

Abstract
Dendritic cells are the most potent antigen-presenting cells, and the possibility of their use for cancer vaccination has renewed the interest in this therapeutic modality. Nevertheless, the ideal immunization protocol with these cells has not been described yet. In this paper we describe the preliminary results of a protocol using autologous tumor and allogeneic dendritic hybrid cell vaccination every 6 weeks, for metastatic melanoma and renal cell carcinoma (RCC) patients. Thirty-five patients were enrolled between March 2001 and March 2003. Though all patients included presented with large tumor burdens and progressive diseases, 71% of them experienced stability after vaccination, with durations up to 19 months. Among RCC patients 3/22 (14%) presented objective responses. The median time to progression was 4 months for melanoma and 5.7 months for RCC patients; no significant untoward effects were noted. Furthermore, immune function, as evaluated by cutaneous delayed-type hypersensitivity reactions to recall antigens and by peripheral blood proliferative responses to tumor-specific and nonspecific stimuli, presented a clear tendency to recover in vaccinated patients. These data indicate that dendritic cell–tumor cell hybrid vaccination affects the natural history of advanced cancer and provide support for its study in less advanced patients, who should, more likely, benefit even more from this approach.
Keywords: Dendritic cells, Hybrid cell vaccination, Melanoma, Renal cell carcinoma
Introduction
Dendritic cells (DCs) are the most effective initiators of an immune response [2, 3], and their generation in vitro has opened new possibilities for cancer therapy. Many experimental data support the enthusiasm generated by these lines of research [8, 15, 18, 20] and have furthered the development of various clinical trials [1, 4, 6, 10, 14, 17, 19, 26, 28].
Crude tumor extracts, tumor cells, selected tumor antigens or peptides, as well as tumor mRNA have been used with DCs as immunization targets in tumor-bearing individuals, each one yielding promising but still not definitive results [11, 24, 29]. Among the various ways of presenting tumor antigens by DC, their fusion with autologous tumor cells seems an attractive approach, since it joins the antigen-processing and immune-stimulatory potential of DCs with the full antigenic spectrum of the tumor cells [30, 31].
In the present report we show the preliminary clinical results of hybrid dendritic cell–tumor cell therapeutic vaccination for metastatic melanoma or renal cell carcinoma (RCC). Patients included in this present study had advanced diseases, large tumor burdens, were mostly negative in cutaneous delayed-type hypersensitivity (DTH) tests against common antigens, and had failed various other treatment approaches. Furthermore, patients entered the protocol with clearly progressive disease that frequently imposed quality-of-life limitations. After vaccination no untoward effects have been observed, most patients experienced clinical benefit and disease stabilization for periods ranging from 4 to more than 20 months, and an overall improvement of immune function occurred.
The promising, but still not completely satisfactory, clinical results obtained and the immune function effects of this dendritic cell–based vaccination method support the further development of such a strategy and should contribute to the design of even better immunotherapeutic approaches for cancer patients.
Patients and methods
Eligibility criteria
Thirty-five patients with histological diagnosis of melanoma or RCC and histological or radiological confirmation of metastatic disease were included in the trial. A primary or metastatic lesion that could be surgically removed and a remaining bidimensionally evaluable metastatic lesion were needed. Patients were further required to have an Eastern Cooperative Oncology Group (ECOG) performance status of 0, 1, or 2; normal or near-normal hematopoietic renal and hepatic function; and to have received no chemotherapy, radiotherapy, or immunotherapy for at least 4 weeks before study enrollment. Patients with uncontrolled metastatic lesions in the brain, hypercalcemia, with other previous or concomitant neoplasia, who were pregnant or lactating, with autoimmune diseases, or were HIV seropositive were excluded. All patients included in the study gave written informed consent. The protocol was approved by the Institutional Ethics Committee and was conducted in the Oncology Center of the Hospital Sírio-Libanês, São Paulo, Brazil.
Clinical protocol, DC generation, tumor cell preparation, and response evaluation
Generation of DCs from peripheral blood
Peripheral blood mononuclear cells (PBMCs) were collected from healthy unrelated volunteers through apheresis performed in a Cobe Spectra Blood Cell Separator 7.0 (Cobe, Lakewood, CO, USA), programmed for mononuclear cell collection, after informed consent of donors. Acid citrate dextrose (ACD) was used as anticoagulant, with an anticoagulant to blood ratio of 1:8–1:11, and one and a half standard blood volemias were processed over 2–4 h. Mononuclear cells were separated further with a density gradient (1.077 g/dl; Lymphoprep; Axis-Shield, Oslo, Norway), resuspended, and seeded in culture flasks in RPMI 1640 culture medium (Gibco, Grand Island, NY, USA), supplemented with 10% fetal calf serum (Gibco, Grand Island, NY, USA). After that, flasks were incubated at 37°C for a period of 4 h, when nonadherent cells were removed, and the RPMI medium was replaced by a serum-free medium (X-Vivo 15; BioWhittaker, Walkersville, MD, USA), containing GM-CSF (50 ng/ml; R&D, Minneapolis, MN, USA) and interleukin 4 (50 ng/ml; R&D, Minneapolis, MN, USA). After 5 days in culture, TNF-α (50 ng/ml; R&D, Minneapolis, MN, USA) was added to the cultures for DC activation. After 2 further days in culture, cells were harvested and used for the generation of the dendritic–tumor hybrid vaccine.
Flow cytometry
Cell preparations (5×106 cells/condition) were labeled with each of the various specific fluorescent antibodies (CD1a, CD14, CD80, CD86, CD83, HLA-ABC, HLA-DR) or controls (Caltag Laboratories, Burlingame, CA, USA) and analyzed in a FACSCalibur cytometer (Becton Dickinson, San Jose, CA, USA) with CellQuest software (Becton Dickinson, San Jose, CA, USA). At least 10,000 events were acquired per antibody analyzed.
Tumor cell preparation
Fresh tumor samples, aseptically obtained from surgical resection of either the primary or a metastatic lesion, were minced, and single-cell suspensions generated by digestion with collagenase type VIII (0.56 mg/ml; Sigma, St Louis, MO, USA) and DNAse I (0.026 mg/ml; Sigma, St Louis, MO, USA), during a 90–120 min incubation at 37°C, under agitation. On average, 1×107 viable cells were obtained for each gram of tissue digested. Cell viability was evaluated by trypan blue exclusion and ranged from 30 to 100%. After recovery, the cells were washed, resuspended in freezing medium (60% RPMI 1640, 30% FCS, and 10% dimethyl sulphoxide; Sigma, St Louis, MO, USA), immediately frozen, and kept in liquid nitrogen until use for hybridization with DCs.
Dendritic cell–tumor cell hybridization and vaccine preparation
At the last day of DC cultures, DCs were harvested, washed, and resuspended in a sterile 5% glucose solution to a concentration of 1×107 cells/ml. Tumor cells were thawed, washed, and also resuspended in a sterile 5% glucose solution to a concentration of 1×107 cells/ml. These two cell suspensions were mixed at equal volumes, and cells were fused by an electric pulse of 1,000 V/cm at 25 μF (applied by a Gene-Pulser II; Bio-Rad, Richmond, CA, USA), after being aligned in an inhomogeneous electrical field (62.5 V/cm) for 15 s. After a 2-min rest in the electroporation cuvette, cells were transferred to a relaxation buffer (100-mM KCL, 3-mM NaCl, 1.25-mM EDTA, 10-mM PIPES, 0.5-mM ATP, adjusted to pH 6.8), where they were kept for a further 5 min [29]. Fusion efficacy was determined in pilot experiments by staining tumor and DCs with CellTracker green and red fluorescence dyes (Molecular Probes, Eugene, OR, USA) followed by flow cytometry counting of double stained hybrids, and ranged between 15 and 30%. Mixtures of both cell preparations not submitted to the electrical pulse did not show double-stained cells. To avoid cell loss, this control of fusion efficacy was not performed in each vaccine preparation. Shortly after fusion, all cells stained with trypan blue, but after the initial rest in the relaxation buffer and further rest in culture medium, they regained the ability to exclude the vital dye. The hybrid cell preparation was centrifuged, resuspended in 1–2 ml of sterile phosphate-buffered saline (pH 7.2) and, after irradiation (200 Gy), injected in the patients. At this moment, cell viability ranged from 60 to 80% of the initial tumor cell viability. Vaccine injections were either intradermic (in the first 12 months of the protocol) or intranodal, under ultrasonographic guidance, and in both cases away from known tumor sites. This change in immunization route was an attempt to increase hybrid cell vaccine access to lymphoid organs, but no comparison of immunization routes was intended.
Vaccine application and response evaluation
Thirteen melanoma and 22 RCC patients were included; 11 melanoma and 19 RCC patients received at least two doses of the hybrid cell vaccine, with a 6-week interval between doses. After the second dose, vaccination continued, with the same interval, until disease progression or until no more tumor cells were available for vaccine preparation. All patients underwent clinical and imaging monitoring at baseline, 6, and 12 weeks after the first dose of the vaccine. Disease evaluation included clinical examination, NMR of brain, CT scans of thorax, abdomen, pelvis, and any other clinically relevant site. After the first two evaluations, patients were clinically evaluated every month, and imaging studies repeated every 12 weeks. Disease progression was defined as ≥25% increase in measurable lesions and/or appearance of new lesions; disease was considered stable when measurable lesions had a less than 50% decrease or less than 25% increase in dimensions; a decrease of >50% was classified as a partial clinical response, and the complete disappearance of lesions as a complete clinical response. Adverse reactions were classified according to the National Cancer Institute toxicity criteria.
Delayed-type hypersensitivity (DTH) reaction tests
Patients were submitted before vaccination and 12 weeks thereafter to DTH reaction tests against common recall antigens (PPD, histoplasmin, candidin, trychophytin). The presence of positive DTH reactions before vaccination was not required for protocol enrolment.
Mixed lymphocyte reactions
Blood was collected from patients on each vaccination day. PBMCs were separated with a density gradient (1.077 g/dl; Lymphoprep; Axis-Shield, Oslo, Norway) and used in mixed lymphocyte reactions against DCs, autologous tumor cells, DC–tumor cell mixtures, and DC-tumor hybrid cells. The reaction to phytohemagglutinin A (PHA; Sigma, St Louis, MO, USA) was also evaluated. Cells were cultured for 5 days, after which their response was evaluated by MTT reduction as described [32].
Results
Thirteen patients with metastatic melanoma and 22 patients with RCC were enrolled in the protocol from March 2001 to March 2003 and vaccinated with dendritic cell–tumor cell hybrids. Of these, two patients with melanoma and three with RCC showed progression immediately after the first dose, and did not receive a second dose of the vaccine, due to the patient’s or to the accompanying physician’s decision. Patients’ characteristics are listed in Table 1.
Table 1.
Demographics of patients enrolled in the study between March 2001 and March 2003
| Patient number | Sex | Age | Metastasis site(s) |
|---|---|---|---|
| Renal cell carcinoma | |||
| 1 | F | 66 | Liver |
| 2 | F | 70 | Pancreas, liver |
| 4 | F | 75 | Lung |
| 5 | F | 29 | Retroperitoneal lymph nodes |
| 6 | M | 45 | Lung, bone |
| 8 | M | 39 | Lung, soft tissue |
| 9 | M | 36 | Lung, bone, soft tissue |
| 10 | M | 44 | Lung |
| 11 | M | 34 | Lung |
| 12 | M | 66 | Liver |
| 13 | M | 66 | Lung, bone |
| 15 | M | 68 | Lung |
| 16 | M | 52 | Liver, retroperitoneal |
| 17 | M | 50 | Lung, retroperitoneal |
| 18 | M | 61 | Liver, bones |
| 19 | M | 53 | Lung |
| 20 | M | 54 | Lung, bones |
| 21 | M | 51 | Liver, retroperitoneal |
| 22 | M | 46 | Lung |
| 14a | M | 70 | Lung, soft tissue, adrenal, contralateral kidney |
| 7a | M | 78 | Skin, central nervous system, bone |
| 3a | F | 32 | Bone |
| Melanoma | |||
| 2 | F | 34 | Skin, lymph nodes, retroperitoneal |
| 3 | F | 42 | Breast, in transit |
| 4 | F | 56 | Lung |
| 6 | F | 51 | Liver, lung |
| 8 | M | 40 | In transit |
| 9 | M | 2 | Soft tissue, retroperitoneal |
| 10 | M | 63 | Lung, in transit |
| 11 | M | 50 | Gastrointestinal tract, skin |
| 12 | M | 36 | Lung, liver, pancreas |
| 13 | M | 44 | Lymph nodes |
| 5a | F | 41 | Lung, in transit |
| 7a | M | 55 | Liver, lung, spleen, bones |
aPatient who received only one dose of the vaccine and therefore was not evaluated further
A total of 145 doses of the vaccine were applied to the patients, with an average number of 4.7 doses/patient. Most patients (71%) experienced disease stabilization with the vaccination, no major adverse reaction occurred, and only two patients presented low-grade fever after vaccine applications. The number of cells in each vaccine dose varied among patients and ranged from 0.6×107 to 3.0×107 tumor cells plus an equal number of DCs (Table 2), depending on the viable tumor cell yield after digestion of the available fresh tumor samples.
Table 2.
Immunization scheme of patients separated according to diagnosis. ID intradermal immunization, IN ultrasound-guided intranodal immunization, SD stable disease, PD progressive disease, OR objective response
| Patient number | Tumor cells/dose (×10−7) | Doses received | Immunization route | Response (at 3 months) | Duration of response (months)a |
|---|---|---|---|---|---|
| Renal cell carcinoma | |||||
| 1 | 1.0 | 5 | ID | SD | 8 |
| 2 | 0.6 | 4 | ID (2) IN (2) | SD | 19+ |
| 4 | 0.6 | 3 | ID | SD | 19+ |
| 5 | 3.0 | 4 | IN | SD | 4 |
| 6 | 0.6 | 8 | ID (6) IN (2) | OR | 21 |
| 8 | 0.6 | 5 | IN | SD | 15+ |
| 9 | 0.6 | 5 | ID | SD | 6 |
| 10 | 1.9 | 8 | ID (1) IN (7) | SD | 18+ |
| 11 | 0.6 | 4 | ID (3) IN (1) | SD | 6 |
| 12 | 2.0 | 2 | IN | SD | 12 |
| 13 | 0.6 | 4 | ID | OR | 5 |
| 15 | 0.6 | 3 | IN | PD | – |
| 16 | 0.6 | 2 | IN | PD | – |
| 17 | 1.5 | 3 | IN | SD | 4 |
| 18 | 0.6 | 3 | IN | SD | 12+ |
| 19 | 1.0 | 3 | ID | SD | 4 |
| 20 | 1.0 | 3 | ID | SD | 4 |
| 21 | 2.0 | 6 | ID (1) IN (5) | OR | 7 |
| 22 | 0.8 | 5 | IN | SD | 6+ |
| 14b | 1.0 | 1 | ID | – | – |
| 7b | 0.6 | 1 | IN | – | – |
| 3b | 0.7 | 1 | IN | – | – |
| Melanoma | |||||
| 1 | 2.0 | 3 | ID | PD | – |
| 2 | 1.1 | 2 | ID | PD | – |
| 3 | 0.6 | 3 | ID (1) IN (2) | SD | 4 |
| 4 | 1.8 | 3 | IN | PD | – |
| 6 | 1.0 | 3 | ID | SD | 4 |
| 8 | 2.3 | 3 | IN | SD | 8 |
| 9 | 1.7 | 10 | ID | SD | 17+ |
| 10 | 1.0 | 9 | ID | SD | 12 |
| 11 | 2.5 | 6 | ID (1) IN (5) | SD | 7 |
| 12 | 1.0 | 8 | ID (6) IN (2) | SD | 11 |
| 13 | 3.0 | 4 | IN | SD | 6 |
| 5 | 0.6 | 1 | ID | – | – |
| 7 | 2.6 | 1 | ID | – | – |
aDuration of response counted from protocol inclusion
bPatient who received only one dose of the vaccine and therefore was not evaluated at 3 months
Immunological effects of vaccination
PBMC activation by any of the stimuli used varied significantly among patients and within samples from the same patient. In spite of this variation, however, a general tendency to improved PBMC responses to all stimuli could be noted in patients over time (Fig. 1a–e). Similarly, among the 18 patients evaluated for this parameter, the frequency of negativity of all four DTH reactions decreased after vaccination from 55.5 to 27.7%, not reaching, however, statistical significance (p=0.17) (Table 3).
Fig. 1a–e.

Evolution of peripheral blood lymphocyte responses during hybrid cell vaccination. Mononuclear cells were isolated from peripheral blood collected before each vaccination dose, every 6 weeks, and their responses to autologous tumor cells (a), dendritic cells (b), tumor–dendritic cell mixtures (c), tumor–dendritic cell hybrids (d), and phytohemagglutinin A (e) were evaluated by MTT reduction. Stimulation indexes were calculated based on MTT reduction by isolated cells
Table 3.
Cutaneous delayed-type hypersensitivity responses to a panel of recall antigens (PPD, candidin, and trychophytin) before and 12 weeks after (two doses) vaccination with the dendritic cell–tumor cell hybrids. Patients who responded to at least one of the antigens, with an induration of at least 4 mm, were considered responders
| Before vaccination | After vaccination | |
|---|---|---|
| Responders | 8 | 13 |
| Nonresponders | 10 | 5 |
Clinical response
Eight of the 11 melanoma patients who received at least two vaccine doses (72.7%) experienced varying intervals of disease stability but no objective remission, while three patients showed evidence of disease progression shortly after the second dose; two patients progressed early and did not receive the second dose of the vaccine (Table 2). Similar to the responses observed in the melanoma patients, 17 of the 19 RCC patients who received two doses of the vaccine (89.5%) experienced disease stability after vaccination (Table 2) that ranged from 3 to more than 20 months. The median overall survival time of the melanoma patients who received at least two doses of the vaccine is 13 months and has not been reached for the RCC patients (with a median follow-up of 20.4 months). The median time to progression (TTP) was not different between diagnoses, and for patients who received two doses of the vaccine, it ranged between 5.6 months (melanoma) and 6.7 months (RCC) (Table 4). When all 35 patients are considered, still no statistical difference is noted, but the median TTP is 4 months for melanoma and 5.7 months for RCC patients.
Table 4.
Time to progression (TTP) of metastatic melanoma and renal cell carcinoma (RCC) patients vaccinated with dendritic cell–tumor cell hybrids every 6 weeks until disease progression or lack of tumor cells for vaccine preparation. OR objective response, SD Stable disease, DP disease progression
| Response | Melanoma | RCC | ||
|---|---|---|---|---|
| n | TTPa | n | TTP | |
| OR | 0 | – | 3 | 6.7 (5–21) |
| SD | 8 | 11.1 (4–17+) | 14 | 8.4 (3.7–19+) |
| DP | 3 | 2.6 (2.3–2.7) | 2 | 2.7 (2.6–2.8) |
| Overall | 11 | 5.6 | 19 | 6.7 |
aMedian time to progression in months (range)
Among the RCC patients who showed disease stability, radiological evidence of actual disease remission was detected in two, who showed a decrease in the number of lung lesions on CT scans. Besides that, one patient who presented a radiological enlargement of a lesion in the renal area and was submitted to reoperation presented a complete pathologic response. The retroperitoneal lesion, as well another one, present in the liver, were removed, shown to be devoid of tumor cells and characterized by a very intense macrophage infiltrate, sometimes organized into granulomas, with the presence of typical giant cells (Fig. 2). This response lasted for 4 months, when he experienced disease progression and dissemination of the tumor in the peritoneal cavity.
Fig. 2a, b.
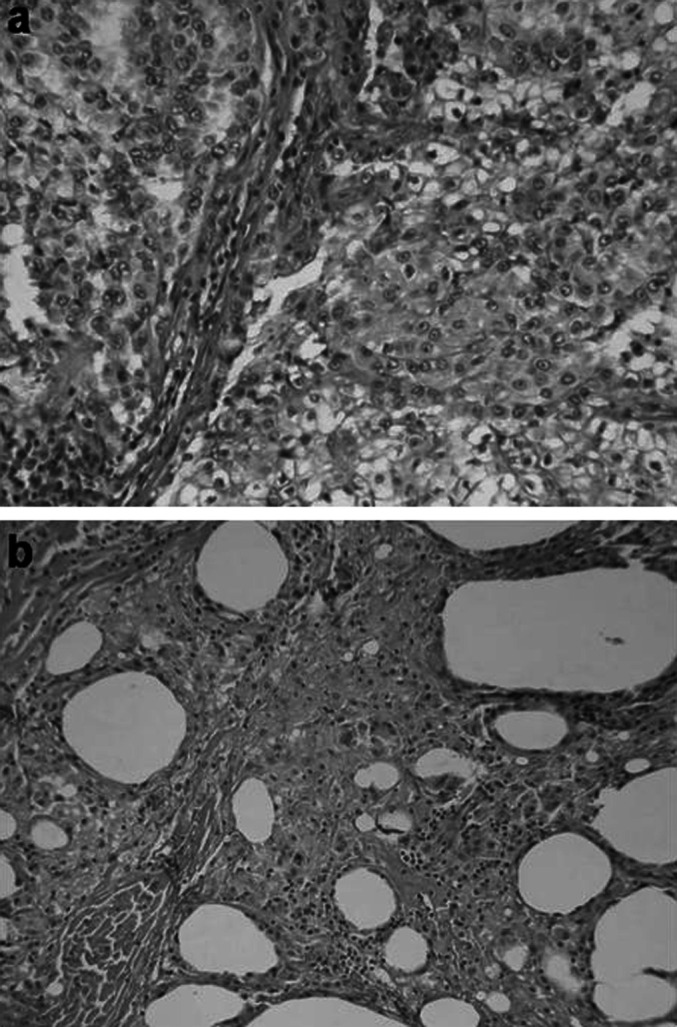
Pathological response to hybrid cell vaccination. Photomicrographies (x400) of renal cell carcinoma (a) removed to prepare tumor cell–dendritic cell hybrid vaccination, and of retroperitoneal lesion removed after two doses of the vaccine (b), showing a very intense macrophage infiltrate, granulomas, typical giant cells, and no tumor cells
Correlation between clinical and immunological response
When TTP was compared among patients, DTH test positivity at protocol enrollment clearly distinguished the patients (Fig. 3a; p=0.0049). No significant difference in TTP, however, was noted between patients who turned positive after vaccination and those who did not (Fig. 3b; p=0.4430). Similarly, no significant association was noted between lymphocyte proliferative responses and clinical response (data not shown).
Fig. 3a, b.

Relevance of delayed-type hypersensitivity (DTH) positivity in the time to progression (TTP) of patients. Patients were submitted to a panel of three DTH tests (PPD, candidin, trychophytin) before and after two doses of hybrid cell vaccination. Patients who were positive for at least one test before vaccination had a significantly longer TTP (p=0.0049) (a), while DTH after vaccination did not discriminate patients as to their TTP (p=0.4430) (b)
Discussion
The present study shows that dendritic cell–tumor cell hybrid vaccination is safe and seems to affect the natural history of advanced melanoma and renal cell carcinoma patients. Vaccinated patients presented an overall recovery of immune function, as evaluated by an increased lymphocyte response to various stimuli and by a tendency to recover DTH responses to recall antigens.
Actually, a compromised immune function is a common feature of advanced cancer [13], and the patients included in the protocol fulfilled this expectation, since most were negative to all DTH challenges used. Very significantly, DTH responses at patient enrollment distinguished patients as to their TTP, confirming the association of poor immune function and disease progression. The fact that both lymphocyte activation and DTH responses showed an increase throughout the vaccination period indicates that the administration of DC-tumor hybrid cells has impacted their immune system. In this regard, the use of allogeneic DCs in the protocol may have played a positive role. The first reason for this would be the allogeneic effect [7], where the T-lymphocyte stimulation caused by the presentation of tumor antigens in the context of the patients’ HLA in the hybrid cell, would be enhanced by the simultaneous presence of the allogeneic donor HLA in the surface of the same cells. However, in cancer patients the use of healthy DC donors might have another role yet. These could provide more effective DCs than it would be possible to obtain from cancer patients, since DCs from cancer patients show functional deficiencies [5, 9, 23] that might negatively affect their function, even when they are generated in vitro [21], as for this protocol.
In spite of the overall impression of immune function improvement with the vaccination, no clear-cut association between immune function and clinical evolution was noted individually. Part of this could be attributed to the heterogeneity of patients to the limited range of clinical responses (mainly disease stability) and to the immune parameters evaluated. The DTH response analysis did not include a tumor-specific test (to save tumor cells for vaccine preparation), and the lymphocyte proliferative responses may have not been sensitive enough to detect shifts in response patterns that might have been induced by vaccination. Actually, the data on lymphocyte proliferation could indicate a general recovery of immune function rather than a tumor-specific vaccine-induced response in the patients. Nevertheless, when for seven patients, the production of interferon γ and interleukin 4 were evaluated in patients’ lymphocyte cultures stimulated with autologous tumor cells, a marginally significant (p=0.15) enhancement of the IFN-γ/IL-4 relation was noted (data not shown), suggesting that such a shift in cytokine production patterns may indeed be induced by vaccination. Anyway, this observation of a lack of association would be in agreement with most reports, since no single immune function parameter has been associated with clinical response to immunotherapeutic approaches in cancer patients. Yet, this is not surprising considering the plethora of possible responses and the limitations of one’s analysis. Indeed, if an association is to be found, it will probably be with complex patterns of immune response, including antigen-presenting cell function, cytokine secretion, lymphocyte recirculation, and subpopulation activation patterns. All these parameters are affected by the presence of tumors and therefore should be considered. As a result, larger patient populations will need to be analyzed before any immune function analysis can be definitively associated with clinical responses to immunotherapy.
Nevertheless, 71% of the vaccinated patients experienced at least disease stabilization, a response routinely considered as clinical benefit in hormone therapy for breast cancer [24]. The fact that major clinical responses were not obtained is not surprising. All patients included in the protocol had very large tumor burdens and, at enrollment, many had very aggressive diseases with evident quality-of-life limitations. In spite of that, some of these patients experienced prolonged periods of disease stability and returned to normal life, without the hurdles of chemotherapy, which, for these patients, would be of very low benefit anyway. When considering the rate of objective responses obtained in the present study for RCC patients (3/22, 14%), it is relevant to note that in a controlled randomized trial for the treatment of RCC [12], a spontaneous remission rate of 6.6% was observed in the placebo group. However, in that same trial, the median TTP was of 1.9 months, compared with the 5.7 months observed in this study.
The results presented here are similar to those obtained in other DC vaccination protocols [4, 16, 17, 19, 22, 27] and support the extension of DC vaccination for patients with less advanced diseases, who should more likely benefit the most from this approach. At the same time, gains in the understanding of DC biology and in vitro generation, patients’ conditioning, and immune response boosting after vaccination should enhance clinical responses even in patients with very advanced diseases. Furthermore, these improvements could provide support for the extension of this immunotherapeutic approach for other types of malignancy.
In conclusion, the data presented here support the use and further study of DC vaccination for the management of advanced cancer patients and, most probably, indicate its use for less advanced patients, who could benefit even more from this approach.
Acknowledgements
This work was partially supported by a research grant from FAPESP (#01/02339-8). A.R.N. received a scholarship from FAPESP (#01/07751-4), and P.C.B.S. received a postdoctoral fellowship from FAPESP (#01/00188-2).
References
- 1.Avigan Clin Breast Cancer Suppl. 2003;4:S158. doi: 10.3816/cbc.2003.s.006. [DOI] [PubMed] [Google Scholar]
- 2.Banchereau Nature. 1998;392:245. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 3.Banchereau Annu Rev Immunol. 2000;18:767. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 4.Banchereau Cancer Res. 2001;61:6451. [PubMed] [Google Scholar]
- 5.Bella Br J Cancer. 2003;89:1463. doi: 10.1038/sj.bjc.6601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang Clin Cancer Res. 2002;8:1021. [Google Scholar]
- 7.Elfenbein J Immunol. 1974;112:2166. [PubMed] [Google Scholar]
- 8.Fallarino Int J Cancer. 1999;80:324. doi: 10.1002/(sici)1097-0215(19990118)80:2<324::aid-ijc25>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 9.Ferrari Cancer Immunol Immunother. 2003;52:359. doi: 10.1007/s00262-002-0365-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fong J Immunol. 2001;166:4254. doi: 10.4049/jimmunol.166.6.4254. [DOI] [PubMed] [Google Scholar]
- 11.Fukao Trends Immunol. 2002;23:231. doi: 10.1016/S1471-4906(02)02198-1. [DOI] [PubMed] [Google Scholar]
- 12.Gleave New Engl J Med. 1998;338:1265. doi: 10.1056/NEJM199804303381804. [DOI] [PubMed] [Google Scholar]
- 13.Hersh Med Clin North Am. 1976;60:623. doi: 10.1016/s0025-7125(16)31902-2. [DOI] [PubMed] [Google Scholar]
- 14.Iwashita Cancer Immunol Immunother. 2003;52:155. doi: 10.1007/s00262-002-0360-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klein J Exp Med. 2000;191:1699. doi: 10.1084/jem.191.10.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marten Hum Gene Ther. 2003;14:483. doi: 10.1089/104303403321467243. [DOI] [PubMed] [Google Scholar]
- 17.Marten Cancer Immunol Immunother. 2002;51:637. doi: 10.1007/s00262-002-0324-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nair Int J Cancer. 1997;70:706. doi: 10.1002/(sici)1097-0215(19970317)70:6<706::aid-ijc13>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 19.Nestle Nat Med. 1998;4:328. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 20.Okada Int J Cancer. 1998;78:196. doi: 10.1002/(sici)1097-0215(19981005)78:2<196::aid-ijc13>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 21.Onishi Clin Immunol. 2002;105:286. doi: 10.1006/clim.2002.5293. [DOI] [PubMed] [Google Scholar]
- 22.O’Rourke Cancer Immunol Immunother. 2003;52:387. doi: 10.1007/s00262-003-0375-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orsini Cancer Res. 2003;63:4497. [PubMed] [Google Scholar]
- 24.Paridaens Ann Oncol. 2003;14:1391. doi: 10.1093/annonc/mdg362. [DOI] [PubMed] [Google Scholar]
- 25.Parmiani J Natl Cancer Inst. 2002;94:805. doi: 10.1093/jnci/94.11.805. [DOI] [PubMed] [Google Scholar]
- 26.Schuler-Thurner J Exp Med. 2002;195:1279. doi: 10.1084/jem.20012100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smithers Cancer Immunol Immunother. 2003;52:41. doi: 10.1007/s00262-002-0318-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Su Cancer Res. 2003;63:2127. [PubMed] [Google Scholar]
- 29.Svane APMIS. 2003;111:818. doi: 10.1034/j.1600-0463.2003.11107813.x. [DOI] [PubMed] [Google Scholar]
- 30.Trefzer Int J Cancer. 2000;85:618. doi: 10.1002/(sici)1097-0215(20000301)85:5<618::aid-ijc4>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 31.Wang J Immunol. 1998;161:5516. [PubMed] [Google Scholar]
- 32.Weichert H, Blechschmidt I, Schroder S, Ambrosius H. The MTT-assay as a rapid test for cell proliferation and cell killing: application to human peripheral blood lymphocytes (PBL) Allerg Immunol (Leipz) 1991;37(3–4):139–144. [PubMed] [Google Scholar]