

Abstract
Patients' autologous macrophages (AM) were used as antigen-presenting cells (APC) in a vaccination protocol against malignant melanoma. AM were administered by various routes, including intralymphatic, since these cells did not express CCR7, a molecule required for APC migration to lymph nodes. Seven HLA-A2 patients with metastatic melanoma—two classified as M1 and five as M3—were included in the study. AM were produced from leukapheresis-separated mononuclear cells by 7-day culture with granulocyte-macrophage colony-stimulating factor. After separation by elutriation, AM were frozen in aliquots and subsequently thawed at monthly intervals, exposed to MAGE-3(271–279) peptide and injected subcutaneously into lymph nodes or into one peripheral lymph vessel. Intradermal tests were performed before and after treatment to determine peptide reactivity. No acute toxicity was observed following injection. One M1 patient had a 7-mm induration intradermal reaction response and was stabilized for 64 weeks. The M3 patients did not show any immunological or clinical response. In 11 patients, the biodistribution of 111In-labeled AM was investigated. There was no clear evidence that AM injected intradermally or subcutaneously left the site of injection. After injection into a lymph vessel of the foot region, scintigraphs showed five to ten popliteal and inguinocrural lymph nodes. This appeared to be the most efficient way to administer rapidly and safely large amounts of peptide-loaded APC into lymph nodes.
Keywords: Melanoma, Immunotherapy, Macrophages, Tumor antigen, Vaccination, Radiolabeling
Introduction
Certain peptidic sequences of melanoma cell proteins can complex with HLA class I molecules to form tumor antigens recognized by cytotoxic T lymphocytes (CTL). Based on this finding, vaccination therapies using professional antigen-presenting cells (APC) exposed to melanoma peptides have been developed. Two types of APC—macrophages [1] and dendritic cells (DC) [2]—have been used for this purpose. Both cell types can be derived in vitro from blood monocytes. Macrophages differentiate after the addition of macrophage colony-stimulating factor (M-CSF) or granulocyte-macrophage colony-stimulating factor (GM-CSF) to the culture medium, while immature DC are produced following the addition of GM-CSF plus IL-4 or GM-CSF plus IL-13 [3].
In previous work, we have compared macrophages with immature DC derived from the same individuals. Although DC express higher levels of HLA-DR than macrophages and are higher activators of mixed lymphocyte reactions, the two cell types are equally effective in activating CTL clones directed against melan-A and NA-17A melanoma peptides [4]. Moreover, DC and macrophages stimulate equally a CTL clone responding to MAGE-3(271–279), although this property is gradually lost when the time between peptide pulse and exposure of APC to CTL is increased [5].
The present work was aimed at evaluating the suitability of GM-CSF-induced macrophages for vaccination protocols. Patients' autologous macrophages (AM) were loaded in vitro with a MAGE-3(271–279) peptide and injected by various routes, including lymph vessels, with a view to evaluating the feasibility and safety of the treatment and to investigating the biodistribution of the cells. The route of injection of APC might be crucial for the induction of an immune response in the regional lymph nodes. Injecting APC into nodes or afferent lymph vessels, as proposed here, first would secure their recruitment and contact with T lymphocytes, and second would shorten the time between the antigen pulse of APC and activation of responding T cells. This factor may be important to consider, since the ability of peptide-pulsed APC to activate CTL drops rapidly and since the peak recruitment in draining lymph nodes of DC injected subcutaneously occurs after about 2 days [6, 7].
Materials and methods
Patients and treatment schedule
Patients with metastatic malignant melanoma were entered into this clinical trial, the end-point of which was to assess the safety and feasibility of the treatment. Eligibility criteria were informed consent, American Joint Committee on Cancer stage IV melanoma [8], at least one line of previous chemotherapy, HLA-A*02.01 tissue group expression, transcription of MAGE-3.A2 gene by the tumor, determined as described previously [9], age between 18 and 75 years, expected survival above 4 months, Karnofsky performance status more than 50, WBC count more than 2500/ml, platelet count more than 100,000/ml, and serum creatinine less than 150 mmol/l.
The treatment schedule included four therapeutic injections of AM suspension. Before these, the patients were injected intradermally with 0.1 ml AM, exposed or not to MAGE-3.A2 antigen, to evaluate reactivity to the antigen. After measuring the response 48 h later, the patients received the therapeutic injections, i.e. four 1-ml subcutaneous injections of AM suspension at the four limb roots. At 2 and 3 months, the patients received 1-ml injections of AM suspension under ultrasound guidance into four different non-melanoma-involved cervical, axillary or inguinal lymph nodes. At 4 months, 5–10 ml of cell suspension was injected into one superficial lymph vessel. Lastly, at 5 months, intradermal injection tests were performed, as above. The lymph vessel injection was adapted from the procedure developed for lymphography. Briefly, after subcutaneous injection of Patent Blue IV (Guerbet, Villepinte, France), a dye taken up by the lymphatic circulation, into the dorsal region of the foot, a vessel was catheterized under local anesthesia. The permeability of the lymph vasculature was checked by introducing an X-ray opacifier (Iopamiron, Schering, Berlin, Germany) before injecting the cell suspension, under controlled pressure (<1 bar). Before the intralymphatic injections, the patients were pretreated with hydroxyzine, midazolam and atropine sulfate. The number of macrophages injected was 106 for intradermal inoculations and all the cells available for other routes. The patients were maintained under observation for 24 h after the therapeutic injections. Before each injection, the patients were questioned as to the possible undesirable effects of the treatment. Whenever possible, measurable lesions were recorded and evaluated at each visit. Toxicity was evaluated by the NCI-CTC (version 2.0) scale. The trial was approved by the Ethical Review Committee (Comité Consultatif de Protection des Personnes dans la Recherche Biomédicale) of Brest and by the Agence Française de Sécurité Sanitaire des Produits de Santé.
Macrophage preparation
Mononuclear cells (MNC) separated by leukapheresis (Cobe Spectra separator) were processed under a controlled atmosphere and cultured in sterile bags, as done before [10]. The material included culture medium, elutriation buffer, sterile bags and tight circuit tubing, kindly provided by IDM (Paris, France). Briefly, platelet contamination was removed by three centrifugation cycles at 200 g and MNC were suspended at 5×106/ml in culture medium supplemented with 250 IU/ml GM-CSF (Leucomax, Schering-Plough) and 2% v/v autologous plasma enriched in calcium, as described by Musson and Henson [11]. Cells seeded in culture bags were pooled on day 7 and processed for elutriation, as described previously [10]. Bacteriological controls were performed on days 0 and 6 of the culture and on all cell suspensions before injection. For cell freezing, elutriation-purified AM were suspended in 4% human serum albumin (HSA) containing 10% dimethyl sulfoxide (DMSO), and aliquoted into seven bags (Hemofreeze), each containing 100 ml cell suspension. The freezing program was: 1°C per minute down to −40°C and 5°C per minute down to −140°C. The first AM bag was used after thawing for cell analysis and bacteriological testing; the others were used for patient treatment. The number and purity of AM injected were determined by Coulter counter analysis, since there was a very clear-cut size difference between AM and other cells.
Macrophage exposure to MAGE-3. A2 peptide
The MAGE-3.A2 peptide sequence 271–279 (flwgpralv) from UCB-Bioproducts (Braine-l'Alleud, Belgium) was suspended in distilled water at 5 mg/ml, aliquoted and kept at −80°C. After thawing, purified AM were suspended in 40 ml X-vivo-10 medium (Boehringer-Ingelheim) and incubated with MAGE-3(271–279) at a final concentration of 50 μg/ml for 2 h at 37°C. They were subsequently washed by centrifugation and resuspended in HSA in a volume adapted to that required for injection. In the case of i.d. injections, control cells were incubated without peptide. The cells were kept at room temperature and injected within 2 h. At various stages from leukapheresis to macrophage exposure to peptides, the cells were counted and viability was determined by trypan blue exclusion. The expression of CD3, CD14, CD16, CD45, CD64 and HLA-DR antigens (Beckman-Coulter monoclonal antibodies) was routinely determined by flow cytometry using isotypic controls to characterize the macrophage populations. Additional investigations were done with anti-CD40, CD80 and CCR7 monoclonal antibodies (Becton Dickinson).
Macrophage radiolabeling
Macrophages were labeled with 111In oxinate (Mallinkrodt, Peten, The Netherlands), as described previously [12]. Briefly, 5×106 to 2×107 cells suspended in 2 ml RPMI-1640 medium were incubated for 15 min with 10 to 15 MBq 111In oxinate with slow mixing. After washing, labeled cells were resuspended in HSA before injection. Cell biodistribution was studied using a gamma camera peaked for two photopeaks of 111In over 72 h. A sample of the 111In solution used for cell labeling was kept and used to calibrate radioactivity counting. Determining the biodistribution of AM was a secondary objective, and was carried out at most twice in a given patient during the six injections performed.
Results
Patient population investigated
The seven patients included had metastatic malignant melanoma, two were classified as M1 and five as M3 [8]. As shown in Fig. 1, all had been treated before with a median of two lines of chemotherapy. Only five patients received the four AM therapeutic injections scheduled due to the rapid progression of the disease, and only three patients received the last intradermal injections and could be evaluated for response to tumor antigen.
Fig. 1.
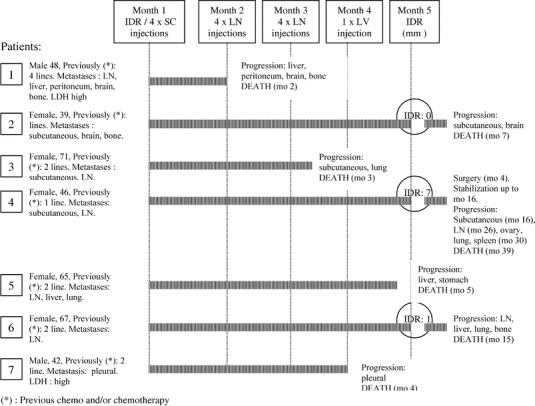
Schematic representation of the seven clinical cases investigated
Characteristics of cells injected
Relatively few MNC were collected per leukapheresis (mean 4.5×109, versus 6–8×109 in healthy volunteers). The mean number of AM separated by elutriation was 0.88×109. These cells (95% viable and 93% pure) were frozen. Thawing and washing of cells resulted in about 50% cell loss but did not markedly impair viability. On average, from one bag of frozen cells, 49.3±18×106 AM were recovered with 88% viability. Table 1 shows the phenotype of whole cell populations at various steps of AM preparation. The 7-day culture in the presence of GM-CSF did not significantly modify the percentage of cells that expressed the markers investigated, except CD16 which was increased. Elutriation, as expected, enriched the cell population in CD14+, CD64+, HLA-DR+ cells. Cryopreservation resulted in a diminution in CD16 expression. Importantly, AM preparations never expressed CCR7 (Fig. 2).
Table 1.
Phenotypic analysis of bulk cell populations at various steps in AM preparation: after leukapheresis, after a 7-day culture in the presence of GM-CSF, after separation by elutriation and after thawing of frozen purified AM
| % CD3+ | % CD14+ | % CD16+ | % CD64+ | % DR+ | |
|---|---|---|---|---|---|
| Apheresis product | 34.7±16.7 | 21.2±7.3 | 16.5±6.5 | 19.2 ±12.2 | 21.5±11.4 |
| End of 7-day culture | 32.4±32.1 | 26.8±9.8 | 31.5±13.0 | 28.0±12.0 | 29.1± 13.4 |
| After elutriation | 5.4±3.8 | 77.8±16.8 | 45.7±43.9 | 80.4±13.9 | 70.0±30.0 |
| Thawed frozen cells | 4.8±4.3 | 86.2±10.8 | 4.0±3.4 | 85.8±10.1 | 86.4±8.8 |
Fig. 2.
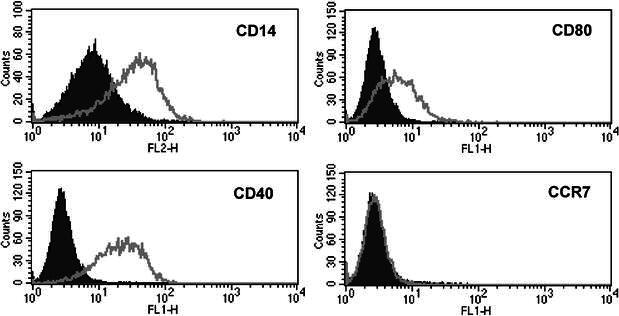
Staining of AM with various monoclonal antibodies. Note the negative reaction with anti-CCR7
Treatment toxicity
No acute toxicity was observed following the injections, especially in nodes and lymphatic vessels. Between the injections, only one grade 3 toxicity was observed: an Enterococcus septicemia that originated from a surgical adenectomy site. Grade 2 side effects were drug fever, vomiting, arthralgia, tumor pain and hyperglycemia. Other side effects were grade 1 or non-tumor-related.
Efficacy
All patients had a negative intradermal reaction (IDR) response to tumor antigen before vaccination. Of the three patients evaluable for IDR response after vaccination, the two M1 patients had an induration of 1 and 7 mm, respectively, and one M3 patient had no detectable response (Fig. 1). A total of 17 tumor sites were evaluated and only one minor response was observed in a patient otherwise progressing. The M1 patient with a 7-mm IDR (patient 4 in Fig. 1) had nodal and subcutaneous involvement which was stabilized and, after surgical removal of his lesions, stabilization lasted for 64 weeks with an overall survival of 128 weeks. The second M1 patient (patient 6) had a 64-week survival from the beginning of treatment. All other patients were in progression at the end of the treatment with a median time to progression of 11.5 weeks and a median overall survival of 20 weeks.
Scintigraphic imaging
When scintigraphic investigations were done, only one of the four subcutaneous or intranodal injections contained radiolabeled cells and was performed in the thigh or in one inguinal node. Likewise, radiolabeled AM not exposed to antigen were injected into the thigh, in contrast to IDR designed to investigate the immune response to the antigen performed in the forearm. In the case of lymph vessel injections, cell radiolabeling was performed whenever possible, but only a fraction of the cells injected were radioactive and mixed with unlabeled cells just prior to injection.
Table 2 shows that 11 injections of radiolabeled AM were performed. On average, 12.7×106 AM were labeled with a 69% labeling efficiency. The radioactivity injected averaged 2.8 MBq. Scintigraphic images suggested that macrophages injected intradermally, subcutaneously or intranodally did not diffuse greatly (Fig. 3). Radioactivity counts were performed on each site of injection 0.5 to 3 h after injection, then 1 day later. They showed losses of radioactivity of 2 to 10% after 24 h for subcutaneous and intradermal injections and possibly more (11 and 26%) after intranodal injections. This could correspond to a spontaneous isotope release from the cells. Marienhagen et al. [13] have calculated that 15% radioactivity is eluted within 24 h from 111In-labeled macrophages maintained in culture. In our hands, even maintained at 4°C, 111In-labeled macrophages released 3 to 10% radioactivity over 24 h.
Table 2.
Results of investigations performed in patients injected with 111In-labeled macrophages
| Injection route | Total number of injections | Injections with 111In-labeled cells | Results (loss of radioactivity after 24 h) |
|---|---|---|---|
| Intradermal | 10 | 2 | 2% and 10% |
| Subcutaneous | 7 | 2 | 4.5% and 8% |
| Intranodal | 12 | 2 | 11% and 26% |
| Intralymphatic | 5 | 5 | Distribution to several lymph nodes |
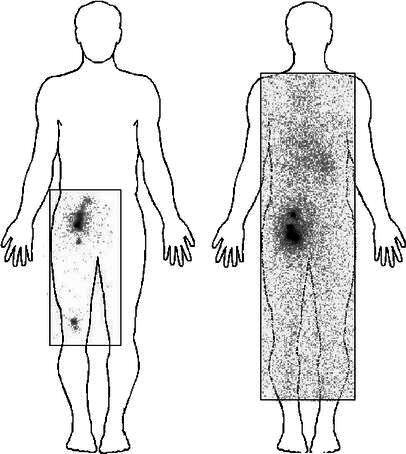
Fig. 3.
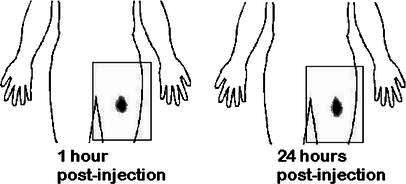
Scintigraphic imaging after intradermal administration of 111In-labeled antigen-loaded macrophages into the thigh. The human silhouette has been overlaid artificially. The rectangles represent the limits of the scintigraph
Scintigraphic imaging did not reveal any migration to draining lymph nodes after intradermal or subcutaneous injections (Fig. 3). Intranodal injections did not show diffusion to neighboring nodes (Fig. 4). In contrast, after injection into a lymph vessel, radioactivity clearly imaged five to ten popliteal and inguinocrural lymph nodes (Fig. 5). Images recorded at various times after injection suggested that AM adhered loosely to the lymph vessel, since variations were observed when comparing images at 2–3 h with later images (Fig. 6). The cells localized rapidly in the nodes and had not gained new nodal sites 2 h after injection. By comparison, the diffusion of the cells was much more restricted than that of the X-ray opacifier in lymphography, although the presence of a few cells in some lymph nodes, undetectable by the method used, cannot be excluded.
Fig. 4.
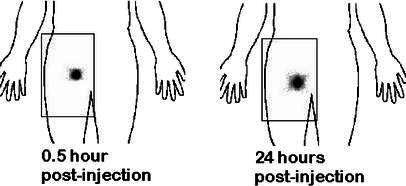
Scintigraphic imaging after intranodal injections of 111In-labeled macrophages. Note the slight local diffusion of radioactivity at 24 h
Fig. 5.
Scintigraphic imaging after injection of 111In-labeled macrophages into lymphatic vessels of the foot. Images taken 24 h after injection show that most radioactivity is retained in the inguinocrural area. In the anterior view, popliteal lymph nodes are visible. In the posterior view, a weakly contrasted picture shows that some—probably unbound—radioactivity has accumulated in the liver
Fig. 6A, B.
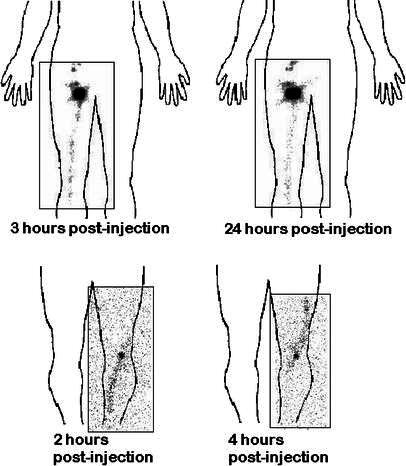
Progression of 111In-labeled macrophages in lymph vessels following intralymphatic injection. A Diminution of radioactivity of the lymph vessel 24 h after injection, and increase in the inguinal node. B Changes on the trajectory of a lymph vessel between 2 and 4 hours
Discussion
The present clinical trial shows that the safety of the treatment was excellent and that the feasibility was acceptable, although the injections into lymph vessels required a radiologist trained in lymphography technology. Intranodal injections were also awkward with small nodes. This trial was of the phase I type, and was not intended to evaluate the efficacy of the treatment. The seven patients included had a particularly poor prognosis and only one was stabilized. The two prolonged survivals observed (15 and 30 months) occurred in the two M1 patients who had IDR indurations of 1 and 7 mm, respectively. This observation is in agreement with the concept of higher efficacy of immunotherapy in regional or non-visceral metastasis [14].
One of the reasons why the trial was stopped after inclusion of only seven patients is that some doubt was cast on the validity of MAGE-3(271–279) as a tumor antigen. MAGE-3 was initially chosen because the gene is frequently transcribed in melanoma [15] and because the peptide induces specific CTL in vitro [16, 17]. After the beginning of the clinical trial, several reports highlighted the weak expression of MAGE-3(271–279) antigen on tumor cell lines. Some melanoma or esophageal cell lines express the antigen [18, 19], sometimes after treatment with interferon gamma [20], while others do not [21]. In a study by Labarrière et al. [22], only 2 of 14 lymph node-derived lymphocyte cultures sampled from melanoma patients lysed target cells loaded with MAGE-3(271–279), without interferon gamma secretion, while 8 of 20 cultures lysed cells expressing Melan-A/Mart-1 and produced interferon gamma. Valmori et al. [23] provided an explanation for these discrepant results by showing that the MAGE-3 sequence 271–279 is altered during the intracellular processing of the protein, except if the enzymatic activity of the proteasome is inhibited. Taken together these data did not encourage the use of MAGE-3(271–279) as a vaccinating peptide, although interesting clinical results have been obtained when combining the peptide with IL-12 injections [24]. MAGE-3(271–279), however, has immunizing properties in vivo [25], as shown in the present work, in the patient with a positive IDR response.
One of the main interests of this work was to compare the biodistribution of APC injected by various routes. In the mouse, a proportion of DC injected subcutaneously, but not those injected intravenously, reached the draining lymph nodes [6, 26]. In the monkey, monocyte-derived immature DC injected subcutaneously completely left the site of injection, in contrast with mature DC, but the same proportion of injected cells (about 1.5%) reached the draining lymph nodes [27, 28]. In humans, the biodistribution of monocyte-derived DC injected subcutaneously seems more uncertain. Morse et al. [29] observed that most immature 111In-labeled DC injected subcutaneously or intradermally remained at the site of injection. A small percentage of cells injected intradermally, but not those injected subcutaneously, migrated rapidly to the draining lymph nodes in two of four patients studied. Thomas et al. [30] also observed an inconstant and partial recruitment into lymph nodes of 99mTc-labeled immature DC injected intradermally. In the present work, no significant migration to the draining lymph nodes after subcutaneous or intradermal injection was found, and anyway the scintigraphic method used would have been unable to detect cells at percentages as low as 1.5%. Literature data indicate that the expression of CCR7 on the APC cell surface might condition the recruitment in lymph nodes [31]. Interestingly, AM did not express this antigen. This justified the intralymphatic and intranodal injections.
Intranodal inoculations showed a slight local diffusion of radioactivity on the scintigraphs, but without clear labeling of neighboring nodes. Although the injections were done under ultrasound guidance, the real localization of the cells injected into the nodes could not be ascertained, and presumably such injections perturbed lymph node intercellular relationships. Injection into an afferent lymph vessel probably respected DC physiology more and preserved lymph node anatomy. Interestingly, AM did not progress in the lymph vasculature at the same rate as the X-ray opacifier and did not cover the same diffusion territory. Scintigraphs taken at various times after injection suggested that AM adhered loosely to lymph vessel endothelium and firmly to the lymphatics that they reached. Comparable conclusions have been drawn by Mackensen et al. [32] who investigated DC prepared from CD34+ cells. After 99mTc labeling, these cells were injected into lymph vessels and the images produced were very similar to ours.
The present work confirms that the injection of APCs into lymph vessels is feasible and safe. The technique might be recommended for monocyte-derived DC vaccination protocols, since only a low or an undetectable proportion of these cells migrate to the draining lymph nodes after subcutaneous or intradermal inoculation. It is not known, however, whether massively increasing the number of cells recruited in the draining lymph nodes will improve immunization. However, experimental data suggest that intranodal is more efficient than subcutaneous injection and that the more cells injected the better [33].
Acknowledgements
We are grateful to Annette Gaudin, Anne Trichet and Sandrine Abgrall for their skilled technical assistance. This work was supported by the Comités départementaux d'Ille-et-Vilaine et du Morbihan de la Ligue nationale contre le Cancer.
References
- 1.Mukherji Proc Natl Acad Sci U S A. 1995;92:8078. [Google Scholar]
- 2.Nestle Nat Med. 1998;4:328. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 3.Morse J Immunother. 1999;22:506. doi: 10.1097/00002371-199911000-00005. [DOI] [PubMed] [Google Scholar]
- 4.Toujas Immunology. 1997;91:635. doi: 10.1046/j.1365-2567.1997.00287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lesimple Res Immunol. 1998;149:663. doi: 10.1016/S0923-2494(99)80036-4. [DOI] [PubMed] [Google Scholar]
- 6.Lappin Immunology. 1999;98:181. doi: 10.1046/j.1365-2567.1999.00850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hermans J Immunol. 2000;164:3095. [Google Scholar]
- 8.Balch Cancer. 2000;88:1484. doi: 10.1002/(SICI)1097-0142(20000315)88:6<1484::AID-CNCR29>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 9.Quillien Anticancer Res. 1997;17:387. [PubMed] [Google Scholar]
- 10.Lesimple J Immunother. 2000;23:675. doi: 10.1097/00002371-200011000-00009. [DOI] [PubMed] [Google Scholar]
- 11.Musson J Immunol. 1979;122:2026. [PubMed] [Google Scholar]
- 12.Quillien Cancer Immunol Immunother. 2001;50:477. doi: 10.1007/s002620100224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marienhagen Nucl Med Commun. 1995;16:357. doi: 10.1097/00006231-199505000-00007. [DOI] [PubMed] [Google Scholar]
- 14.Boon Melanoma Res. 2001;11:538. [Google Scholar]
- 15.Brasseur Int J Cancer. 1995;63:375. doi: 10.1002/ijc.2910630313. [DOI] [PubMed] [Google Scholar]
- 16.Van Eur J Immunol. 1994;24:3038. doi: 10.1002/eji.1830241218. [DOI] [PubMed] [Google Scholar]
- 17.Tjandrawan J Immunother. 1998;21:149. [Google Scholar]
- 18.Kawashima Hum Immunol. 1998;59:1. doi: 10.1016/S0198-8859(97)00255-3. [DOI] [PubMed] [Google Scholar]
- 19.Kanaoka J Surg Oncol. 1999;71:16. doi: 10.1002/(sici)1096-9098(199905)71:1<16::aid-jso4>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 20.Takaki Int J Oncol. 1998;12:1103. doi: 10.3892/ijo.12.5.1103. [DOI] [PubMed] [Google Scholar]
- 21.Chaux J Immunol. 1999;163:2928. [PubMed] [Google Scholar]
- 22.Labarriere Int J Cancer. 1998;78:209. doi: 10.1002/(SICI)1097-0215(19981005)78:2<209::AID-IJC15>3.3.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 23.Valmori J Exp Med. 1999;189:895. doi: 10.1084/jem.189.6.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gajewski TF, Fallarino F, Ashikari A, Sherman M (2001) Immunization of HLA-A2+ melanoma patients with MAGE-3 or MelanA peptide-pulsed autologous peripheral blood mononuclear cells plus recombinant human interleukin. Clin Cancer Res 7 [3 Suppl]:895s [PubMed]
- 25.Schuler-Thurner J Immunol. 2000;165:3492. doi: 10.4049/jimmunol.165.6.3492. [DOI] [PubMed] [Google Scholar]
- 26.Eggert Cancer Res. 1999;59:3340. [PubMed] [Google Scholar]
- 27.Barratt-Boyes J Immunol. 1997;158:4543. [PubMed] [Google Scholar]
- 28.Barratt-Boyes Adv Exp Med Biol. 1997;417:71. doi: 10.1007/978-1-4757-9966-8_12. [DOI] [PubMed] [Google Scholar]
- 29.Morse Cancer Res. 1999;59:56. [PubMed] [Google Scholar]
- 30.Thomas J Melanoma Res. 1999;5:474. doi: 10.1097/00008390-199910000-00007. [DOI] [PubMed] [Google Scholar]
- 31.Sallusto Immunol Rev. 2000;177:134. doi: 10.1034/j.1600-065x.2000.17717.x. [DOI] [PubMed] [Google Scholar]
- 32.Mackensen Cancer Immunol Immunother. 1999;48:118. doi: 10.1007/s002620050555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lambert Cancer Res. 2001;61:641. [PubMed] [Google Scholar]