

Abstract
Purpose
In the field of cancer immunotherapy research, the targeting of effector cells with specific antibodies is a very promising approach. Recent advances in genetic engineering have made it possible to prepare immunoglobulin fragments consisting of variable domains using bacterial expression systems.
Methods
We have produced an anti-epidermal growth-factor receptor (EGFR) × anti-CD3 bispecific diabody (Ex3 diabody) in an Escherichia coli (E. coli) expression system with refolding method. The Ex3 diabody targets lymphokine-activated killer cells with a T-cell phenotype (T-LAK cells) to EGFR positive bile duct carcinoma cells with dramatic enhancement of cytotoxicity in vitro. This specific killing of EGFR-positive cells was completely inhibited by parental mAb IgGs directed to EGFR and the CD3 antigen.
Results
When T-LAK cells were cultured with EGFR-positive tumor cells in the presence of Ex3 diabody, they produced much higher levels of IFN-γ, GM-CSF, and TNF-α than in its absence, this being a possible mechanism underlying specific antitumor activity. The Ex3 diabody showed good stability when tested at 37°C for 48 h, and also markedly inhibited tumor growth of bile duct carcinoma xenografts in severe combined immunodeficient (SCID) mice. When Ex3 diabody (20 μg/mouse) was administrated intravenously, together with T-LAK cells and interleukin-2 (IL-2), complete cure of tumors were observed in three of six mice, and the other three showed marked retardation of tumor growth.
Conclusion
The Ex3 diabody can be considered a highly promising reagent for study of specific targeting immunotherapy against bile duct and other EGFR-positive carcinomas.
Keywords: Bispecific diabody, Refolding system, Bile duct carcinoma, EGF receptor, Targeting immunotherapy
Introduction
Targeting of effector cells with specific antibodies is a very promising approach in cancer immunotherapy. Bispecific antibodies (BsAbs) that bind to a tumor-associated antigen (TAA) and lymphocytes have significant potential utility for adoptive immunotherapy as a tool to arm cytotoxic lymphocytes. Several examples have been reported to have marked antitumor effects in vitro and in vivo; however, preparation of BsAbs by chemical conjugation or hybrid hybridoma (quadroma) methods [6] is very time-consuming and laborious. Furthermore, their down-sizing and/or humanization are necessary [69] in order to avoid human anti-mouse antibody (HAMA) reactions [44].
Recent advances in genetic engineering have made it possible to prepare immunoglobulin fragments using bacterial expression systems [7]. For generation of recombinant BsAbs, diabodies [25, 60], dimeric Ab fragments consisting of two non-covalently associated single-chain Abs (scFvs), are attractive reagents. In order to form two binding sites, the variable domains of two Abs (A and B) are arranged with VHA-VLB (the VH domain of Ab A and the VL domain of Ab B) on one chain and VHB-VLA (the VH domain of Ab B and the VL domain of Ab A) on the other [1, 25, 60]. Diabodies are the smallest BsAbs available, and the distance between the two antigen-binding sites is less than half of IgG [49]. This compactness contributes to low immunogenicity and high tumor penetration [72]; therefore, diabodies are promising reagents for targeted cancer immunotherapy. Recently, we constructed an Mx3 (anti-MUC1 × anti-CD3) diabody, which enhances antitumor activity of T-LAK cells [60]. In addition, a SEA D227A fusion Mx3 diabody (the anti-MUC1 × anti-CD3 diabody genetically fused with mutated superantigen staphylococcal enterotoxin A (SEA D227A) was constructed. The fusion diabody, as expected, showed great enhancement of the antitumor activity of T-LAK cells [61].
Previous reports have demonstrated that epidermal growth factor receptor (EGFR) is widely expressed on various solid malignant tumors including bile duct cancer [48, 58]. Signaling through EGFR is considered to regulate tumor cell functions, such as cell-cycle progression, inhibition of apoptosis, angiogenesis, tumor cell motility, adhesion, and invasion [13, 18, 19, 32, 36, 50, 64, 67, 70]. Furthermore, overexpression of EGFR on cancer cells is now known to be closely associated with malignancy, metastatic phenotype and a poor prognosis [12, 17, 20, 37, 47, 68]. As a targeting molecule for cancer immunotherapy, EGFR may be a key, since it is connected directly to signaling pathways [15, 21, 29, 43, 45, 46, 55, 65, 71].
From the above considerations, we developed an anti-EGFR × anti-CD3 bispecific diabody (Ex3 diabody). In this study, we demonstrated that the refolded Ex3 diabody exerts strong bispecific binding to both EGFR-positive TFK-1 cells and CD3-positive T-LAK cells. In a cytotoxicity assay, Ex3 diabody greatly enhanced antitumor effects of both T-LAK cells and freshly isolated peripheral blood mononuclear cells (PBMCs), when EGFR-positive bile duct carcinomas were tested, the efficacy being approximately 100-fold greater than that with our previous SEA D227A-Mx3 diabody [61]. The Ex3 diabody also enhanced T-LAK cytotoxicity against other EGFR positive, but not negative, cell lines, indicating specificity. Furthermore, immunotherapy with Ex3 diabody combined with T-LAK cells in bile duct carcinoma-xenografted SCID mice resulted in pronounced tumor growth inhibition. For adoptive immunotherapy using T-LAK cells, this stable Ex3 diabody offers a powerful reagent with a clinical potential, when this diabody is completely humanized.
Materials and methods
mAbs and hybridoma cell lines
For construction of the diabody, 528 and OKT3 hybridoma cell lines were used as the source of V-region genes. The 528 mAb [31] was a mouse IgG2a directed to the human EGFR antigen, OKT3 mAb was a mouse IgG2a directed to the human CD3ε chain [57, 66], and MUSE11 mAb a mouse IgG1 directed at human MUC1 antigen on tumor cells (produced by Dr. Hinoda, Sapporo Medical University, Sapporo, Japan) [24]. MUSE11 and OKT8 mAbs were used for inhibition assays.
Cloning of the VH and VL genes of the anti-EGFR (528) mAb
The VH and VL genes of 528 hybridoma cells were cloned by reverse transcription (RT)-PCR. Briefly, total mRNA was isolated from 528 hybridoma cells using the Isogene system (Nippon Gene, Tokyo, Japan). Then, mRNA was reverse transcribed with a first-strand cDNA synthesis kit (Pharmacia) and the VL and VH genes were amplified by PCR using the primer mix reported previously [38]. The detailed methods will be published elsewhere. Briefly, primers were redesigned to contain a restriction enzyme sequence to obtain 528 scFv fragments in the orientation VH-(Gly4Ser)3-VL, compatible with the pRA OH-OL (OKT-3 scFv, VH-(Gly4Ser)3-VL) expression vector previously constructed in our laboratory. Seventeen 5’ primers specifically designed to hybridize to the 5’ ends of mouse antibody light chain leader sequences and four 3’ primers hybridizing with the 5’ end of mouse κ light chain constant region were used to clone the VL gene. Nineteen 5’ primers specifically designed to hybridize to the 5’ ends of mouse antibody heavy chain leader sequences and four 3’ primers hybridizing with the 5’ end of the mouse IgG heavy chain constant region were used to clone the VH gene [38]. Finally, 4 for the VL gene and 9 for the VH gene were selected. Among various VL and VH combinations, one combination showed particularly strong affinity for EGFR.
Construction of expression vectors
The procedure for the construction of two kinds of hetero scFv expression vectors was as described previously [22, 60]. The procedure for 5H-OL and OH-5L, a hetero scFv combining the heavy and light chain sequences of 528 and OKT3 hybridomas with a (Gly4Ser) linker, utilized the pRA vector, constructed by exchanging the SSI terminator of the pSNE4 vector [3] for the T7 terminator. Construction of the expression vectors, pRA MH-OL and pRA OH-ML, hetero scFv of MUSE11 and OKT3, was as described previously [60]. Then 5H and 5L, the heavy and light chain sequences of the Fv region of 528 mAb IgG, were replaced with the MH and ML using NcoI-EagI or EcoRV-SacII restriction enzymes (Fig. 1A).
Fig. 1.

A Construction of the two kinds of hetero scFv expression vectors. The VH and VL region genes of 528 are designated as 5H and 5L, and VH and VL region genes of OKT3 as OH and OL, respectively. c-myc a sequence encoding an epitope recognized by 9E10 mAb; 6×His a sequence encoding six C-terminal histidine residues, pel-B a signal peptide sequence of bacterial pectate lyase; rbs a sequence encoding the ribosome binding region. B The 12.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and C Western blotting analysis under reducing conditions of 5H-OL (lanes 1–3) and OH-5L (lanes 5–7). Molecular size markers (in kilodaltons) are shown on the left. Lanes 1 and 5: proteins in the bacterial supernatant fraction; lanes 2 and 6: proteins in the intracellular soluble fraction; lanes 3 and 7: proteins in the intracellular insoluble fraction; lane 4: refolded Ex3 diabody
Expression and refolding of Ex3 diabody
Expression and refolding of the Ex3 diabody were performed using the methods developed for preparation of the anti-MUC1 × anti-CD3 diabody [60, 63]. In brief, E. coli strain BL21 (DE3) pLysS transformed with the expression vector was grown at 28°C in 2xYT broth. In order to induce protein production, 1 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG) was added to the culture and the cells were grown overnight. From 200 ml of culture, bacterial supernatant (BS), intracellular soluble (ICS), and intracellular insoluble (ICIS) fractions were obtained as follows. The BS fraction was first separated from the culture medium by centrifugation (2000 g, 35 min). Then, the cell pellets were resuspended in 80 ml phosphate-buffered saline (PBS), ultrasonicated at 150 W for 15 min and centrifuged at 4500 g for 20 min. The ICS fraction was recovered from the supernatant. Then the separated intracellular insoluble (ICIS) fraction was solubilized with 10 ml of 6 M guanidine hydrochloride in PBS (Gu-HCl/PBS) overnight at 4°C.
Purification and refolding of diabody
After solubilization of ICIS fractions containing the majority of 5H-OL and OH-5L, purification was carried out separately. Each protein was applied to a 2 ml TALON metal affinity resin column (Clontech, Palo Alto, Calif.), followed by extensive washing with 6 M Gu-HCl/PBS containing 1 mM imidazole. Thereafter, 5H-OL and OH-5L were eluted with 6 M Gu-HCl/PBS containing 500 mM imidazole. Purified 5H-OL and OH-5L were diluted to a concentration of approximately 0.1 mg/ml. For refolding of the Ex3 diabody, 5H-OL and OH-5L were mixed stoichiometrically and dialyzed for phased-guanidine removal with Gu-HCl/PBS, with addition of L-arginine as previously reported [60, 63]. Finally, the sample was dialyzed against PBS (pH 7.9) in order to remove L-arginine, which has in itself immunostimulatory and antitumor effects [4, 40, 53, 54, 59]. Then it was centrifuged at 4500 g for 20 min at 4°C to remove insoluble material. Thereafter, gel filtration chromatography was performed using a Superdex 200 column (Pharmacia Biotech, Uppsala, Sweden) as described below, and the dimeric molecules were collected. Then, the concentrated sample was filtered through a 0.22-μm ultrafiltration membrane (Millipore, Tokyo, Japan).
SDS-PAGE and Western blotting
One-milliliter aliquots of culture supernatant were used for the analysis. The total proteins in each fraction, precipitated with 6% trichloroacetic acid (TCA) and 0.083% deoxycholate, were applied to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) [39] under reducing conditions, and stained with Coomassie brilliant blue R-250. Then, the proteins in the gels were blotted onto nitrocellulose membranes (Amersham, Little Chalfont, Buckinghamshire, UK) and treated with blocking buffer (PBS containing 0.05% Tween 20 and 4% skim milk) at room temperature for 1 h. Thereafter, they were incubated with peroxidase-conjugated anti-His tag mAb (Santa Cruz Biotechnology, Santa Cruz, Calif.), followed by signal enhancement using the ECL Detection System (Amersham).
Purification of Ex3 BsAb
The Ex3 BsAb was generated by chemical recombination of IgG fragments as described by Brennan et al. [9]. In brief, 528 and OKT3 mAb IgGs were converted into F(ab’)2 fragments by limited proteolysis with pepsin. F(ab’)2 fragments were generated by a mild reduction with 0.5 mM dithiothereitol (DTT). The OKT3 Fab’ fragments were modified with 5, 5’-dithio-bis-2-nitro-benzoic acid (DTNB). The F(ab’)2 BsAb was generated by conjugation of the Fab’-TNB (thio-bis 2-nitro-benzoic acid) derivative with the hinge-SH groups of the second Fab’ fragment. Removal of intermediate products (Fab’, Fab’-TNB) from the BsAb was performed using gel filtration as described below.
Gel-filtration chromatography
Gel-filtration was achieved using a Superdex 200 column (Pharmacia Biotech, Uppsala, Sweden) connected to a fast protein liquid chromatography (FPLC) system. The elution buffer was PBS, pH 7.9. Molecular mass was calibrated with Combithek (Boehringer, Mannheim, Germany) calibration proteins for gel chromatography. Samples adjusted to 0.2 mg/ml were loaded on a Superdex 200 column at a flow rate of 3.0 ml/min, and 3.0 ml fractions were collected.
Cell lines and transfectants
Human bile duct carcinoma (TFK-1, OCUCh-LM1, HuCC-T1), human lung carcinoma (OBA-LK-1, A549, NCI-H69), human breast cancer (MCF-7, CRL-1500, SK-BR-3), human epidermoid cancer (A431), Chinese hamster ovary (CHO), murine mammary gland cancer (MM46) cell lines were used in this study. The TFK-1 and OBA-LK1 cell lines were established in our laboratory [56] and cultured with RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin (designated below as the culture medium).
Preparation and stimulation of effector cells
For induction of T-LAK cells, PBMCs isolated by density-gradient centrifugation from a healthy volunteer were cultured for 48 h in culture medium supplemented with 100 IU/ml recombinant human IL-2, kindly supplied by Shionogi Pharmaceutical Co. (Osaka, Japan) at a cell density of 1×106/ml in a culture flask (A/S Nunc, Roskilde, Denmark) precoated with OKT3 mAb (10 μg/ml). The proliferated cells were then transferred to another flask and expanded in culture medium containing 100 IU/ml IL-2 for 2–3 weeks, as reported previously [30, 60, 61]. Surface marker analysis showed that more than 90% cells were positive for CD3 and CD8, but CD56 was almost negative [30]. In some cases, PBMCs isolated by density-gradient centrifugation were immediately used for 51Cr release assay in the presence of Ex3 diabody.
Flow-cytometric analysis
Test cells (1×106) were first incubated on ice with 10 μg of recombinant Ab for 30 min as the first Ab. After washing with PBS plus 0.1% NaN3, they were exposed to 9E10 anti-c-myc mAb (Santa Cruz Biotechnology, Calif.) as the second Ab and then FITC-conjugated anti-mouse IgG as the third Ab for 30 min on ice. The stained cells were analyzed by flow cytometry (FACS Calibur, Becton Dickinson, San Jose, Calif.).
Blocking test
TFK-1 or T-LAK cells (1×106 cells) were incubated on ice with or without excess competing Ex3 diabody (20 μg) for 30 min. After washing with PBS plus 0.1% NaN3, they were incubated with 0.25 μg 528 mAb IgG or 0.1 μg of OKT3 IgG for 30 min on ice, further washed, and incubated on ice with FITC-conjugated anti-mouse IgG. Flow cytometric analysis was then performed as described above.
Absorption test
For the absorption test, 20-μg aliquots of Ex3 diabody were incubated with 5.0×106 TFK-1 or T-LAK cells on ice for 30 min. After centrifugation, the supernatant was collected and half was added to TFK-1 or T-LAK cells (1.0×106 cells) for detection of residual antibody activity. After incubation on ice for 30 min, the cells were exposed to the second (9E10 mAb) and the third (FITC-conjugated anti-mouse IgG) antibody. Flow cytometric analysis was performed as described above.
In vitro stability test
Ten-microgram aliquots of Ex3 diabody in 500 μl of culture medium were incubated at 37°C for 1–48 h. Then, reactivity with T-LAK and TFK-1 cells (1×106 cells) was analyzed by flow cytometry and compared with that of freshly prepared Ex3 diabody (10 μg).
Attachment of T-LAK cells mediated by Ex3 diabody
T-LAK cells and TFK-1 cells were co-cultured at an E:T ratio of 5:1 in a A/2 96-well microplate (Costar, Cambridge, Mass.) in the presence or absence of Ex3 diabody (1.0 μg/ml) at 37°C. Cell attachment was examined microscopically during co-cultivation for 1–12 h. A blocking test was also performed using equal amounts of parental (528 and OKT3) or irrelevant (MUSE11 or OKT8) IgGs.
In vitro cytotoxicity assay
Cytotoxic T-LAK cell effector function was measured using a standard 51Cr release assay. In brief, 1×104 51Cr-labeled target cells were added to various numbers of effector cells at effector-target cell ratios ranging from 100:1 to 1:1 in U-shaped 96-well microtiter plates (Microwell 96U, Nalgen Nunc International). For evaluation of recombinant Ab, various dilutions of Ex3 diabody (0.1 ng/ml to 10 μg/ml in a final volume of 200 μl) were added. After 6-h incubation at 37°C, 100-μl aliquots of supernatant were removed and evaluated for 51Cr-release in a γ-counter (ARC2000, Aloka, Japan). The percentage cytotoxicity to cancer cells was calculated as follows: [(experimental 51Cr release−spontaneous 51Cr release)/(maximum 51Cr release−spontaneous 51Cr release)]×100 [16].
Inhibition of specific lysis in 51 Cr release assay
A blocking test using parental mAb IgGs (528 or OKT3) and irrelevant IgGs (MUSE11; anti-MUC1 mAb or OKT8; anti-CD8 mAb) was also performed as follows: 1×104 51Cr-labeled target cells were added to effector cells at effector-target cell ratios of 20:1 in U-shaped 96-well microtiter plates. Then, 1 μg/ml of Ex3 diabody and various concentrations of parental or irrelevant IgGs (0.1–10 μg/ml) were added in a final volume of 200 μl. After culture for 6 h at 37°C, percentage lysis of target cells was calculated.
Enzyme-linked immunosorbent assay (ELISA)
TFK-1 cells (1×104 cells/well) were cultured for 24 h in 96-well plates at 37°C. Then, the wells were washed with culture medium, and T-LAK cells (5×104 cells/well) were added and co-cultured at 37°C with or without Ex3 diabody at the final concentration of 10–1000 ng/ml. After 6–48 h of co-culture, the supernatants were harvested and applied for ELISA with human IFN-γ, GM-CSF, TNF-α and IL-2 ELISA kit (Endogen, Woburn, Mass.), following the manufacturer’s instructions. A blocking test using an equal amount of parental 528 or OKT3 mAb IgG was also performed.
In vivo tumor model
Female 5-week-old SCID mice (Fox CHASE C.B.-17/Icr-Scid Jcr) purchased from Japan Clea (Tokyo, Japan), were inoculated with 5x106 TFK-1 cells subcutaneously into the dorsal thoracic wall on day 0. Treatment was initiated on day 10 thereafter. When the mice, whose tumors grew to approximately 5 mm in diameter, were randomly divided into nine groups. Animals whose tumors did not reach 5 mm in diameter were excluded from the experiments. For four consecutive days, the mice received i.v. 2×107 T-LAK cells sensitized with the Ex3 diabody or BsAb via the tail vein, together with IL-2 (500 IC/mouse) in 0.15 ml PBS. Before administration, T-LAK cells were preincubated with 2 or 20 μg of Ex3 diabody, 2 μg of Ex3 BsAb (made of chemical conjugation), or 2 μg of 528 mAb IgG at 4°C for 30 min, and 500 IU of IL-2 were added. The cell suspension was then injected without washing. Tumor size was measured with a caliper weekly for 9 weeks, and approximate tumor volume (V, in cubic millimeters) was calculated from linear measurements of the width (A, in millimeters) and length (B, in millimeters) as follows: V=(A2×B)/2.
Results
Expression of hetero scFvs in E. coli
The two hetero scFvs, OH-5L, and 5H-OL were separately produced using E. coli strain BL21 (DE3) pLysS harboring the plasmid pRA-OH-5L or pRA-5H-OL (Fig. 1A). Results of SDS-PAGE (Fig. 1B) and Western blotting (Fig. 1C) using anti-His tag Ab showed each gene product to exist mainly in the intracellular fractions. To obtain Ex3 diabodies from the ICIS fractions, we employed refolding of insoluble diabody. For this, 5H-OL and OH-5L were mixed stoichiometrically and dialyzed for phased-guanidine removal with Gu-HCl/PBS with addition of L-arginine as previously reported [60, 63]. Although removal of L-arginine by dialysis against PBS led to significant aggregation of proteins (~50% of total proteins), final yields of Ex3 diabodies amounted to approximately 1 mg from 1-l culture of either scFv.
Flow cytometric analysis
Binding of the refolded Ex3 diabody to each antigen was confirmed by flow cytometry. Strong reactivity was observed with TFK-1 cells (EGFR positive) and T-LAK cells (CD3 positive; Fig. 2A,B), and its specificity was almost identical with the parental mAb IgGs (528 or OKT3). The affinity of Ex3 diabody for CD3 was lower than that of the parental mAb. Previous reports showed the affinity of anti-CD3 scFv and other diabody (anti-TAA × anti-CD3) to CD3 to usually be lower than that of parental mAb, but this did not prove to be a disadvantage for arming lymphocytes to tumor cells [11, 33]. The Ex3 diabody did not react with CHO cells (EGFR, CD3 negative; data not shown).
Fig. 2A–J.
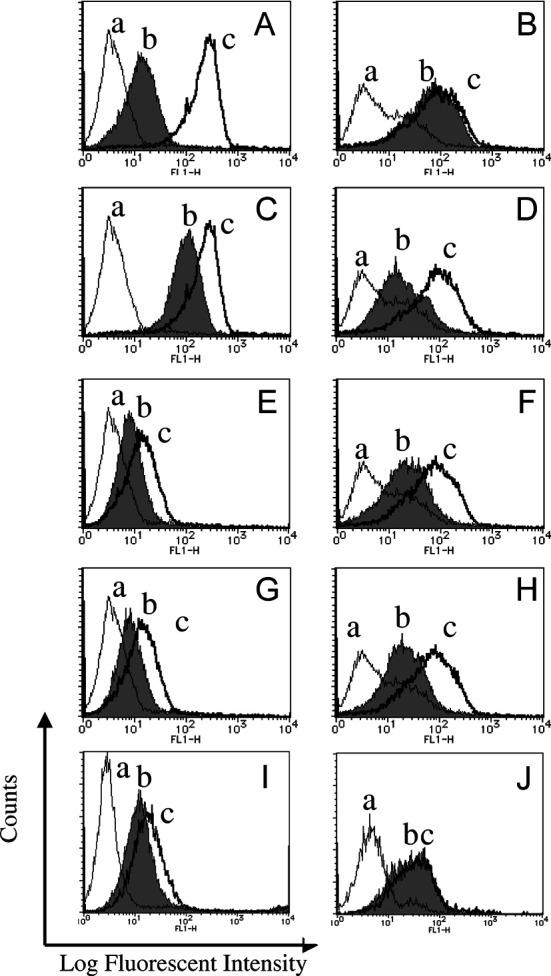
Flow cytometry analysis of Ex3 diabody tested with T-LAK cells (left) and TFK-1 cells (right). A,B Reactivity of the Ex3 diabody (a negative control profile; b profiles of cells reacted with Ex3 diabody; and c profiles of cells reacted with either OKT3 parental IgG for T-LAK or 528 IgG for TFK-1 cells). C,D Blocking test by competing Ex3 diabody [a negative control profile; b profiles of cells pretreated with Ex3 diabody first and reacted with parental mAb IgGs (OKT-3 for T-LAK and 528 for TFK-1, respectively), then FITC conjugated anti-mouse IgG was used; and c profiles of cells reacted with parental IgG (OKT-3 to T-LAK, 528 to TFK-1 cells, respectively)]. E–H Cross absorption of Ex3 diabody with T-LAK cells or TFK-1 cells (a negative control profile; b profile of cells reacted with the supernatant after absorption with T-LAK cells or TFK-1 cells; and c profile of cells reacted with same amount of Ex3 diabody without absorption). E,G Ex3 diabody absorbed with T-LAK and TFK-1 cells, respectively. Residual antibody activity was tested with T-LAK cells. F,H Ex3 diabody absorbed with T-LAK and TFK-1 cells, respectively. Residual antibody activity was tested with TFK-1 cells. I,J In vitro stability test of Ex3 diabody (a negative control profile; b profile of cells reacted with the Ex3 diabody incubated at 37°C for 48 h; c profile of cells reacted with freshly prepared Ex3 diabody). Ex3 diabody tested with I T-LAK cells and J TFK-1 cells
To confirm the specificity of Ex3 diabody for EGFR and CD3, blocking tests were performed. Flow cytometric analysis showed that competing Ex3 diabody could significantly inhibit the binding of parental mAbs (528 and OKT3) to TFK-1 or T-LAK cells (Fig. 2C,D).
An absorption test was performed to examine the bispecificity of the Ex3 diabody (Fig. 2E–H). Ex3 diabody was incubated with sufficient TFK-1 or T-LAK cells. After collecting each supernatant, the absorbed diabody was added to other TFK-1 or T-LAK cells separately. If Ex3 diabody did consist of mixture of two monovalent scFv molecules, the remaining diabody after absorption with TFK-1 cells should react with T-LAK cells, and that absorbed with T-LAK should react with TFK-1 cells at the same level as previously. In our experiment, the reactivity of diabody absorbed with TFK-1 cells was apparently reduced when it was tested with not only TFK-1 but also T-LAK cells. Similarly, that of diabody absorbed with T-LAK cells was greatly reduced when tested with TFK-1 and T-LAK cells, clearly demonstrating our Ex3 diabody to be bispecific for EGFR and CD3, not mixtures of two scFvs.
In vitro stability of Ex3 diabody was analyzed by flow cytometry after incubation in RPMI-1640 culture medium containing 10% FBS at 37°C for 48 h. Similar reactions were observed with T-LAK cells and TFK-1 cells as with freshly prepared Ex3 diabody (Fig. 2I,J). Freezing at −80°C for more than 3 months did not change reactivity of the diabody.
Attachment of T-LAK cells mediated by Ex3 diabody
In order to investigate the ability of Ex3 diabody to direct T-LAK cells against TFK-1 in vitro, T-LAK cells and TFK-1 cells were co-cultured at an E:T ratio of 5:1 in a A/2 96-well microplate in the presence or absence of Ex3 diabody, then cells were examined microscopically. When Ex3 diabody was added to co-cultures of TFK-1 and T-LAK cells, T-LAK cells started to attach to TFK-1 cells after 2 h (rosette formation; Fig. 3A) and TFK-1 cells were surrounded entirely after 12 h of cultivation (Fig. 3B). On the other hand, this was not observed in the absence of Ex3 diabody (Fig. 3C,D) and was blocked by equal amounts of parental 528 or OKT3 mAb IgGs (Fig. 3E,F), but not by irrelevant mAbs (Fig. 3G,H). From these results, we conclude that the Ex3 diabody can direct T-LAK cells against TFK-1 cells quite specifically.
Fig. 3A–H.
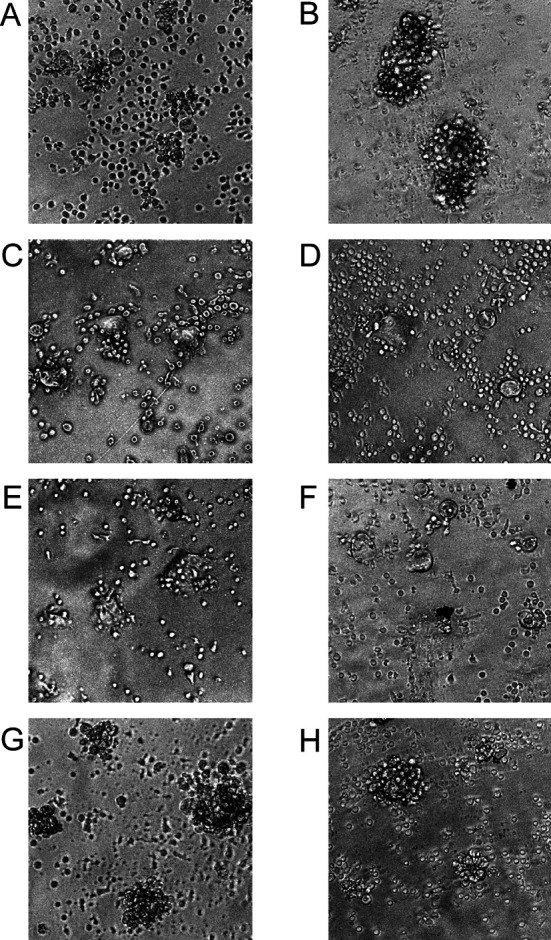
Phase-contrast inverted micrographs of attachment of T-LAK to TFK-1 cells mediated by Ex3 diabody. TFK-1 cells were co-cultured with T-LAK cells at 37°C at an E:T ratio of 5 for 2 or 12 h in the presence (A 2 h, B 12 h) or absence (C 2 h, D 12 h) of Ex3 (1.0 μg/ml) diabody. A blocking test was performed using 1.0 μg/ml of parental (E 528, F OKT3) or irrelevant IgGs (G MUSE11, H OKT8). E–H, twelve hours after co-culture of TFK-1 and T-LAK cells
Cytotoxicity assay in vitro
The ability of the Ex3 diabody to induce tumor cell lysis by redirecting T-LAK cells or freshly isolated PBMCs was evaluated by 6-h 51Cr release assay. Lysis of EGFR-positive TFK-1 cells in the presence of T-LAK cells was specifically triggered by Ex3 diabody in a dose-dependent manner at an E:T ratio of 10:1 (Fig. 4A). This resulted in 90% of specific killing at a concentration of 1 μg/ml Ex3 diabody with T-LAK cells (closed squares) at an E:T ratio of 100:1, but was lower when they were co-cultured with PBMCs (closed triangles) instead of T-LAK cells. In the absence of Ex3 diabody, T-LAK cells alone (open squares) or PBMCs alone (open triangles) could not kill TFK-1 cells effectively (Fig. 4B). The mixture of equal amounts of 528 scFv and OKT3 scFv did not show any such enhancing effects of cytotoxicity, of which value was identical to T-LAK cell control. Tumor cell lysis was also observed with other EGFR-positive bile duct carcinoma cells, namely OCUCh-LM1 (triangles) and HuCC-T1 (circles) cells (Fig. 4C), but not EGFR-negative cells (Table 1). These results indicated that the strong killing effect is specifically dependent on expression of EGFR on the target cells.
Fig. 4A–C.

Enhanced cytotoxicity of T-LAK cells or peripheral blood mononuclear cells (PBMCs) by Ex3 diabody in 6-h 51Cr release assay. A The TFK-1 cells were co-cultured with T-LAK cells at an E:T ratio of 10 in combination with various concentrations of Ex3 diabody. * p<0.01, ** p<0.001, and *** p<0.0001 for each group vs the control (without Ex3 diabody). B The TFK-1 cells were co-cultured with T-LAK cells (squares) or PBMCs (triangles) at various E:T ratios in the presence (closed symbols) or absence (open symbols) of Ex3 diabody (1.0 μg/ml). ** p<0.01, *** p<0.001, and **** p<0.0001 for the percentage of specific lysis by T-LAK cells or PBMCs in the presence of 1 μg/ml of Ex3 diabody vs in the absence of Ex3 diabody for each group. C Cytotoxicity to three bile duct carcinoma cell lines. TFK-1 (squares), OCUCh-LM1 (triangles), HuCC-T1 (circles) cells were co-cultured with T-LAK cells at various E:T ratios in the presence (closed symbols) or absence (open symbols) of Ex3 diabody (1.0 μg/ml). **, ##, †† indicate p<0.01, ***, ###, ††† indicate p<0.001, and ****, ####, †††† indicate p<0.0001 for the percentage of specific lysis of TFK-1, OCUCh-LM1, and HuCC-T1, respectively, in the presence of 1 μg/ml of Ex3 diabody vs in the absence of Ex3 diabody for each group. Data are representative of at least three independent experiments, using different donors, with similar results. Statistical analysis was carried out using the Student’s t test
Table 1.
Epidermal growth-factor receptor (EGFR)-dependent enhancement of T-LAK cytotoxicity (6-h 51Cr release assay) by Ex3 diabody
| Target cells | T-LAK alonea
(E:T ratio=25:1) |
T-LAK with Ex3 diabodyb
(E:T ratio=25:1, 1 μg/ml) |
Enhancement | |
|---|---|---|---|---|
| Cytotoxicityc | Origin | |||
| EGFR positive | ||||
| TFK-1 | 29.00±0.86 | 76.39±0.86 | 2.64±0.04**** | Bile duct |
| OCUCh-LM1 | 21.93±0.57 | 64.96±0.33 | 2.96±0.04**** | Bile duct |
| HuCC-T1 | 15.15±0.79 | 63.78±0.30 | 4.23±0.11**** | Bile duct |
| OBA-LK | 44.09±0.75 | 87.11±2.26 | 1.98±0.03**** | Lung (large cell carcinoma) |
| A549 | 14.15±2.61 | 66.84±4.68 | 5.14±0.60*** | Lung (adenocarcinoma) |
| CRL1500 | 8.91±1.07 | 45.23±2.90 | 5.22±0.35*** | Breast |
| SK-BR-3 | 39.92±1.18 | 84.18±8.06 | 2.11±0.11** | Breast |
| A431 | 15.99±1.99 | 81.23±6.28 | 5.22±0.36*** | Epidermoid cancer |
| EGFR negative | ||||
| MCF-7 | 16.83±0.67 | 10.59±0.35 | 0.62±0.02** | Breast |
| NCI-H69 | 54.95±2.28 | 46.27±4.53 | 0.84±0.04 | Lung (small cell carcinoma) |
| Other species | ||||
| CHO-K1 | 42.92±5.22 | 49.13±2.41 | 1.18±0.08 | Chinese hamster ovarian cell |
aPercentage of specific lysis of target cells in the absence of 1.0 μg/ml Ex3 diabody
bPercentage of specific lysis of target cells in the presence of 1.0 μg/ml Ex3 diabody
cThe ratio of cytotoxicity of T-LAK cells in the presence of Ex3 diabody vs T-LAK alone
Data are mean±SE from three independent experiments
**p<0.01; ***p<0.001; ****p<0.0001. Percentage of specific lysis of T-LAK in the presence of Ex3 diabody (1.0 μg/ml) vs T-LAK alone. E:T ratio=25:1
Inhibition of specific lysis by parental mAb IgG
To confirm the specificity of Ex3 diabody, we examined whether this lysis was blocked by parental mAb IgGs, which reacted with target cells better than the diabody, when examined by flow cytometry. TFK-1 cells were co-cultured with T-LAK cells at an E:T ratio of 20 in the presence or absence of Ex3 diabody at the final concentration of 1.0 μg/ml. Predictably, the cytotoxicity was markedly diminished by adding equal amounts of parental mAb IgG [528 (closed triangles) or OKT3 (closed squares)], to almost the same low level as with T-LAK controls without Ex3 diabody (crosses). Such inhibition was not observed when irrelevant MUSE11 (open triangles) or OKT8 (open squares) mAbs (Fig. 5) were added. Each mAb IgG alone did not have any effect on cytotoxicity or growth inhibition (data not shown). These results clearly indicated that the lysis was induced in an EGFR- and CD3-specific manner. In the absence of effector cells, Ex3 diabody, 528 mAb IgG, or OKT3 mAb IgG in itself had no effects on tumor cell death or growth (data not shown).
Fig. 5.

Inhibition of Ex3 diabody activity (6-h 51Cr release assay) by parental or irrelevant mAb IgGs. The TFK-1 cells were co-cultured with T-LAK cells at an E:T ratio of 20 in the presence of Ex3 diabody (1.0 μg/ml) in combination with parental IgG (closed triangle 528; closed square OKT3) or irrelevant IgG (open triangle, MUSE11; open square OKT8) at various concentrations (0.1–10.0 μg/ml). Cytotoxic activity of T-LAK cells against TFK-1 in the absence of Ex3 diabody is also shown (+): ## p<0.01 for cytotoxic activity of T-LAK cells with vs without Ex3 diabody. *** p<0.001 and **** p<0.0001 for percentage of specific lysis of T-LAK induced by Ex3 diabody (1.0 μg/ml) in the presence of parental IgG vs in the absence of parental IgG. The E:T ratio was 20. Data are representative of four independent experiments, using T-LAK cells from different donors, with similar results
Cytokine production
Cytokine production by T-LAK cells mediated by Ex3 diabody was analyzed. TFK-1 cells were cultured in 96-well plates at 37°C for 24 h. Then T-LAK cells were added to them at an E:T ratio of 5:1 with or without Ex3 diabody at various concentrations (10 ng/ml to 1 μg/ml). A blocking test using parental mAb IgG was also performed. After 48 h, the culture supernatants were harvested and analyzed for production of IFN-γ, GM-CSF, TNF-α, and IL-2 by ELISA. T-LAK cells alone produced IFN-γ at quite a low level. When they were incubated in the presence of Ex3 diabody, moderate levels were produced. This was unaffected by co-culture with allogeneic TFK-1 cells. On the other hand, co-culture of T-LAK cells with TFK-1 cells in the presence of Ex3 diabody resulted in a statistically significant increase of IFN-γ production in a dose-dependent manner. Additionally, production of the cytokine was inhibited when an equal amount of parental mAb IgG was added (Fig. 6A).
Fig. 6A–H.

Cytokine production by T-LAK cells was determined by enzyme-linked immunosorbent assay. The concentration of A IFN-γ, B GM-CSF, C TNF-α, and D IL-2 in supernatants obtained from several culture conditions: 1 T-LAK cells alone; 2 T-LAK cells+Ex3 diabody (1 μg/ml); 3 T-LAK cells co-cultured with TFK-1 at an E:T ratio of 5 (without Ex3 diabody); 4–6 T-LAK cells co-cultured with TFK-1 cells in the presence of Ex3 diabody (10 ng/ml, 100 ng/ml, 1.0 μg/ml, respectively); 7, 8 1.0 μg/ml of parental mAb IgG (528, OKT3, respectively) were added to the co-culture of T-LAK cells and TFK-1 in the presence of Ex3 diabody (1.0 μg/ml). The mean values for each condition are given in white characters: ** p<0.01; *** p<0.001; **** p<0.0001. Statistical analysis was carried out using the Student’s t test. Columns mean values; bars SE. Time course of the E IFN-γ, F GM-CSF, G TNF-α, and H IL-2. Change in supernatant concentration in the presence of the Ex3 diabody (closed circle 0.01 μg/ml; closed square 0.1 μg/ml; closed triangle 1.0 μg/ml) during 6–48 h co-cultivation of T-LAK and TFK-1 cells. ** p<0.01, *** p<0.001, and **** p<0.0001 for each cytokine concentration vs that with 0.01 μg/ml Ex3 diabody for each group. Statistical analysis was carried out using the Student’s t test. Columns mean cytokine values, bars SE
Similarly, remarkable dose-dependent increased production of GM-CSF (Fig. 6B), TNF-α (Fig. 6C) was observed when T-LAK cells were co-cultured with TFK-1 cells in the presence of Ex3 diabody, compared with other conditions. In the case of IL-2, its concentration in the supernatant after 48-h co-cultivation was slightly increased when co-cultured with TFK-1 cells in the presence of low Ex3 diabody (Fig. 6D). On the other hand, there was no effect on the amounts of IL-4 or IL-10 (data not shown).
Sequential analysis of cytokine production
We also analyzed the concentrations of IFN-γ, GM-CSF, TNF-α, and IL-2 in the culture supernatant of T-LAK and TFK-1 cells in the presence of Ex3 diabody (0.01–1.0 μg/ml) after co-culture for 6, 12, 24, and 48 h. The concentrations of IFN-γ and GM-CSF increased with time (Fig. 6E,F). On the other hand, the concentration of TNF-α and IL-2 rapidly increased in the first 12 h and decreased thereafter (Fig. 6G,H).
Experimental therapy in xenografted SCID mice
Xenografted SCID mice (Fig. 7) were divided into seven groups (six mice per group) treated as follows: group A, injection of buffer (PBS) alone; group B, injection of T-LAK cells alone; group C, injection of T-LAK cells with Ex3 diabody (20 μg/mouse); group D, injection of T-LAK cells with Ex3 diabody (2 μg/mouse); group E, injection of T-LAK cells with chemically conjugated Ex3 bispecific antibodies (BsAbs; 2 μg/mouse); group F, injection of T-LAK cells with 528 IgG (2 μg/mouse); group G, injection of 528 IgG (2 μg/mouse) alone.
Fig. 7.

Results of in vivo adoptive immunotherapy for xenografted TFK-1 tumors in severe combined immunodeficient (SCID) mice. The TFK-1 cells (5.0×106) were inoculated subcutaneously into SCID mice 10 days before treatment. Then, T-LAK cells (2.0×107) were injected intravenously via the tail vein on 4 consecutive days (0~3) together with IL-2 (500 IU/mouse) in 200 μl phosphate-buffered saline (PBS). Group A, injection of buffer (PBS) alone (open circles); group B, injection of T-LAK cells alone (closed circles); group C, injection of T-LAK cells with Ex3 diabody (20 μg; closed squares); group D, injection of T-LAK cells with Ex3 diabody (2 μg; open squares); group E, injection of T-LAK cells with Ex3 BsAb (2 μg; closed triangles); group F, injection of T-LAK cells with 528 IgG (2 μg; closed diamonds); group G, injection of 528 IgG (2 μg) alone (open diamonds). ** p<0.01, *** p<0.001, **** p<0.0001 for each group vs the control group (group A). Statistical analysis was carried out using the Student’s t test
In the first trial 21 tumor-established mice were divided into seven groups (three mice per group). Mice whose tumors did not reach 5 mm in diameter were excluded from the experiments. The second experimental trial was performed, similarly; therefore, each group consisted of six mice in all. Compared with non-treatment group (group A), T-LAK cells alone (group B) did not have any effect on tumor growth. On the other hand, when T-LAK cells were injected with Ex3 diabody (groups C and D) or Ex3 BsAb (group E), significant inhibition of tumor growth was observed from the early phase onwards, which continued throughout the experimental period. Even at 10 weeks after administration, tumor growth in group C–E was remarkably suppressed. Complete disappearance of the tumors was observed in three of six mice in group C and one of six in groups D and E. The Ex3 diabody or Ex3 BsAb alone did not show any effects (data not shown). Therapy with 528 mAb IgG alone (group G) resulted in moderate tumor growth inhibition, but 528 mAb IgG plus T-LAK cells did not show additive influence (group F).
Discussion
Despite much-improved surgical techniques, the long-term survival of patients with bile duct carcinoma is still poor. One reason is that micro-metastases or residual cancer cells in the lymphogenous or perineural spaces persist because bile duct carcinoma is already too advanced by the time surgery is performed [30]; thus, as effective adjuvant therapy is necessary to eliminate residual bile duct carcinoma, we have focused on adoptive immunotherapy using T-LAK cells with BsAb [30]. Recent advances in genetic and protein engineering have led to new formats for recombinant BsAbs. A small recombinant BsAb, termed a diabody, was firstly developed by Holliger et al. [25] and several molecules have now been constructed [1, 26, 27, 34]. The use of the anti-CD3 × anti-tumor associated antigen (TAA) bispecific diabody is an attractive and highly specific approach in antitumoral immunotherapy [25]. In addition, diabodies have other merits such as easy humanization, increased tumor penetration, decreased immunogenicity due to the small molecular size compared with chemically prepared BsAb, and convenient production in high yields using bacterial expression systems. The anti-TAA × anti-CD3 diabody can be used not only to direct T cells toward tumors, but also to stimulate T cells. Previous reports documented this ability in terms of artificial signaling via the CD3 antigen, which mimics the physiological antigen-specific activation of T lymphocytes by MHC-bound antigen [27]. To date, several kinds of bispecific diabodies directed to tumor and T cell or natural killer (NK) cell antigens have been constructed [1, 2, 11, 26, 27, 33, 34, 35, 73] and some diabodies have exhibited marked inhibition of tumor growth in vitro and in vivo [2, 11, 22, 35, 73]. Using bacterial expression, however, some proteins cannot be expressed in soluble form. Recently, we succeeded in producing diabodies using a refolding system [60, 63], proving dimerization and bispecificity. Our previous study showed that the administration of SEA D227A fusion anti-MUC1 × anti-CD3 diabody (SEA D227A-Mx3 diabody) together with T-LAK cells greatly enhanced cytotoxicity against TFK-1 in vitro and in vivo [61]; however, SEA may also elicit anti-SEA antibodies and this may limit the repeated administration of SEA D227A-Mx3 diabody for cancer therapy; therefore, we tried to construct some new diabodies reactive with TAA and CD3, and evaluated their efficacy to augment the cytotoxicity of T-LAK cells without the help of fused superantigen. Among several anti-TAA × CD3 diabodies we constructed, the anti-EGFR × anti-CD3 diabody most particularly enhanced the cytotoxicity of T-LAK cells against EGFR-positive TFK-1 cells.
Epidermal growth-factor receptor is widely expressed on various solid malignant cells, such as squamous cell carcinomas of the head and neck, non-small cell lung tumors, and breast, colon, pancreatic, kidney, ovarian, bladder, and bile duct carcinomas [48, 58], and therefore is a promising target for cancer therapy. Indeed, a number of reports of inhibition of the EGFR pathway with various molecules, such as anti-EGFR antibodies, EGFR tyrosine kinase inhibitors, and antisense EGFR oligonucleotides, have provided evidence of a block of cell cycle progression, resulting in apoptosis in various cancer cell lines [15, 21, 29, 43, 45, 46, 55, 65, 71].
Refolded Ex3 diabody has a dimeric structure, strongly reacting with both EGFR and CD3 (Fig. 2), and its specificity and bispecificity could be here confirmed with by parental mAb IgG-blocking and absorption tests. Additionally, Ex3 diabody was proved to maintain the same level of the reactivity with EGFR and CD3 after incubation at 37°C for 48 h in RPMI-1640 culture medium (Fig. 2I,J) and freezing–thawing (data not shown), indicating high stability. When this diabody was stored at 4°C for 60 days, it showed almost same reactivity as fresh one, by flow cytometry and cytotoxicity. With the Mx3 diabody and the mSEA-Mx3 diabody, such high stability was not observed; therefore, the Ex3 diabody is suitable for clinical application to target T-LAK cells to EGFR expressing tumor cells. Indeed, agglutination of T-LAK cells around target tumor cells was observed microscopically after co-culture with EGFR-positive TFK-1 cells at 37°C in the presence of Ex3 diabody. This agglutination was inhibited by parental mAb, but not by irrelevant mAb, supporting the specificity of the Ex3 diabody for EGFR and CD3 (Fig. 3).
The effector cells in this study were not specific CTLs but non-specific T-LAK cells (CD3+, CD8+, CD56−) or freshly isolated PBMCs; however, Ex3 diabody greatly enhanced the cytotoxic activity of T-LAK cells against EGFR-positive TFK-1 cells in a dose-dependent manner (Fig. 4A), specific killing reaching 80–90% at an E:T ratio of 50 in the presence of Ex3 diabody at 1 μg/ml (Fig. 4B). Furthermore, only 50 ng/ml of Ex3 diabody was sufficient to obtain 100% growth inhibition of TFK-1 cells, when combined with T-LAK cells at an E:T ratio of 5 for 48 h using MTS, 3-(4,5-dimethylthiazole-2-yl)-5-(3-carcoxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazorium, inner salt, assay kit (CellTiter 96TM aqueous non-radioactive cell proliferation assay; Promega, Madison, Wis.; data not shown). This tumor growth inhibition test (MTS test) has been shown to reflect very well the results of in vivo experiments [5, 16]. The antitumor activity of T-LAK was not enhanced at all by mixtures of equal amounts of 528 scFv and OKT3 scFv, in the MTS tumor growth inhibition assay even at 100 ng/ml, whereas Ex3 diabody showed more than 95% growth inhibition at the same concentration.
The Ex3 diabody also greatly enhanced the cytotoxicity of T-LAK cells against other EGFR-positive cells, but not against EGFR-negative cells (Fig. 4; Table 1). The enhancement was furthermore completely inhibited by parental 528 and OKT3 mAb IgGs (Fig. 5), presumably due to not only the difference in affinity between the recombinant diabody and mAb IgG, but also because it is directed to an epitope common to the Ex3 diabody. This also indicates effector cells in the freshly isolated PBMCs are not NK cells but T cells.
Addition of Ex3 diabody to the culture medium of T-LAK cells could induce approximately two- to tenfold higher production of IFN-γ, GM-CSF, TNF-α, and IL-2 by T-LAK cells compared with the culture supernatant of T-LAK cells alone (Fig. 6A–D), indicating that Ex3 diabody can stimulate T-LAK cells by artificial signaling via CD3, which mimics the physiological antigen-specific activation of T cells [27]. IFN-γ, GM-CSF, and TNF-α production by T-LAK cells was markedly increased when co-cultured with TFK-1 cells in the presence of Ex3 diabody in a dose-dependent manner. While IL-2 production was slightly increased in the presence of Ex3 diabody (10 ng/ml) in the first 12 h but diminished thereafter (Fig. 6H). This was probably due to the time difference of cytokine production after stimulation with Ex3 diabody.
In the xenograft tumor model (Fig. 7), administration of Ex3 diabody alone or Ex3 BsAb alone did not exert any effect, but with co-injection of T-LAK cells, significant tumor growth inhibition was observed. Especially, complete disappearance of tumors was observed in three of six mice (group C, 20 μg Ex3 diabody plus T-LAK). Therapy with 528 mAb IgG (group G), which contained Fc region, itself had only a slight effect on tumor growth in vitro, but caused considerable inhibition in xenografted SCID mice.
The efficacy of Ex3 diabody was evaluated here using allogeneic T-LAK cells or PBMCs in vitro and in vivo. Regarding whether the strong killing was induced only by the allogeneic reaction, it has been reported that enhanced cytotoxicity against autologous tumor cells was detected when autologous T cells were targeted by BsAb at the same levels as when allogeneic T cells were used [5, 8, 10, 27], indicating the reliability of our approach for targeting immunotherapy in the clinical setting.
Epidermal growth-factor receptor is also expressed in normal tissues, though at a very low level compared with malignant tissues; namely, 40,000–100,000 receptors per normal cell, as opposed to approximately 2,000,000 per malignant cell, were reported [14, 23, 28, 51, 74]. It is possible that a small number of T-LAK cells might be targeted against normal cells; however, a surprisingly low rate of adverse side effects associated with EGFR blockade in combination with chemotherapy have been reported [28]. Malignant tumor cells faced with inadequate cell-matrix contacts critically depend on EGFR activation for survival, rendering them more susceptible to apoptosis induction by EGFR blockade.
In the present study, administration of Ex3 diabody or parental 528 mAb IgG alone to TFK-1 cells had little effect on tumor cell growth or cell death at the concentration of 0.1–10.0 μg/ml in vitro, although previous reports showed the growth inhibition by 528 mAb [41, 42, 62]. It is unclear whether the enhanced cytotoxicity with T-LAK cells was simply due to their activation and improved targeting to EGFR-positive tumor cells, or to synergistic effects of both retargeting and EGFR signal pathway blocking, e.g., tyrosine kinase inhibition. Compared with other recombinant diabodies (anti-TAA × CD3) reported previously, the Ex3 diabody demonstrated extremely strong enhancement, the efficacy being approximately 100-fold greater than that with our previous SEA D227A-Mx3 diabody [61]; thus, marked antitumor activity can be attributed to the stability of Ex3 diabody, and EGFR blocking.
Combination of Ex3 diabody with vaccine therapy or costimulatory molecules, such as anti-CD28 mAb, B7.1 or B7.2, and 4–1BBL, may further enhance the cytotoxicity of T-LAK cells as in the case with BsAb Mx3 and 4–1BBL [75]. Further studies are now in progress to explore this event.
Conclusion
In conclusion, we have developed and produced an anti-EGFR × anti-CD3 bispecific diabody with bacterial expression using a refolding system. It dramatically enhances the cytotoxicity of T-LAK cells specifically against EGFR-positive tumor cells and has advantages over chemically synthesized BsAbs, such as of humanization by gene engineering, low-cost, high-yield, easy procedures, with low immunogenicity and high penetrability. The results are highly suggestive of utility of humanized Ex3 diabody for adoptive immunotherapy of patients with bile duct carcinomas or other cancers expressing EGFR. We have succeeded in producing humanized Ex3 diabody, of which attractive results will be reported soon.
Acknowledgements
We thank F. Koizumi for her help in preparation of the manuscript and H. Saeki for valuable advice for this study.
References
- 1.Adams Br J Cancer. 1998;77:1405. doi: 10.1038/bjc.1998.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arndt Blood. 1999;94:2562. [PubMed] [Google Scholar]
- 3.Asano R, Takemura S, Tsumoto K, Sakurai N, Teramae A, Ebara S, Katayose Y, Shinoda M, Suzuki M, Imai K, Matsuno S, Kudo T, Kumagai I. Functional construction of the anti-mucin core protein (MUC1) antibody MUSE11 variable regions in a bacterial expression system. J Biochem (Tokyo) 2000;127:673. doi: 10.1093/oxfordjournals.jbchem.a022656. [DOI] [PubMed] [Google Scholar]
- 4.Barbul Nutrition. 1990;6:53. [Google Scholar]
- 5.Beun J Immunol. 1993;150:2305. [PubMed] [Google Scholar]
- 6.Bohlen Cancer Res. 1993;53:4310. [PubMed] [Google Scholar]
- 7.Boss Nucleic Acids Res. 1984;12:3791. doi: 10.1093/nar/12.9.3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brandl Exp Hematol. 1999;27:1264. doi: 10.1016/S0301-472X(99)00072-7. [DOI] [PubMed] [Google Scholar]
- 9.Brennan Science. 1982;229:81. [Google Scholar]
- 10.Canevari J Natl Cancer Inst. 1995;87:1463. doi: 10.1093/jnci/87.19.1463. [DOI] [PubMed] [Google Scholar]
- 11.Cochlovius J Immunol. 2000;165:888. doi: 10.4049/jimmunol.165.2.888. [DOI] [PubMed] [Google Scholar]
- 12.D’Amico J Thorac Cardiovasc Surg. 1999;117:736. doi: 10.1016/s0022-5223(99)70294-1. [DOI] [PubMed] [Google Scholar]
- 13.de J Pathol. 1998;184:53. doi: 10.1002/(SICI)1096-9896(199801)184:1<53::AID-PATH6>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 14.Ennis Cancer Invest. 1991;9:553. doi: 10.3109/07357909109018953. [DOI] [PubMed] [Google Scholar]
- 15.Fan Cancer Res. 1993;53:4637. [PubMed] [Google Scholar]
- 16.Ferrini Int J Cancer. 1985;36:337. [PubMed] [Google Scholar]
- 17.Fischer-Colbrie Anticancer Res. 1997;17:613. [PubMed] [Google Scholar]
- 18.Gibson J Biol Chem. 1999;274:17612. doi: 10.1074/jbc.274.25.17612. [DOI] [PubMed] [Google Scholar]
- 19.Giordano J Cell Biochem. 1998;70:1. doi: 10.1002/(SICI)1097-4644(19980701)70:1<1::AID-JCB1>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 20.Glynne-Jones Hum Pathol. 1996;27:688. doi: 10.1016/s0046-8177(96)90399-8. [DOI] [PubMed] [Google Scholar]
- 21.He J Natl Cancer Inst. 1998;90:1080. doi: 10.1093/jnci/90.14.1080. [DOI] [PubMed] [Google Scholar]
- 22.Helfrich Int J Cancer. 1998;76:232. doi: 10.1002/(SICI)1097-0215(19980413)76:2<232::AID-IJC11>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 23.Herbst Cancer. 2002;94:1593. doi: 10.1002/cncr.10372. [DOI] [PubMed] [Google Scholar]
- 24.Hinoda J Clin Lab Anal. 1993;7:100. doi: 10.1002/jcla.1860070206. [DOI] [PubMed] [Google Scholar]
- 25.Holliger Proc Natl Acad Sci USA. 1993;90:6444. [Google Scholar]
- 26.Holliger Protein Eng. 1996;9:299. doi: 10.1093/protein/9.3.299. [DOI] [PubMed] [Google Scholar]
- 27.Holliger Cancer Res. 1999;59:2909. [PubMed] [Google Scholar]
- 28.Kari Cancer Res. 2003;63:1. [PubMed] [Google Scholar]
- 29.Karnes Gastroenterology. 1998;114:930. doi: 10.1016/s0016-5085(98)70312-9. [DOI] [PubMed] [Google Scholar]
- 30.Katayose Cancer Res. 1996;56:4205. [PubMed] [Google Scholar]
- 31.Kawamoto Proc Natl Acad Sci USA. 1983;80:1337. [Google Scholar]
- 32.Kerbel Mol Med. 1998;4:286. [PMC free article] [PubMed] [Google Scholar]
- 33.Kipriyanov Protein Eng. 1997;10:445. doi: 10.1093/protein/10.4.445. [DOI] [PubMed] [Google Scholar]
- 34.Kipriyanov Int J Cancer. 1998;77:763. doi: 10.1002/(SICI)1097-0215(19980831)77:5<763::AID-IJC16>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 35.Kipriyanov J Immunol. 2002;169:137. doi: 10.4049/jimmunol.169.1.137. [DOI] [PubMed] [Google Scholar]
- 36.Kiyokawa J Biol Chem. 1997;272:18656. doi: 10.1074/jbc.272.30.18656. [DOI] [PubMed] [Google Scholar]
- 37.Klijn Endocr Rev. 1992;13:3. [Google Scholar]
- 38.Krebber J Immunol Methods. 1997;201:35. doi: 10.1016/S0022-1759(96)00208-6. [DOI] [PubMed] [Google Scholar]
- 39.Laemmli Nature. 1970;227:680. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 40.Lieberman Ann Surg. 1992;1992:157. doi: 10.1097/00000658-199202000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Masui Cancer Res. 1984;44:1002. [PubMed] [Google Scholar]
- 42.Masui Cancer Res. 1986;46:5592. [PubMed] [Google Scholar]
- 43.Modjtahedi Int J Oncol. 1998;13:335. doi: 10.3892/ijo.13.2.335. [DOI] [PubMed] [Google Scholar]
- 44.Mountain Biotechnol Genet Eng Rev. 1992;10:1. doi: 10.1080/02648725.1992.10647886. [DOI] [PubMed] [Google Scholar]
- 45.Moyer Cancer Res. 1997;57:4838. [PubMed] [Google Scholar]
- 46.Nagane Cancer Res. 1996;56:5079. [PubMed] [Google Scholar]
- 47.Niikura Int J Gynecol Pathol. 1997;16:60. doi: 10.1097/00004347-199701000-00010. [DOI] [PubMed] [Google Scholar]
- 48.Nonomura Liver. 1988;8:157. doi: 10.1111/j.1600-0676.1988.tb00985.x. [DOI] [PubMed] [Google Scholar]
- 49.Perisic Structure. 1994;2:1217. doi: 10.1016/s0969-2126(94)00123-5. [DOI] [PubMed] [Google Scholar]
- 50.Perry Prostate. 1998;35:117. doi: 10.1002/(SICI)1097-0045(19980501)35:2<117::AID-PROS5>3.3.CO;2-5. [DOI] [Google Scholar]
- 51.Petrides Cancer Res. 1990;50:3934. [PubMed] [Google Scholar]
- 52.Renard Am J Pathol. 2002;160:113. doi: 10.1016/S0002-9440(10)64355-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reynolds Surgery. 1988;104:142. [Google Scholar]
- 54.Reynolds Ann Surg. 1990;211:202. doi: 10.1097/00000658-199002000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodeck J Cell Sci. 1997;110:113. doi: 10.1242/jcs.110.2.113. [DOI] [PubMed] [Google Scholar]
- 56.Saijyo Tohoku J Exp Med. 1995;177:61. doi: 10.1620/tjem.177.61. [DOI] [PubMed] [Google Scholar]
- 57.Salmeron J Immunol. 1991;147:3047. [PubMed] [Google Scholar]
- 58.Salomon Crit Rev Oncol Hematol. 1995;19:183. doi: 10.1016/1040-8428(94)00144-I. [DOI] [PubMed] [Google Scholar]
- 59.Takeda Cancer Res. 1975;35:2390. [PubMed] [Google Scholar]
- 60.Takemura Protein Eng. 2000;13:583. doi: 10.1093/protein/13.8.583. [DOI] [PubMed] [Google Scholar]
- 61.Takemura Cancer Immunol Immunother. 2002;51:33. doi: 10.1007/s00262-001-0245-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Teramoto Cancer. 1996;77:1639. doi: 10.1002/(SICI)1097-0142(19960415)77:8<1639::AID-CNCR33>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 63.Tsumoto J Immunol Methods. 1998;219:119. doi: 10.1016/S0022-1759(98)00127-6. [DOI] [PubMed] [Google Scholar]
- 64.Turner Clin Exp Metastasis. 1996;14:409. doi: 10.1007/BF00123400. [DOI] [PubMed] [Google Scholar]
- 65.Uckun Clin Cancer Res. 1998;4:901. [PubMed] [Google Scholar]
- 66.Van J Immunol. 1980;124:2708. [Google Scholar]
- 67.Verbeek FEBS Lett. 1998;425:145. doi: 10.1016/s0014-5793(98)00224-5. [DOI] [PubMed] [Google Scholar]
- 68.Volm Br J Cancer. 1998;77:663. doi: 10.1038/bjc.1998.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.von Curr Opin Oncol. 1996;8:493. doi: 10.1097/00001622-199611000-00009. [DOI] [PubMed] [Google Scholar]
- 70.Wells Adv Cancer Res. 2000;78:31. doi: 10.1016/s0065-230x(08)61023-4. [DOI] [PubMed] [Google Scholar]
- 71.Wu J Clin Invest. 1995;95:1897. doi: 10.1172/JCI117871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu Immunotechnology. 1996;2:21. doi: 10.1016/1380-2933(95)00027-5. [DOI] [PubMed] [Google Scholar]
- 73.Xiong Cancer Lett. 2002;177:29. doi: 10.1016/S0304-3835(01)00758-3. [DOI] [PubMed] [Google Scholar]
- 74.Yao Cancer Res. 1988;48:6753. [PubMed] [Google Scholar]
- 75.Yoshida Cancer Immunol Immunother. 2003;52:97. doi: 10.1007/s00262-002-0334-y. [DOI] [PMC free article] [PubMed] [Google Scholar]