

Abstract
Cell migration is of paramount importance in physiological processes such as immune surveillance, but also in the pathological processes of tumor cell migration and metastasis development. The factors that regulate this tumor cell migration, most prominently neurotransmitters, have thus been the focus of intense investigation. While the majority of neurotransmitters have a stimulatory effect on cell migration, we herein report the inhibitory effect of the endogenous substance anandamide on both tumor cell and lymphocyte migration. Using a collagen-based three-dimensional migration assay and time-lapse videomicroscopy, we have observed that the anandamide-mediated signals for CD8+ T lymphocytes and SW 480 colon carcinoma cells are each mediated by distinct cannabinoid receptors (CB-Rs). Using the specific agonist docosatetraenoylethanolamide (DEA), we have observed that the norepinephrine-induced migration of colon carcinoma cells is inhibited by the CB1-R. The SDF-1–induced migration of CD8+ T lymphocytes was, however, inhibited via the CB2-R, as shown by using the specific agonist JWH 133. Therefore, specific inhibition of tumor cell migration via CB1-R engagement might be a selective tool to prevent metastasis formation without depreciatory effects on the immune system of cancer patients.
Keywords: Anandamide, Cannabinoid receptors, Cell migration, T lymphocytes, Tumor cells
Introduction
Cannabinoids are a class of hydrophobic substances found in Cannabis sativa. The most prominent substrate is Δ9-tetrahydrocannabinol, which induces psychoactive effects upon intake. The fervent search for a specific cannabinoid receptor ended in 1988, when the existence of a specific receptor in the rat brain was confirmed. Devane and colleagues gave it the designation CB1-R [6]. A second receptor, the peripheral cannabinoid receptor CB2-R, was discovered in macrophages of the margin zone of the spleen, and was cloned by Munro and co-workers [27]. This CB2-R has since been found in lymph nodes, Peyer’s plaques of the small intestine, and leukocytes. Both receptors CB1-R and CB2-R are expressed on leukocytes [20, 28, 31, 35].
Both cannabinoid receptors are Gi/o-protein-coupled transmembrane receptors, and the subsequent signaling pathways negatively regulate the adenylyl cyclase and activate the mitogen-activated protein kinase [23, 30]. The discovery of these receptors led to the discovery of the first endogenous ligand to the CB1-R shortly thereafter, which was found in pigeon brain [7]. This ligand was termed “anandamide,” based on “ananda,” the Sanskrit word for bliss, and its amide-containing chemical structure. This ligand is an arachidonic acid derivate, also called arachidonoylethanolamide. Anandamide binds to the CB2-R, as well, but with less affinity than to the CB1-R [32]. In accordance with the aforementioned intracellular signal transduction pathways activated by CB-R engagement, anandamide inhibits the forskolin-stimulated adenylyl cyclase, thereby reducing the cellular cAMP production [32].
In human breast cancer and prostate cancer cells, anandamide inhibits proliferation via the CB1-R [4, 24]. The CB1-R engagement leads to an inhibition of cAMP generation, as described above, and leads to the suppression of receptor tyrosine kinase signaling, as was shown by the inhibition of prolactin- and nerve growth factor-induced cell proliferation [4]. With regard to the immune system, anandamide has an anti-inflammatory function and plays a role in the reduction of chronic pain. For example, anandamide was found to inhibit neutrophil recruitment [3]. Furthermore, there is an inhibitory effect of anandamide in bronchoalveolar lavage fluid of lipopolysaccharide-treated mice on tumor necrosis factor α production [3]. Macrophages themselves produce anandamide and related substances, e.g. palmitoyl ethanolamide (PEA) and 2-arachidonoyl glycerol (2-AG), upon stimulation with ionomycin, and also contribute to the homeostasis of endocannabinoids by inactivating these substances through several pathways [5].
The influence of neurotransmitters on the migration of tumor cells and leukocytes has recently been the subject of systematic examination [12]. Norepinephrine is one of the most potent inducers for the migration of tumor cells, stimulating the migration of SW 480 colon carcinoma cells and MDA-MB-468 breast carcinoma cells via the β2-adrenoceptors [8, 17, 22]. This effect is abolished by the γ-aminobutyric acid (GABA), mediated through the metabotropic GABAB receptor [17]. The GABAB receptor is coupled to the adenylyl cyclase via Gi proteins and exerts its inhibiting effect on migration via the down-regulation of cAMP. Since anandamide has a similar effect on the adenylyl cyclase, we investigated the role of anandamide on the migration of tumor cells and leukocytes.
Methods and materials
Cultivation of tumor cell lines
Cells of the human colon carcinoma cell line SW 480 (American Type Culture Collection, Rockville, MD) were cultured at 37°C in a humidified atmosphere in an antibiotic-free Leibovitz L-15 culture medium (PAA Laboratories, Linz, Austria), containing 10% active fetal calf serum (PAA Laboratories).
Lymphocyte isolation
Human CD8+ T lymphocytes were isolated from heparinized peripheral blood as described previously [10]. In short, the peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll-Hypaque density gradient centrifugation (ICN, Meckenheim, Germany), followed by a positive selection of the CD8+ T lymphocytes using immunomagnetic beads coated with mouse antihuman CD8 antibody (Abs) (Dynabeads; Dynal, Hamburg, Germany). The cell-bound beads were detached with polyclonal anti-mouse Fab Abs (Detachabead; Dynal).
Isolated T lymphocytes were maintained overnight in RPMI culture medium (PAA Laboratories), 10% heat-inactivated fetal calf serum and 1% penicillin/streptomycin (50 U/ml and 50 µg/ml; Gibco, Eggenstein-Leopoldshafen, Germany) at 37°C in a humidified atmosphere containing 5% CO2.
Neutrophil granulocyte isolation
Neutrophil granulocytes were isolated as previously described [11]. In short, the PBMCs were isolated using Ficoll-Hypaque density gradient centrifugation. The pellet containing neutrophil granulocytes and erythrocytes was mixed with platelet-depleted serum of the same donor, and subsequently diluted 1:1.3 with a high molecular weight dextran solution (Macrodex; Fresenius, Bad Homburg, Germany) containing 0.01-M EDTA. After 3 h the supernatant containing the neutrophil granulocytes was separated from the pellet containing the erythrocytes. Remaining erythrocytes were removed by a hypotonic lysis with 0.3% sodium chloride for 2 min on ice. The purified neutrophil granulocytes were used immediately after isolation.
Flow cytometry
The presence of CB1-R and CB2-R has been proven on the surface of leukocytes [20, 28, 31, 35], but not on colon carcinoma cells. Therefore, we analyzed the expression of these two receptors flow cytometrically using a FacsCalibur flow cytometer (Becton Dickinson, Heidelberg, Germany). We incubated 1×105 cells with 5 µg/ml of the primary antibody (H-150 for the CB1-R, Santa Cruz Biotechnologies, Santa Cruz, CA, USA; the anti-CB2-R antibody was derived from Calbiochem) for 10 min at room temperature. After washing we incubated the cells with 5 µg/ml of a fluorescein-isothiocyanate (FITC)-conjugated anti-rabbit antibody (Coulter-Immunotech, Hamburg, Germany). Nonspecific binding was determined by an isotypic control rabbit antibody (Coulter-Immunotech).
In addition, flow cytometry was used to assess the viability of the cells immediately after the end of the migration experiments using propidium iodide staining as described previously [22].
Cell migration assay
The cell migration assay and the subsequent evaluation of the migration paths were performed as described previously [8, 14]. In short, 5×104 SW 480 colon carcinoma cells or 2×105 leukocytes were mixed with 150 μl of a carbonate-buffered collagen solution containing minimum essential Eagle’s medium (Flow, McLean, VA, USA). This mixture was inserted into self-constructed migration chambers [14] and allowed to polymerize for 30 min at 37°C. To investigate the regulation of cell migration, the pharmacological substances (neurotransmitters and agonists) were mixed with the collagen solution prior to polymerization, and the residual chamber volume was filled with solutions of these substances after polymerization. The migration of the cells was recorded via time-lapse videomicroscopy.
After recording, the migration paths of 30 randomly selected cells were digitized in 15-min intervals for tumor cells and 1-min intervals for leukocytes. The migratory activity was calculated for each step as the part of the cells (percentage) which was locomotory active. All migratory activities shown in the figures are mean values of three independent experiments (for the leukocyte experiments, the cells of three different blood donors were used); in the text we provide the mean values and standard deviations (SD) at the steady stage level of the observation period. Statistical significance of changes was calculated using Student’s t-test.
Agonists
Anandamide, JWH 133, DEA, HU 210 and human stromal cell–derived factor 1 (SDF-1) alpha were obtained from Biotrend Chemikalien, Germany. Norepinephrine and fMLP (formyl-methionyl-leucyl-phenylalanine) were from Sigma-Aldrich, Deisenhofen, Germany.
Results
Tumor cell migration
Norepinephrine induces migration of SW 480 colon carcinoma cells and MDA-MB-468 breast carcinoma cells [8, 22]. We now report that this induced tumor cell migration is completely inhibited by anandamide in SW 480 cells (Fig. 1). Both CB1-R and CB2-R were expressed on the surface of these cells, whereas the expression of the CB2-R was very low (Fig. 1A). These results were confirmed by immunoblotting (data not shown). Norepinephrine induced migration from 28±5% spontaneously locomoting cells to 54±8% locomoting cells after treatment with 10 µM norepinephrine. Anandamide (40 nM) alone had no effect (29±4% locomoting cells), but inhibited norepinephrine-induced migration (36±5%; p<0.05; Fig. 1B). This inhibitory effect was also observed in MDA-MB-468 breast carcinoma cells, and with HU 210, a tetrahydrocannabinol-related, non–arachidonic acid agonist to both CB-Rs (data not shown). We investigated the receptor responsible for this inhibitory function of anandamide by receptor-specific agonists. We used DEA (40 nM) as a specific agonist for the CB1-R, and JWH 133 (10 nM) as a specific agonist for the CB2-R. The differences in the concentrations that were used result from the different affinities described for the agonists [1, 15]. DEA (Fig. 1C), but not JWH 133 (Fig. 1D) inhibited the norepinephrine-induced migration. After treatment with DEA, the norepinephrine-induced migration was reduced significantly (p<0.005) from 62±4% down to 37±5% locomoting cells (Fig. 1C), whereas treatment with JWH 133 had no effect (60±10% locomoting cells with norepinephrine vs 62±6% locomoting cells with norepinephrine and JWH 133; Fig. 1D). Thus, the inhibitory function of anandamide on the migration of the tumor cells is mediated by the CB1-R.
Fig. 1A–D.
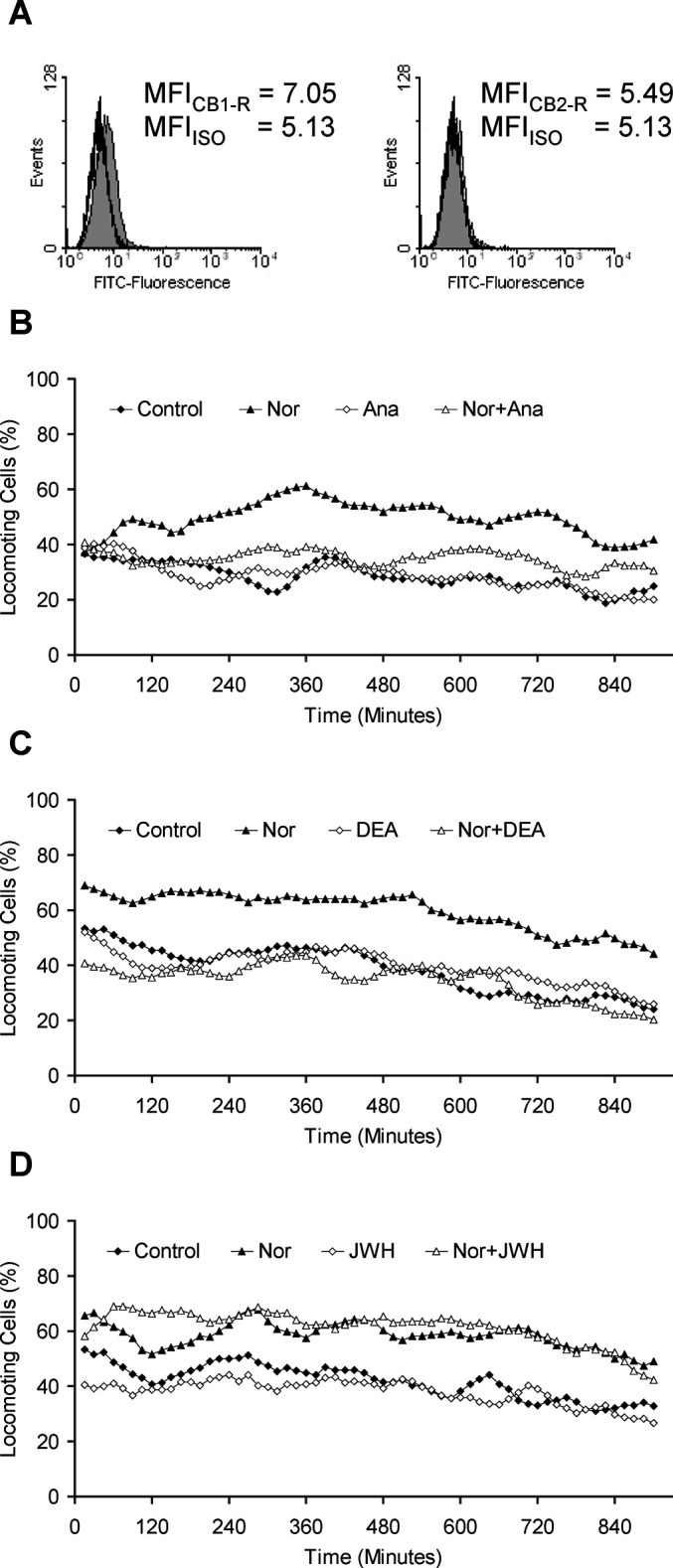
Expression of cannabinoid receptors and effects of anandamide on the migration of SW 480 colon carcinoma cells. A The FITC-fluorescence of specifically bound antibodies against CB1-R and CB2-R (gray area) was compared with an isotypic control antibody (black line). MFI mean fluorescence intensity. B,C,D The migration of the cells was induced by 10 µM norepinephrine. Anandamide (B) or the CB1-R–specific agonist DEA (C) was added to the collagen mixture at concentrations of 40 nM. The CB2-R–specific agonist JWH 133 (D) was used at 10 nM
Migration of leukocytes
Chemokines are the most important regulators for the migration of leukocytes. These peptides bind to serpentine receptors similar to those neurotransmitters, which regulate migration [12]. We have reported previously that the chemokine SDF-1 induces the migration of CD8+ T lymphocytes [11]. Here we observed that this induced migration of T lymphocytes was inhibited by anandamide, similar to the effect observed in tumor cells (Fig. 2A): the SDF-1–induced migration was significantly reduced (p<0.05) from 77±19% to 55±25% locomoting cells in combination with anandamide, whereas this endogenous cannabinoid alone had no effect on the migration of the lymphocytes. There was no significant difference between the spontaneous migration (5±3%) of the cells and the cells treated with anandamide (3±1%). In contrast to our results with SW 480 colon carcinoma cells, the specific CB1-R agonist DEA did not reduce the SDF-1–induced migratory activity of the lymphocytes (Fig. 2B). JWH 133, the specific CB2-R agonist, did however reduce the SDF-1–induced migration by 20% (from 58±17% to 36±11%; Fig. 1C).
Fig. 2A–C.

Effects of anandamide on the migration of T lymphocytes. The migration of the cells was induced by 1 μg/ml SDF. Anandamide (A) or DEA (B) was added at concentrations of 40 nM. JWH 133 (C) was added to the collagen at a concentration of 10 nM
Neutrophil granulocytes are integral features of the innate immune system and can recognize the formylated peptide fMLP, a bacterial breakdown product, via a serpentine receptor (fMLP-R). Neutrophil granulocytes respond to fMLP with an increase of migratory activity from 6±2% to 71±4% locomoting cells (Fig. 3) [11]. The addition of anandamide to these cells had no effect on the spontaneous locomotion (8±7% locomoting cells), nor on the fMLP-induced migration (68±11% locomoting cells).
Fig. 3.

Effects of anandamide (40 nM) on the migration of fMLP (10 nM) induced neutrophil granulocytes
Discussion
Recent findings in cell migration research have shown that not only is the migration of leukocytes strongly influenced by ligands to serpentine receptors, i.e., chemokines and neurotransmitters, but the migration of tumor cells also depends on such signals from the environment. Thus has the concept of metastasis formation shifted from a solely genetically determined standpoint to an understanding of the complex interplay of signal substances of the immune system and the neuroendocrine system with tumor cells [12]. Chemokines have been shown to promote the migratory activity of tumor cells [21] and are even responsible for the localization of metastases [26]. Neurotransmitters likewise induce migration of tumor cells and provoke a chemotactic response, as shown for norepinephrine [8]. While numerous reports now provide evidence for a promigratory function of neurotransmitters, only very limited information is available about neurotransmitters with an inhibitory effect. We have previously shown that the main inhibitory neurotransmitter of the brain, GABA, inhibits the norepinephrine-induced migratory activity of breast [8] and colon [17] carcinoma cells. GABA exerts its effect on migration via the metabotropic GABAB receptor, which is intracellularly coupled to inhibitory Gi proteins. Its activation in turn leads to a down-regulation of adenylyl cyclase activity [17]. Based on these results, Ortega has suggested a possible use of GABA or GABAB agonists (e.g., baclofen, a drug used in the treatment of epilepsy) for the pharmacological prevention of metastasis formation [29].
Anandamide alone had no effect on the migration of both tumor cells and leukocytes. These results are in accordance with findings of Kishimoto and co-workers on HL-60 leukemia cells and monocytes [19], whereas ananamide was shown to have a stimulatory effect on embryonic kidney cell [33], microglial cell migration [34], and in myeloid leukemia cells transfected with the CB2-R gene [16]. However, in our experiments, anandamide inhibited the norepinephrine-induced migration of colon carcinoma cells and the SDF-1–induced migration of T lymphocytes. Such functional differences between neuronal cells and leukocytes have been observed for GABA, too [2, 12], although there is unanimity about the engagement of Gi proteins by both neurotransmitters in either cell types. Anandamide is an endogenous ligand for the two identified cannabinoid receptors CB1-R and CB2-R. Like the GABAB receptor, CB1-R and CB2-R are coupled to Gi proteins, and a down-regulation of adenylyl cyclase activity has been shown as well [23, 30]. The aforementioned stimulatory effects as observed by Jorda and co-workers [16] might be due to the overexpression of the receptors in the transfected cells. Such overexpression might lead to the interaction of the receptor not only to Gi, but also to Gs proteins as was shown on the example of the α2-adrenoceptor [9]. Thus, we assume that anandamide exerts its inhibitory effect shown herein on cell migration via intracellular signal transduction pathways similar to GABA. In a broader, conceptual sense, this leads to the hypothesis that other neurotransmitters, whose serpentine receptors are also coupled to Gi proteins, generally act as inhibitors for the migration of tumor cells, by a down-regulation of adenylyl cyclase activity, while stimulatory chemokines and neurotransmitters (e.g., norepinephrine) increase the activity of this enzyme [13, 17]. In this study, we have used the most potent inducers of migration available to us for each cell type, but the principle of the dual control of the adenylyl cyclase also holds true for other initiators and inhibitors.
The most striking consequence of these results in regard to a possible therapeutic approach using cannabinoid receptor agonists in the inhibition of tumor cell migration is the fact that the inhibitory effect in tumor cells is mediated via the CB1-R, but in T lymphocytes via the CB2-R; neutrophil granulocytes were not at all affected by anandamide. Thus, specific CB1-R agonists such as the endogenously produced DEA [30], might be a selective tool for the pharmacological inhibition of metastasis formation, without the immunosuppressive effect observed in cannabinoid therapy [5, 18]. In addition, anandamide has been shown to have antiproliferative and apoptosis-inducing properties in metastatic prostate cancer cells by inducing ceramide production [25]. Although it is unlikely that such signaling pathways of long-term treatment contribute to the immediate effect of migration inhibition, such impacts on other cell functions would make anandamide or CB1-R–specific analogs even more effective tools in cancer treatment.
Acknowledgements
This study was supported by the Fritz Bender Foundation, Munich, Germany, and the Bruno and Helene Joester Foundation, Cologne, Germany. We thank Beate Mainusch for excellent technical assistance and Theodore L. Drell IV for critical reading of the manuscript.
References
- 1.Barg Eur J Pharmacol. 1995;287:145. doi: 10.1016/0014-2999(95)00487-4. [DOI] [PubMed] [Google Scholar]
- 2.Behar Cereb Cortex. 2001;11:744. doi: 10.1093/cercor/11.8.744. [DOI] [PubMed] [Google Scholar]
- 3.Berdyshev Life Sci. 1998;63:PL125. doi: 10.1016/s0024-3205(98)00324-5. [DOI] [PubMed] [Google Scholar]
- 4.De Proc Natl Acad Sci U S A. 1998;95:8375. doi: 10.1073/pnas.95.14.8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Chem Phys Lipids. 2000;108:191. doi: 10.1016/s0009-3084(00)00196-1. [DOI] [PubMed] [Google Scholar]
- 6.Devane Mol Pharmacol. 1988;34:605. [PubMed] [Google Scholar]
- 7.Devane Science. 1992;258:1946. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 8.Drell Breast Cancer Res Treat. 2003;80:63. doi: 10.1023/A:1024491219366. [DOI] [PubMed] [Google Scholar]
- 9.Eason J Biol Chem. 1992;267:15795. [PubMed] [Google Scholar]
- 10.Entschladen J Immunol. 1997;159:3203. [PubMed] [Google Scholar]
- 11.Entschladen Cell Immunol. 2000;199:104. doi: 10.1006/cimm.1999.1605. [DOI] [PubMed] [Google Scholar]
- 12.Entschladen Cancer Immunol Immunother. 2002;51:467. doi: 10.1007/s00262-002-0300-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Entschladen Lancet Oncol. 2004;5:254. doi: 10.1016/S1470-2045(04)01431-7. [DOI] [PubMed] [Google Scholar]
- 14.Friedl J Immunol Methods. 1993;165:157. doi: 10.1016/0022-1759(93)90341-4. [DOI] [PubMed] [Google Scholar]
- 15.Huffman Bioorg Med Chem. 1999;7:2905. doi: 10.1016/s0968-0896(99)00219-9. [DOI] [PubMed] [Google Scholar]
- 16.Jorda Ann N Y Acad Sci. 2003;996:10. [Google Scholar]
- 17.Joseph Cancer Res. 2002;62:6467. [PubMed] [Google Scholar]
- 18.Kaminski Biochem Pharmacol. 1994;48:1899. doi: 10.1016/0006-2952(94)90588-6. [DOI] [PubMed] [Google Scholar]
- 19.Kishimoto J Biol Chem. 2003;278:24469. doi: 10.1074/jbc.M301359200. [DOI] [PubMed] [Google Scholar]
- 20.Klein J Leukoc Biol. 2003;74:486. doi: 10.1189/jlb.0303101. [DOI] [PubMed] [Google Scholar]
- 21.Lang Int J Cancer. 2002;99:673. doi: 10.1002/ijc.10424. [DOI] [PubMed] [Google Scholar]
- 22.Masur Cancer Res. 2001;61:2866. [PubMed] [Google Scholar]
- 23.Matsuda Nature. 1990;346:561. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 24.Melck Endocrinology. 2000;141:118. doi: 10.1210/endo.141.1.7239. [DOI] [PubMed] [Google Scholar]
- 25.Mimeault Prostate. 2003;56:1. doi: 10.1002/pros.10190. [DOI] [PubMed] [Google Scholar]
- 26.Muller Nature. 2001;410:50. [Google Scholar]
- 27.Munro Nature. 1993;365:61. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 28.Nong Adv Exp Med Biol. 2001;493:229. doi: 10.1007/0-306-47611-8_27. [DOI] [PubMed] [Google Scholar]
- 29.Ortega Trends Pharmacol Sci. 2003;24:151. doi: 10.1016/S0165-6147(03)00052-X. [DOI] [PubMed] [Google Scholar]
- 30.Pertwee Curr Med Chem. 1999;6:635. [PubMed] [Google Scholar]
- 31.Roth Chem Phys Lipids. 2002;121:229. doi: 10.1016/S0009-3084(02)00159-7. [DOI] [PubMed] [Google Scholar]
- 32.Slipetz Mol Pharmacol. 1995;48:352. [PubMed] [Google Scholar]
- 33.Song J Pharmacol Exp Ther. 2000;294:204. [PubMed] [Google Scholar]
- 34.Walter J Neurosci. 2003;23:1398. [Google Scholar]
- 35.Yuan J Neuroimmunol. 2002;133:124. doi: 10.1016/S0165-5728(02)00370-3. [DOI] [PubMed] [Google Scholar]