROS) (P30 AG10161, R01 AG15819), the Texas Alzheimer's Research and Care Consortium (TARCC) (funded by the Darrell K Royal Texas Alzheimer's Initiative), Vanderbilt University/Case Western Reserve University (VAN/CWRU)(R01 AG019757, R01 AG021547, R01 AG027944, R01 AG028786, P01 NS026630, and Alzheimer's Association), the Washington Heights‐Inwood Columbia Aging Project (WHICAP) (RF1 AG054023), the University of Washington Families (VA Research Merit Grant, NIA: P50AG005136, R01AG041797, NINDS: R01NS069719), the Columbia University HispanicEstudio Familiar de Influencia Genetica de Alzheimer (EFIGA) (RF1 AG015473), the University of Toronto (UT) (funded by Wellcome Trust, Medical Research Council, Canadian Institutes of Health Research), and Genetic Differences (GD) (R01 AG007584). The CHARGE cohorts are supported in part by National Heart, Lung, and Blood Institute (NHLBI) infrastructure grant HL105756 (Psaty), RC2HL102419 (Boerwinkle) and the neurology working group is supported by the National Institute on Aging (NIA) R01 grant AG033193.
The CHARGE cohorts participating in the ADSP include the following: Austrian Stroke Prevention Study (ASPS), ASPS‐Family study, and the Prospective Dementia Registry‐Austria (ASPS/PRODEM‐Aus), the Atherosclerosis Risk in Communities (ARIC) Study, the Cardiovascular Health Study (CHS), the Erasmus Rucphen Family Study (ERF), the Framingham Heart Study (FHS), and the Rotterdam Study (RS). ASPS is funded by the Austrian Science Fond (FWF) grant number P20545‐P05 and P13180 and the Medical University of Graz. The ASPS‐Fam is funded by the Austrian Science Fund (FWF) project I904), the EU Joint Programme‐Neurodegenerative Disease Research (JPND) in frame of the BRIDGET project (Austria, Ministry of Science) and the Medical University of Graz and the Steiermärkische Krankenanstalten Gesellschaft. PRODEM‐Austria is supported by the Austrian Research Promotion agency (FFG) (Project No. 827462) and by the Austrian National Bank (Anniversary Fund, project 15435). ARIC research is carried out as a collaborative study supported by NHLBI contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C). Neurocognitive data in ARIC is collected by U012U01HL096812, 2U01HL096814, 2U01HL096899, 2U01HL096902, 2U01HL096917 from the NIH (NHLBI, NINDS, NIA and NIDCD), and with previous brain MRI examinations funded by R01‐HL70825 from the NHLBI. CHS research was supported by contracts HHSN268201200036C, HHSN268200800007C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, and grants U01HL080295 and U01HL130114 from the NHLBI with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided by R01AG023629, R01AG15928, and R01AG20098 from the NIA. FHS research is supported by NHLBI contracts N01‐HC‐25195 and HHSN268201500001I. This study was also supported by additional grants from the NIA (R01s AG054076, AG049607 and AG033040 and NINDS (R01 NS017950). The ERF study as a part of EUROSPAN (European Special Populations Research Network) was supported by European Commission FP6 STRP grant number 018947 (LSHG‐CT‐2006‐01947) and also received funding from the European Community's Seventh Framework Programme (FP7/2007‐2013)/grant agreement HEALTH‐F4‐2007‐201413 by the European Commission under the programme “Quality of Life and Management of the Living Resources” of 5th Framework Programme (no. QLG2‐CT‐2002‐01254). High‐throughput analysis of the ERF data was supported by a joint grant from the Netherlands Organization for Scientific Research and the Russian Foundation for Basic Research (NWO‐RFBR047.017.043). The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, the Netherlands Organization for Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), and the municipality of Rotterdam. Genetic data sets are also supported by the Netherlands Organization of Scientific Research NWO Investments (175.010.2005.011, 911‐03‐012), the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, the Research Institute for Diseases in the Elderly (014‐93‐015; RIDE2), and the Netherlands Genomics Initiative (NGI)/Netherlands Organization for Scientific Research (NWO) Netherlands Consortium for Healthy Aging (NCHA), project 050‐060‐810. All studies are grateful to their participants, faculty and staff. The content of these manuscripts is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the U.S. Department of Health and Human Services.
The FUS cohorts include: the Alzheimer's Disease Centers (ADC) ( P30 AG019610, P30AG013846, P50 AG008702, P50 AG025688, P50 AG047266, P30 AG010133, P50 AG005146, P50 AG005134, P50 AG016574, P50 AG005138, P30 AG008051, P30 AG013854, P30AG008017, P30 AG010161, P50 AG047366, P30 AG010129, P50 AG016573, P50 AG016570, P50 AG005131, P50 AG023501, P30 AG035982, P30 AG028383, P30 AG010124, P50AG005133, P50 AG005142, P30 AG012300, P50 AG005136, P50 AG033514, P50 AG005681,and P50 AG047270), Alzheimer's Disease Neuroimaging Initiative (ADNI) (U19AG024904), Amish Protective Variant Study (RF1AG058066), Cache County Study (R01AG11380, R01AG031272, R01AG21136, RF1AG054052), Case Western Reserve University Brain Bank (CWRUBB) (P50AG008012), Case Western Reserve University Rapid Decline (CWRURD) (RF1AG058267, NU38CK000480), Cuban American Alzheimer's Disease Initiative (CuAADI) (3U01AG052410), Estudio Familiar de Influencia Genetica en Alzheimer (EFIGA) (5R37AG015473, RF1AG015473, R56AG051876), Genetic and Environmental Risk Factors for Alzheimer Disease Among African Americans Study (GenerAAtions) (2R01AG09029, R01AG025259, 2R01AG048927), Gwangju Alzheimer and Related Dementias Study (GARD) (U01AG062602), Hussman Institute for Human Genomics Brain Bank (HIHGBB) (R01AG027944, Alzheimer's Association Identification of Rare Variants in Alzheimer Disease), Ibadan Study of Aging (IBADAN) (5R01AG009956), Mexican Health and Aging Study (MHAS)(R01AG018016), Multi‐Institutional Research in Alzheimer's Genetic Epidemiology (MIRAGE) (2R01AG09029, R01AG025259, 2R01AG048927), Northern Manhattan Study (NOMAS) (R01NS29993), Peru Alzheimer's Disease Initiative (PeADI)(RF1AG054074), Puerto Rican 1066 (PR1066) (Wellcome Trust (GR066133/GR080002), European Research Council (340755)), Puerto Rican Alzheimer Disease Initiative (PRADI) (RF1AG054074), Reasons for Geographic and Racial Differences in Stroke (REGARDS) (U01NS041588), Research in African American Alzheimer Disease Initiative (REAAADI) (U01AG052410), Rush Alzheimer's Disease Center (ROSMAP) (P30AG10161, P30AG72975, R01AG15819, R01AG17919, U01AG46152, U01AG61356), University of Miami Brain Endowment Bank (MBB), and University of Miami/Case Western/North Carolina A&T African American (UM/CASE/NCAT) (U01AG052410, R01AG028786).
The four LSACs are: the Human Genome Sequencing Center at the Baylor College of Medicine (U54 HG003273), the Broad Institute Genome Center (U54HG003067), The American Genome Center at the Uniformed Services University of the Health Sciences (U01AG057659), and the Washington University Genome Institute (U54HG003079).
Biological samples and associated phenotypic data used in primary data analyses were stored at Study Investigators institutions, and at the National Cell Repository Associated Phenotypic Data used in primary and secondary data analyses were provided by Study Investigators, the NIA funded Alzheimer's Disease Centers (ADCs), and the National Alzheimer's Coordinating Center (NACC, U01AG016976) and the National Institute on Aging Genetics of Alzheimer's Disease Data Storage Site (NIAGADS, U24AG041689) at the University of Pennsylvania, funded by NIA This research was supported in part by the Intramural Research Program of the National Institutes of health, National Library of Medicine. Contributors to the Genetic Analysis Data included Study Investigators on projects that were individually funded by NIA, and other NIH institutes, and by private U.S. organizations, or foreign governmental or nongovernmental organizations.
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; Cere Spir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck &Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; PiramalImaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics.The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
1.
Collaborators
Alzheimer's Disease Neuroimaging Initiative
Maria Carrillo (Alzheimer's Association), Eric M. Reiman, Kewei Chen (Banner Alzheimer's Institute), Donna Masterman (Biogen), Robert C. Green (Brigham and Women's Hospital/Harvard Medical School), Carole Ho (Denali Therapeutics), Adam Fleisher (Eli Lilly), Andrew J. Saykin, Kwangsik Nho, Liana G. Apostolova, Shannon L. Risacher (Indiana University School of Medicine), Jonathan Jackson (Massachusetts General Hospital), Arvin Forghanian‐Arani, Bret Borowski, Chad Ward, Christopher Schwarz, Clifford R. Jack, Jr., David Jones, Jeff Gunter, Kejal Kantarci, Matthew Senjem, Prashanthi Vemuri, Robert Reid, Ronald Petersen (Mayo Clinic), John K. Hsiao (National Institute of Health), William Potter (William Potter), Eliezer Masliah, John K. Hsiao, Laurie Ryan, Marie Bernard, Nina Silverberg (National Institute on Aging), Adrienne Kormos, Cat Conti, Dallas Veitch, Derek Flenniken, Diana Truran Sacrey, Mark Choe, Miriam Ashford, Stephanie Rossi Chen (NCIRE / The Vererans Health Research Institute), Kelley Faber, Kelly Nudelman, Kristi Wilme, Tatiana M. Foroud (NCRAD/Indiana University School of Medicine), John Q. Trojanowki, Leslie M. Shaw, Magdalena Korecka, Michal Figurski (Perelman School of Medicine, University of Pennsylvania), Zaven Khachaturian (Prevent Alzheimer's Disease 2020), Lisa Barnes (Rush University), Ian Malone, Nick C. Fox (University College London), Laurel Beckett, Michael W. Weiner, William Jagust (University of California), Susan Landau, William Jagust (University of California, Berkeley), Alexander Knaack, Charles DeCarli, Danielle Harvey, Evan Fletcher, Laurel Beckett (University of California, Davis), Hector González (University of California, San Diego), Chengshi Jin, Duygu Tosun‐Turgut, John Neuhaus, Juliet Fockler, Rachel Nosheny (University of California, San Francisco), Robert A. Koeppe (University of Michigan), John Q. Trojanowki, Leslie M. Shaw, Paul A. Yushkevich, Sandhitsu Das (University of Pennsylvania), Chet Mathis (University of Pittsburgh), Arthur W. Toga, Caileigh Zimmerman, Devon Gessert, Elizabeth Shaffer, Garrett Miller, Godfrey Coker, Gustavo Jimenez, Jennifer Salazar, Jeremy Pizzola, Karen Crawford, Lindsey Hergesheimer, Michael Donohue, Michael Rafii, Olusegun Adegoke, Paul Aisen, Payam Mahboubi, Rema Raman, Sarah Walter, Scott Neu, Shelley Moore, Stephanie Smith, Taylor Clanton, Yuliana Cabrera, Michael Donohue, Neda Jahanshad, Paul Thompson, Sophia I. Thomopoulos, Talia M. Nir, Karen Crawford (University of Southern California), Tom Montine (University of Washington), Ozioma Okonkwo (University of Wisconsin), Li Shen (UPenn School of Medicine), Richard Perrin, Erin Franklin, Erin Householder, Haley Bernhardt, John C. Morris, Lisa Taylor‐Reinwald, Nigel J. Cairns, Richard J. Perrin (Washington University). The full collaborator list is available in https://nam04.safelinks.protection.outlook.com/?url = https%3A%2F%2Fadni.loni.usc.edu%2Fwp‐content%2Fuploads%2Fhow_to_apply%2FADNI_Acknowledgement_List.pdf&data = 05%7C01%7Ckatsumata.yuriko%40uky.edu%7C7484c356d76d4ad9649e08dbe6eadb29%7C2b30530b69b64457b818481cb53d42ae%7C0%7C0%7C638357668786321944%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata = 531kgVlDLBCXf5dKUyfLL47T50BRyWMeedhgsKXWpAc%3D&reserved = 0
National Alzheimer's Coordinating Center
Sarah Biber, Jessica Welsch, Hannah Rosentreter, Brittany Fair, Amy Young, Brittany Hale, Valerie Hockens, Faith Buckley, Aileen Ida, Jane Xia, Emily Almeda, Kari A. Stephens, Dean K. Shibata, Kwun Chuen Gary Chan, Will Longstreth, Yen‐Chi Che, Jessica Culhane, Zack Miller, Sarah Yasuda, Sean Mooney, Janene Hubbard, Ben Keller, Zach Stark, Joylee Wu, Laura Mcleod, Chandima Hewanadungodage, Brendan Smith
Katsumata Y, Fardo DW, Shade LM, et al. Genetic associations with dementia‐related proteinopathy: Application of item response theory. Alzheimer's Dement. 2024;20:2906–2921. 10.1002/alz.13741
Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp‐content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
Contributor Information
Yuriko Katsumata, Email: katsumata.yuriko@uky.edu.
The Alzheimer's Disease Neuroimaging Initiative:
Maria Carrillo, Eric M. Reiman, Kewei Chen, Donna Masterman, Robert C. Green, Carole Ho, Adam Fleisher, Andrew J. Saykin, Kwangsik Nho, Liana G. Apostolova, Shannon L. Risacher, Jonathan Jackson, Arvin Forghanian-Arani, Bret Borowski, Chad Ward, Christopher Schwarz, Clifford R. Jack, David Jones, Jeff Gunter, Kejal Kantarci, Matthew Senjem, Prashanthi Vemuri, Robert Reid, Ronald Petersen, John K. Hsiao, William Potter, Eliezer Masliah, John K. Hsiao, Laurie Ryan, Marie Bernard, Nina Silverberg, Adrienne Kormos, Cat Conti, Dallas Veitch, Derek Flenniken, Diana Truran Sacrey, Mark Choe, Miriam Ashford, Stephanie Rossi Chen, Kelley Faber, Kelly Nudelman, Kristi Wilme, Tatiana M. Foroud, John Q. Trojanowki, Leslie M. Shaw, Magdalena Korecka, Michal Figurski, Zaven Khachaturian, Lisa Barnes, Ian Malone, Nick C. Fox, Laurel Beckett, Michael W. Weiner, William Jagust, Susan Landau, William Jagust, Alexander Knaack, Charles DeCarli, Danielle Harvey, Evan Fletcher, Laurel Beckett, Hector González, Chengshi Jin, Duygu Tosun‐Turgut, John Neuhaus, Juliet Fockler, Rachel Nosheny, Robert A. Koeppe, John Q. Trojanowki, Leslie M. Shaw, Paul A. Yushkevich, Sandhitsu Das, Chet Mathis, Arthur W. Toga, Caileigh Zimmerman, Devon Gessert, Elizabeth Shcrer, Garrett Miller, Godfrey Coker, Gustavo Jimenez, Jennifer Salazar, Jeremy Pizzola, Karen Crawford, Lindsey Hergesheimer, Michael Donohue, Michael Rafii, Olusegun Adegoke, Paul Aisen, Payam Mahboubi, and Rema Raman
The National Alzheimer's Coordinating Center:
Sarah Walter, Scott Neu, Shelley Moore, Stephanie Smith, Taylor Clanton, Yuliana Cabrera, Michael Donohue, Neda Jahanshad, Paul Thompson, Sophia I. Thomopoulos, Talia M. Nir, Karen Crawford, Tom Montine, Ozioma Okonkwo, Li Shen, Richard Perrin, Erin Franklin, Erin Householder, Haley Bernhardt, John C. Morris, Lisa Taylor-Reinwald, Nigel J. Cairns, Richard J. Perrin, Sarah Biber, Jessica Welsch, Hannah Rosentreter, Brittany Fair, Amy Young, Brittany Hale, Valerie Hockens, Faith Buckley, Aileen Ida, Jane Xia, Emily Almeda, Kari A. Stephens, Dean K. Shibata, Kwun Chuen Gary Chan, Will Longstreth, Yen-Chi Che, Jessica Culhane, Zack Miller, Sarah Yasuda, Sean Mooney, Janene Hubbard, Ben Keller, Zach Stark, Joylee Wu, Laura Mcleod, Chandima Hewanadungodage, and Brendan Smith
REFERENCES
- 1. Armstrong RA, Lantos PL, Cairns NJ. Overlap between neurodegenerative disorders. Neuropathology. 2005;25:111‐124. [DOI] [PubMed] [Google Scholar]
- 2. Kovacs GG, Milenkovic I, Wohrer A, et al. Non‐Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community‐based autopsy series. Acta Neuropathol. 2013;126:365‐384. [DOI] [PubMed] [Google Scholar]
- 3. Jellinger KA. The enigma of mixed dementia. Alzheimers Dement. 2007;3:40‐53. [DOI] [PubMed] [Google Scholar]
- 4. Kovacs GG, Alafuzoff I, Al‐Sarraj S, et al. Mixed brain pathologies in dementia: the BrainNet Europe consortium experience. Dement Geriatr Cogn Disord. 2008;26:343‐350. [DOI] [PubMed] [Google Scholar]
- 5. Kryscio RJ, Abner EL, Jicha GA, et al. Self‐reported memory complaints: a comparison of demented and unimpaired outcomes. J Prev Alzheimers Dis. 2016;3:13‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nelson PT, Trojanowski JQ, Abner EL, et al. New old pathologies: AD, PART, and cerebral age‐related TDP‐43 with sclerosis (CARTS). J Neuropathol Exp Neurol. 2016;75:482‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nho K, Saykin AJ. Alzheimer's disease neuroimaging I, Nelson PT. Hippocampal sclerosis of aging, a common Alzheimer's disease ‘Mimic’: risk genotypes are associated with brain atrophy outside the temporal lobe. J Alzheimers Dis. 2016;52:373‐383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Neltner JH, Abner EL, Jicha GA, et al. Brain pathologies in extreme old age. Neurobiol Aging. 2016;37:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jellinger KA, Alafuzoff I, Attems J, et al. PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol. 2015;129:757‐762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nelson PT, Jicha GA, Schmitt FA, et al. Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol . 2007;66:1136‐1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ighodaro ET, Abner EL, Fardo DW, et al. Risk factors and global cognitive status related to brain arteriolosclerosis in elderly individuals. J Cereb Blood Flow Metab. 2017;37:201‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karanth S, Nelson PT, Katsumata Y, et al. Prevalence and clinical phenotype of quadruple misfolded proteins in older adults. JAMA Neurol. 2020;77:1299‐1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. James BD, Wilson RS, Boyle PA, Trojanowski JQ, Bennett DA, Schneider JA. TDP‐43 stage, mixed pathologies, and clinical Alzheimer's‐type dementia. Brain. 2016;139:2983‐2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brenowitz WD, Hubbard RA, Keene CD, et al. Mixed neuropathologies and estimated rates of clinical progression in a large autopsy sample. Alzheimers Dement. 2017;13:654‐662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Montine TJ, Koroshetz WJ, Babcock D, et al. Recommendations of the Alzheimer's disease‐related dementias conference. Neurology. 2014;83:851‐860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Whitwell JL, Jack CR Jr, Parisi JE, et al. Rates of cerebral atrophy differ in different degenerative pathologies. Brain. 2007;130:1148‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jack CR Jr, Knopman DS, Chetelat G, et al. Suspected non‐Alzheimer disease pathophysiology–concept and controversy. Nat Rev Neurol. 2016;12:117‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cai L, Choi K, Hansen M, Harrell L. Item response theory. Annu Rev Stat Appl. 2016;3:297‐321. [Google Scholar]
- 19. Rasch G. Studies in mathematical psychology: I. Probabilistic models for some intelligence and attainment tests. Nielsen & Lydiche; 1960. [Google Scholar]
- 20. Muraki E. A generalized partial credit model: application of an EM algorithm. Appl Psychol Meas. 1992;16:159‐176. [Google Scholar]
- 21. Fardo D, Celedon JC, Raby BA, Weiss ST, Lange C. On dichotomizing phenotypes in family‐based association tests: quantitative phenotypes are not always the optimal choice. Genet Epidemiol. 2007;31:376‐382. [DOI] [PubMed] [Google Scholar]
- 22. Bhandari M, Lochner H. Effect of continuous versus dichotomous outcome variables on study power when sample sizes of orthopaedic randomized trials are small. Arch Orthop Trauma Surg. 2002;122:96‐98. [DOI] [PubMed] [Google Scholar]
- 23. Altman DG, Royston P. The cost of dichotomising continuous variables. BMJ. 2006;332:1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. Religious orders study and rush memory and aging project. J Alzheimers Dis. 2018;64:S161‐S189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Katsumata Y, Abner EL, Karanth S, et al. Distinct clinicopathologic clusters of persons with TDP‐43 proteinopathy. Acta Neuropathol. 2020;140:659‐674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thal DR, Rub U, Orantes M, Braak H. Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791‐1800. [DOI] [PubMed] [Google Scholar]
- 27. Braak H, Braak E. Alzheimer's disease affects limbic nuclei of the thalamus. Acta Neuropathol. 1991;81:261‐268. [DOI] [PubMed] [Google Scholar]
- 28. Mirra SS. The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer's disease: a commentary. Neurobiol Aging. 1997;18:S91‐94. [DOI] [PubMed] [Google Scholar]
- 29. Mirra SS, Heyman A, McKeel D, et al. The consortium to establish a registry for Alzheimer's disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479‐486. [DOI] [PubMed] [Google Scholar]
- 30. Leung YY, Valladares O, Chou YF, et al. VCPA: genomic variant calling pipeline and data management tool for Alzheimer's Disease Sequencing Project. Bioinformatics. 2019;35:1768‐1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Naj AC, Lin H, Vardarajan BN, et al. Quality control and integration of genotypes from two calling pipelines for whole genome sequence data in the Alzheimer's disease sequencing project. Genomics. 2019;111:808‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Danecek P, Bonfield JK, Liddle J, et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10:giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Purcell S, Neale B, Todd‐Brown K, et al. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet. 2007;81:559‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second‐generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Core Tea R, m . R: a language and environment for statistical computing. 3.4.4 ed. Vienna, Austria: R. Foundation for Statistical Computing; 2021.
- 36. Chalmers RP. mirt: a multidimensional item response theory package for the R environment. Journal of Statistical Software. 2012;48:1‐29. [Google Scholar]
- 37. Zhou X, Stephens M. Genome‐wide efficient mixed‐model analysis for association studies. Nat Genet. 2012;44:821‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics. 2010;26:2190‐2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Obenchain V, Lawrence M, Carey V, Gogarten S, Shannon P, Morgan M. VariantAnnotation: a bioconductor package for exploration and annotation of genetic variants. Bioinformatics. 2014;30:2076‐2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Team BC, Maintainer BP, TxDb.Hsapiens.UCSC.hg38.knownGene: Annotation package for TxDb object(s). R package version 3.4.6. ed2019.
- 41. Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome‐wide association scan results. Bioinformatics. 2010;26:2336‐2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10:e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wallace C. A more accurate method for colocalisation analysis allowing for multiple causal variants. PLoS Genet. 2021;17:e1009440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Consortium GT, Laboratory DA, et al. Genetic effects on gene expression across human tissues. Nature. 2017;550:204‐213. Coordinating Center ‐Analysis Working G, Statistical Methods groups‐Analysis Working G, Enhancing Gg, Fund NIHC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ng B, White CC, Klein HU, et al. An xQTL map integrates the genetic architecture of the human brain's transcriptome and epigenome. Nat Neurosci. 2017;20:1418‐1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aoki N, Murray ME, Ogaki K, et al. Hippocampal sclerosis in Lewy body disease is a TDP‐43 proteinopathy similar to FTLD‐TDP Type A. Acta Neuropathol. 2015;129:53‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dickson DW, Baker M, Rademakers R. Common variant in GRN is a genetic risk factor for hippocampal sclerosis in the elderly. Neuro‐degenerative diseases. 2010;7:170‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Murray ME, Cannon A, Graff‐Radford NR, et al. Differential clinicopathologic and genetic features of late‐onset amnestic dementias. Acta Neuropathol. 2014;128:411‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Katsumata Y, Shade LM, Hohman TJ, et al. Multiple gene variants linked to Alzheimer's‐type clinical dementia via GWAS are also associated with non‐Alzheimer's neuropathologic entities. Neurobiol Dis. 2022;174:105880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dugan AJ, Nelson PT, Katsumata Y, et al. Analysis of genes (TMEM106B, GRN, ABCC9, KCNMB2, and APOE) implicated in risk for LATE‐NC and hippocampal sclerosis provides pathogenetic insights: a retrospective genetic association study. Acta Neuropathol Commun. 2021;9:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bellenguez C, Kucukali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat Genet. 2022;54:412‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lambert JC, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tsuang D, Leverenz JB, Lopez OL, et al. GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology. 2012;79:1944‐1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guerreiro R, Ross OA, Kun‐Rodrigues C, et al. Investigating the genetic architecture of dementia with Lewy bodies: a two‐stage genome‐wide association study. Lancet Neurol. 2018;17:64‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang HS, Yu L, White CC, et al. Evaluation of TDP‐43 proteinopathy and hippocampal sclerosis in relation to APOE epsilon4 haplotype status: a community‐based cohort study. Lancet Neurol. 2018;17:773‐781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wennberg AM, Tosakulwong N, Lesnick TG, et al. Association of apolipoprotein E epsilon4 with transactive response DNA‐binding protein 43. JAMA Neurol. 2018;75:1347‐1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Brown DF, Dababo MA, Bigio EH, et al. Neuropathologic evidence that the Lewy body variant of Alzheimer disease represents coexistence of Alzheimer disease and idiopathic Parkinson disease. J Neuropathol Exp Neurol. 1998;57:39‐46. [DOI] [PubMed] [Google Scholar]
- 58. Nelson PT, Kryscio RJ, Jicha GA, et al. Relative preservation of MMSE scores in autopsy‐proven dementia with Lewy bodies. Neurology. 2009;73:1127‐1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jellinger KA. Neuropathological spectrum of synucleinopathies. Mov Disord. 2003;18(6):S2‐12. Suppl. [DOI] [PubMed] [Google Scholar]
- 60. Song Y, Lin F, Ye CH, et al. Rare, low‐frequency and common coding variants of ARHGEF28 gene and their association with sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2020;87:138. e1‐ e6. [DOI] [PubMed] [Google Scholar]
- 61. Ma Y, Tang L, Chen L, et al. ARHGEF28 gene exon 6/intron 6 junction mutations in Chinese amyotrophic lateral sclerosis cohort. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:309‐311. [DOI] [PubMed] [Google Scholar]
- 62. Tamaki Y, Urushitani M. Molecular dissection of TDP‐43 as a leading cause of ALS/FTLD. Int J Mol Sci. 2022:23:12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Droppelmann CA, Keller BA, Campos‐Melo D, Volkening K, Strong MJ. Rho guanine nucleotide exchange factor is an NFL mRNA destabilizing factor that forms cytoplasmic inclusions in amyotrophic lateral sclerosis. Neurobiol Aging. 2013;34:248‐262. [DOI] [PubMed] [Google Scholar]
- 64. Keller BA, Volkening K, Droppelmann CA, Ang LC, Rademakers R, Strong MJ. Co‐aggregation of RNA binding proteins in ALS spinal motor neurons: evidence of a common pathogenic mechanism. Acta Neuropathol. 2012;124:733‐747. [DOI] [PubMed] [Google Scholar]
- 65. Chang P, Heier C, Qin W, Han L, Huang F, Sun Q. Molecular identification of transmembrane protein 68 as an endoplasmic reticulum‐anchored and brain‐specific protein. PLoS One. 2017;12:e0176980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang Y, Zeng F, Zhao Z, et al. Transmembrane protein 68 functions as an MGAT and DGAT enzyme for triacylglycerol biosynthesis. Int J Mol Sci. 2023:24:2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sol J, Jove M, Povedano M, et al. Lipidomic traits of plasma and cerebrospinal fluid in amyotrophic lateral sclerosis correlate with disease progression. Brain Commun. 2021;3:fcab143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rajaratnam S, Soman AP, Phalguna KS, et al. Integrated omic analysis delineates pathways modulating toxic TDP‐43 protein aggregates in amyotrophic lateral sclerosis. Cells. 2023:12:1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Courage C, Jackson CB, Hahn D, et al. SDHA mutation with dominant transmission results in complex II deficiency with ocular, cardiac, and neurologic involvement. Am J Med Genet A. 2017;173:225‐230. [DOI] [PubMed] [Google Scholar]
- 70. Na U, Yu W, Cox J, et al. The LYR factors SDHAF1 and SDHAF3 mediate maturation of the iron‐sulfur subunit of succinate dehydrogenase. Cell Metab. 2014;20:253‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lucini CB, Braun RJ. Mitochondrion‐dependent cell death in TDP‐43 proteinopathies. Biomedicines. 2021;9:376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jiang L, Ngo ST. Altered TDP‐43 structure and function: key insights into aberrant RNA, mitochondrial, and cellular and systemic metabolism in amyotrophic lateral sclerosis. Metabolites. 2022:12:709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jackson CB, Nuoffer JM, Hahn D, et al. Mutations in SDHD lead to autosomal recessive encephalomyopathy and isolated mitochondrial complex II deficiency. J Med Genet. 2014;51:170‐175. [DOI] [PubMed] [Google Scholar]
- 74. Ohlenbusch A, Edvardson S, Skorpen J, et al. Leukoencephalopathy with accumulated succinate is indicative of SDHAF1 related complex II deficiency. Orphanet J Rare Dis. 2012;7:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ghezzi D, Goffrini P, Uziel G, et al. SDHAF1, encoding a LYR complex‐II specific assembly factor, is mutated in SDH‐defective infantile leukoencephalopathy. Nat Genet. 2009;41:654‐656. [DOI] [PubMed] [Google Scholar]
- 76. Krzyzewska IM, Lauffer P, Mul AN, et al. Expression quantitative trait methylation analysis identifies whole blood molecular footprint in fetal alcohol spectrum disorder (FASD). Int J Mol Sci. 2023:24:6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information