

Abstract
l-Ascorbic acid (vitamin C) has been reported to play a role in the treatment and prevention of cancer. However, its specific mechanistic pathways remain obscure. This study was carried out to identify the sodium ascorbate–induced apoptotic pathway in B16F10 murine melanoma cells. Sodium ascorbate was found to induce the apoptosis of B16F10 murine melanoma in a time- and dose-dependent manner, and this was prevented by pretreatment with N-acetyl-l-cysteine (NAC), a well-known antioxidant. In fact, sodium ascorbate–treated B16F10 melanoma cells showed increased intracellular reactive oxygen species generation (ROS) levels. These results indicate that sodium ascorbate induced apoptosis in B16F10 murine melanoma cells by acting as a prooxidant. We examined the involvement of caspase-8 using a specific caspase-8 inhibitor (z-IETD-fmk) on the sodium ascorbate–induced apoptotic pathway. Cell death was found not to be inhibited by z-IETD-fmk treatment, indicating that sodium ascorbate–induced apoptosis is not mediated by caspase-8. In addition, we detected a reduction in the mitochondrial membrane potential during apoptosis and confirmed cytochrome-c release from mitochondria by immunoblotting. Taken together, it appears that the induction of a prooxidant state by sodium ascorbate and a subsequent reduction in mitochondrial membrane potential are involved in the apoptotic pathway of B16F10 murine melanoma cells, and that this occurs in a caspase-8–independent manner.
Keywords: Vitamin C, Melanoma, Apoptosis
Introduction
Melanoma is one of the most malignant tumors in humans. They escape from immune surveillance and spread more rapidly than any other tumors using several mechanisms including MHC down-regulation, increasing ROS levels, and increasing FasL expression [6, 11, 12, 18]. Sodium ascorbate can increase its susceptibility to immune surveillance and cause the induction of melanoma death through the modulation of some of these mechanisms [20, 23, 24].
Cell death can be categorized as being due to necrosis and apoptosis. Necrosis does not require any new proteins [1]. In contrast, apoptosis is executed by the activation of cytosolic cystein proteases, the caspases, and it shows violent blebbing of the nucleus, DNA fragmentation, and apoptotic body formation as its characteristics [22, 31]. Among 14 caspases, caspase-8 is known to act as an initiator of the apoptotic process by activating downstream effector caspases (caspase-3, -6, and -7) directly [19, 27] or by decreasing the mitochondrial membrane potential, which is followed by the secretion of cytochrome c and apoptosis-inducing factor (AIF) [16, 32, 33].
Sodium ascorbate enhances the human immune system by increasing interferon production and phagocytic activity [25, 29]. Furthermore, it is highly toxic especially to malignant melanoma [2], but the specific mechanism is still uncertain. It is only accepted that sodium ascorbate inhibits the invasion and growth of malignant melanoma, in the presence of metal ions [3, 4, 17]. In this study, therefore, we attempted to determine the sodium ascorbate–induced apoptotic pathway in B16 murine melanoma cells.
Materials and methods
Cells
The B16F10 murine melanoma cells used in this study were maintained in continuous log phase growth and cultured in RPMI 1640 medium supplemented with 2 mM l-glutamine, 100-U/ml penicillin, 100-μg/ml streptomycin, and 10% heat-inactivated fetal bovine serum (FBS), hereafter referred to as complete medium (CM).
Induction and measurement of apoptosis
B16F10 melanoma cells treated with or without sodium ascorbate were washed twice with cold phosphate buffered saline (PBS), and then resuspended in ×1 binding buffer at a concentration of 1×106 cells/ml. One hundred microliters of the solution (1×105 cells) was transferred to a 5-ml culture tube. Then, 5 μl of Annexin V-FITC was added, and cells were incubated at room temperature for 15 min in the dark with gentle vortexing. Then, 400 μl of ×1 binding buffer was added to each tube. The Annexin V-FITC apoptosis detection kit was purchased from Pharmingen (San Diego, CA).
Detection of reactive oxygen species generation (ROS)
B16F10 melanoma cells (1×104 cells/well) were incubated in a 96-well plate. Cells were then further incubated with 50 μM of 2',7'-dichlorofluorescein diacetate (DCFH-DA; Eastman Kodak, Rochester, NY) at 37°C. After incubation, cells were analyzed with a Cytofluor 2350 plate reader (Millipore, Bedford, MA) with excitation at 485 nm and emission at 530 nm.
Inhibitor studies
Jurkat and B16F10 melanoma cells (1×106 cells/ml) were pretreated for 30 min at 37°C with z-IETD-fmk. Targets were treated with antihuman Fas Ab (2 ng/ml) and 10 mM of sodium ascorbate for 24 h at 37°C, and apoptosis was determined using an Annexin V-FITC apoptosis detection kit.
Measurement of mitochondrial membrane potential
B16F10 melanoma cells (1×106 cells/ml) treated with or without 10 mM of sodium ascorbate were incubated for 4 h, washed twice with cold PBS, and then 0.1 μM of DiOC6 was added. Cells were incubated at 37°C for another 15 min, and analyzed by flow cytometry.
Analysis of cytochrome-c release
Jurkat cells (5x106 cells/ml) were treated with antihuman Fas Ab (2 ng/ml), and B16F10 melanoma cells (5x106 cells/ml) were treated with 10 mM of sodium ascorbate for 24 h at 37°C. The cells were then washed twice with PBS and resuspended in 200 μl of digitonin lysis buffer (75-mM NaCl, 1-mM NaH2PO4, 8-mM Na2HPO4, 250-mM sucrose, 190-μg/ml digitonin). Digitonin is a weak nonionic detergent, which at low concentrations selectively renders the plasma membrane permeable, causing it to release cytosolic components but to leave other organelles intact. After 5 min on ice, the cells were spun for 5 min at 14,000 rpm and 4°C in a microcentrifuge. Supernatants were transferred to fresh tubes and the concentration of cytosolic proteins was measured by the BCA method. Thirty micrograms of each sample was then added to SDS-loading buffer (0.5 M Tris-HCl [pH 6.8], 1 M 2-ME, 10% [w/v] SDS, 10% [v/v] glycerol, 0.05% [w/v] bromophenol blue) and boiled for 10 min. The boiled samples were then loaded into 15% polyacrylamide gels, electrophoresed, and transferred to nitrocellulose membranes. Membranes were blocked overnight at 4°C in PBS containing 0.1% (v/v) Tween-20 and 5% (w/v) nonfat milk proteins. Blocked membranes were then incubated with a monoclonal antihuman cytochrome-c Ab (1 μg/ml) for 1 h at room temperature, washed three times (5 min each) with PBS containing 0.1% Tween-20, and incubated with a HRP-conjugated antimouse IgG secondary Ab (1:5,000). Cytochrome c was detected on blots by enzyme-linked chemiluminescence.
Statistical analysis
The statistical significance was determined by using ANOVA at p<0.05. All results were given as a 2-tailed p value.
Results
Sodium ascorbate induces the apoptosis of B16F10 murine melanoma cells
It is known that sodium ascorbate has a cytotoxic effect on malignant melanoma cells in the presence of metal ions [3, 4, 17]. First, we investigated whether sodium ascorbate can induce the apoptosis of B16F10 murine melanoma cells. The induction of apoptosis was assessed by phosphatidylserine externalization using FITC-annexin V binding. The data shown in Fig. 1, confirms that apoptosis was induced from 1 h after treatment with sodium ascorbate and peaked at 3 h. Even though sodium ascorbate is known to act as a prooxidant in the presence of metal ions, its role as a prooxidant has not been clarified. To clarify this, we measured the intracellular reactive oxygen species (ROS) levels in B16F10 murine melanoma cells after treatment with sodium ascorbate. Sodium ascorbate was found to increase intracellular ROS levels (Fig. 2A), and sodium ascorbate–induced apoptosis was found to be completely blocked by pretreatment with N-acetyl-l-cysteine (Fig. 2B). Collectively, these results show that sodium ascorbate induces the apoptosis of B16F10 murine melanoma cells and that in this respect it behaves as a prooxidant.
Fig. 1.

Kinetics of sodium ascorbate–induced apoptosis. Cells were incubated with media (control) or sodium ascorbate (10 mM) for various time periods as indicated, at 37°C in a 5% CO2 humidified incubator. After incubation, cells were harvested and apoptosis was detected by Annexin V-FITC analysis. Results are representative of more than five experiments
Fig. 2A, B.
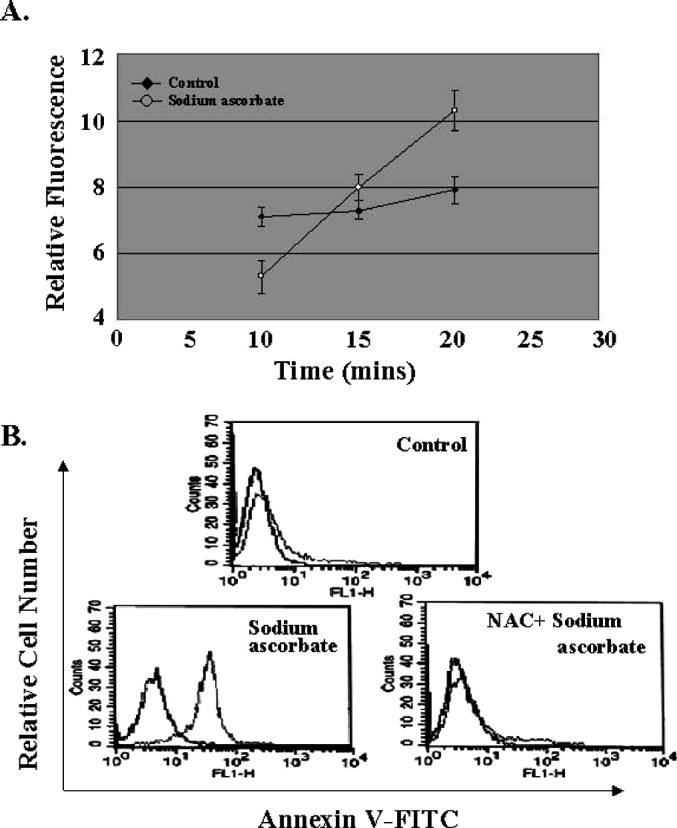
Effect of sodium ascorbate on apoptosis as a prooxidant. A Effect of sodium ascorbate on ROS levels of B16F10 melanoma cells. Cells (2×104 cells/well) were placed in wells of a 96-well microtiter plate followed by an addition of 50-μM 2', 7'-dichlorofluorescein diacetate at 37°C in a 5% CO2 humidified incubator, and then analyzed with a Cytofluor 2350 plate reader with excitation at 485 nm and emission at 530 nm. Results are representative of more than five experiments, each performed in triplicate. The data are shown as the mean ± SD. B Effect of the antioxidant, N-acetyl-l-cysteine on sodium ascorbate–induced apoptosis. B16F10 melanoma cells were incubated with medium (control), sodium ascorbate (10 mM), or sodium ascorbate/N-acetyl-l-cysteine (10 mM) for 4 h. After incubation, cells were collected and stained with Annexin V-FITC to detect apoptosis of melanoma cells. Results are representative of more than five experiments
Caspase-8 is not involved in sodium ascorbate–induced apoptosis
It is now widely accepted that the apoptotic events occurring within target cells arise after the proteolytic activation of endogenous cysteine proteases called caspases [19, 27]. Of the 14 members of this family, caspase-8 plays an important role in the initiation of apoptosis. To examine the involvement of caspase-8 in sodium ascorbate–induced apoptosis in melanoma, we made use of a specific inhibitor of caspase-8 (z-IETD-fmk). In general, it is known that anti-Fas Abs induce the apoptosis on Jurkat cells activating caspase-8 [21]. In contrast, sodium ascorbate–induced apoptosis was not blocked by z-IETD-fmk pretreatment (Fig. 3). However, we detected the activation of the effector caspase, caspase-3 (data not shown). Therefore, our results suggest that sodium ascorbate induces the apoptosis of B16F10 murine melanoma cells in a caspase-8–independent manner.
Fig. 3A–F.
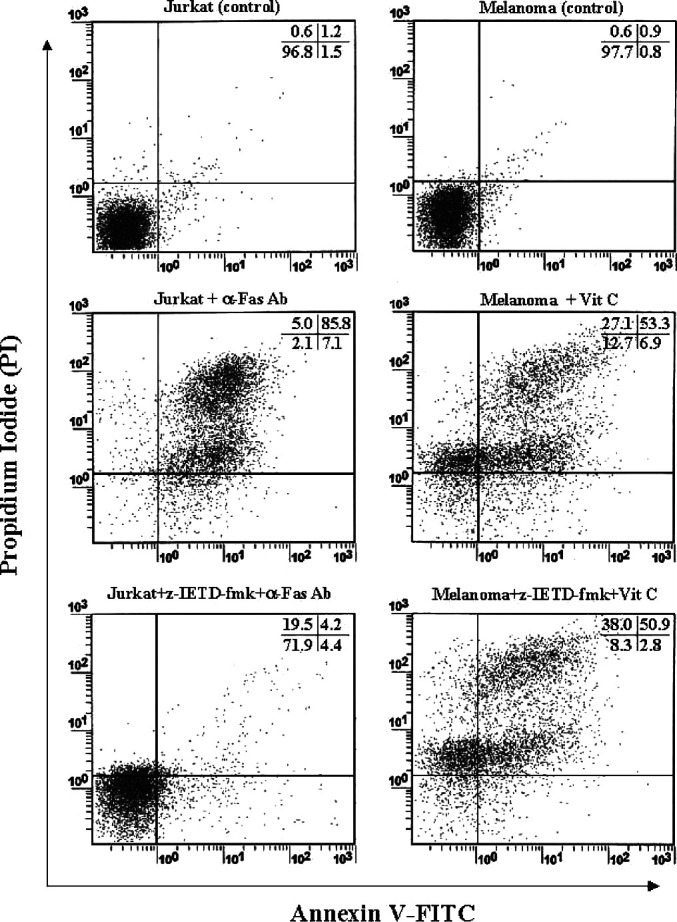
Caspase-8 is not involved in the sodium ascorbate–induced apoptosis. Jurkat and B16F10 melanoma cells (1×106 cells/ml) were pretreated for 30 min at 37 C with z-IETD-fmk (20 mM). Following this incubation, targets were treated with antihuman Fas Ab (2 ng/ml) for 24 h at 37°C and 10 mM of Sodium ascorbate for 4 h at 37°C, and then apoptosis was determined using Annexin V-FITC apoptosis detection kit. A Jurkat cells (control), B Jurkat cells + anti-Fas Ab (2 ng/ml), C Jurkat cells + z-IETD-fmk (20 mM) + anti-Fas Ab (2 ng/ml), D B16F10 melanoma cells (control), E B16F10 melanoma cells + sodium ascorbate (10 mM), F B16F10 melanoma cells + z-IETD-fmk (20 mM) + sodium ascorbate (10 mM). Results are representative of more than five experiments
Sodium ascorbate induces the loss of mitochondrial membrane potential (Δψm) and the release of cytochrome c in melanoma (B16F10 cells)
The loss of Δψm is a common early event of apoptosis induced by a variety of stimuli [9, 10, 14]. We monitored the integrity of Δψm during the apoptosis induced by sodium ascorbate using the potential-sensitive dye DiOC6 by flow cytometry [14]. As shown in Fig. 4A, no leakage of DiOC6 was observed in the absence of sodium ascorbate. However, we confirmed the leakage of DiOC6 and the shift of peak fluorescence intensity in the melanoma cells treated with sodium ascorbate. Thus, we conclude that sodium ascorbate induces the disruption of Δψm, during the apoptotic process. Cytosolic cytochrome c promotes the formation of a complex with Apaf-1 and procaspase-9, called apoptosome, and then facilitates the cleavage and activation of caspase-9 and caspase-3 [16]. Thus, the release of cytochrome c from mitochondria appears to be an essential apoptotic event, which occurs after the loss of mitochondrial membrane potential. It has been reported previously that cytochrome c is released from Jurkat cells after treatment with anti-Fas MAb [21] and we confirmed that in our results. In addition, we detected the release of cytochrome c from melanoma after exposure to sodium ascorbate (Fig. 4B). Taken together, our results show that sodium ascorbate induces the apoptosis of B16F10 murine melanoma by disrupting Δψm and causing the release of cytochrome c.
Fig. 4.
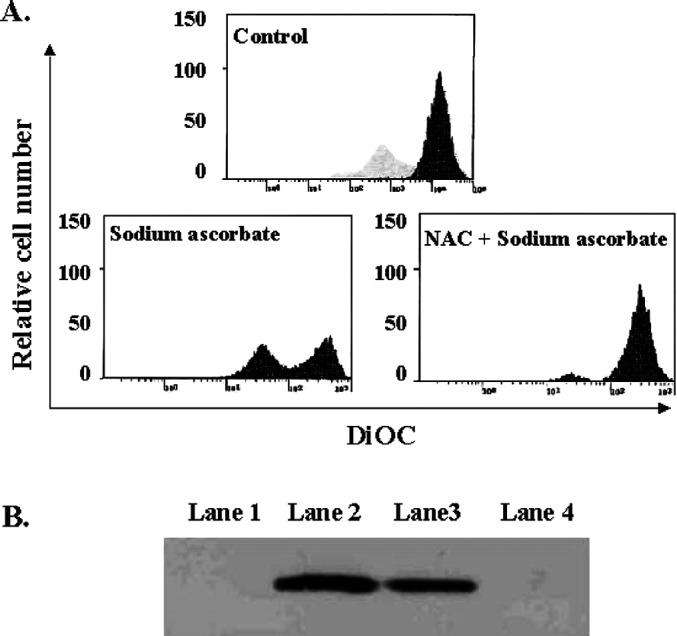
Sodium ascorbate induces apoptosis on B16 murine melanoma cells via the decrease of mitochondrial potential and the release of cytochrom C. A. Measurement of mitochondrial membrane potential: 10 mM of N-acetyl-l-cysteine pretreated or untreated B16F10 melanoma cells were cultured with 10 mM of sodium ascorbate and then stained with DiOC6. Results are representative of three experiments. B Cytochrome-c release induced by sodium ascorbate; Jurkats were incubated without (Lane 1) or with (Lane 2) anti-Fas Ab for 24 h at 37°C. B16F10 murine melanoma cells were incubated with (Lane 3) or without (Lane 4) 10 mM of sodium ascorbate for 4 h at 37°C. Cells were washed in PBS and resuspended in digitonin lysis buffer for 5 min on ice followed by centrifugation. Fifty microliters of cytosolic proteins in SDS-loading buffer were loaded onto 15% polyacrylamide gels. Following SDS-PAGE, proteins were transferred to nitrocellulose membranes and probed with a monoclonal anti-cytochrome-c Ab (1 μg/ml). After the addition of a biotin conjugated-antimouse Ab (1:5,000), proteins were detected using ECL. Results are representative of more than five experiments
Discussion
L-ascorbic acid (vitamin C) has been reported to play a role in the treatment and the prevention of cancer, especially melanoma [2, 3, 4, 8]. Even though many efforts have been devoted to discover its mode of action, it is still obscure. It has previously been reported that sodium ascorbate enhances prostaglandin synthesis, which inhibits melanoma growth [7]. In this study, we attempted to clarify the specific mechanism of cell death induced by sodium ascorbate, and we found that sodium ascorbate induces the apoptosis of B16F10 murine melanoma by disrupting Δψm and causing the release of cytochrome c.
We had already confirmed the cytotoxic effect of sodium ascorbate against B16F10 in our previous study (data not shown). In addition, we also confirmed that sodium ascorbate induces apoptotic cell death in B16F10 murine melanoma cells by confirming apoptotic features, such as phosphatidylserine externalization as shown in Fig. 1. This means that sodium ascorbate might not act as an antioxidant, but as a prooxidant, because it readily scavenges nitrogen species and reactive oxygen such as hydrogen peroxide, hydroxyl radical, and superoxide anion when it acts as a primary antioxidant in plasma and within a cell. It may thereby block oxidative damage to important biological molecules including DNA, proteins, and lipids. As evidence of this, intracellular reactive oxygen species (ROS) levels were shown to be increased in B16F10 murine melanoma cells after treatment with sodium ascorbate (Fig. 2A). Indeed, the blockade of the apoptotic cell death by pretreatment of N-acetyl-l-cysteine (NAC), a well-known antioxidant, helps explain the role of sodium ascorbate as a prooxidant (Fig. 2B).
It has been known that there are two kinds of apoptotic pathway, type I and type II. The former is mediated by the activation of caspase-8 and the latter is mediated by decreased mitochondria membrane potential [13]. Sodium ascorbate–induced apoptosis was not blocked by pretreatment of the caspase-8 specific inhibitor (z-IETD-fmk). This suggests that it does not act through the caspase-8–dependent pathway. It is believed that cytochrome c released from mitochondria is linked with the onset of apoptosis induced by a variety of stimuli, including TNF, Fas, and chemotherapeutic drugs [9, 10, 14]. In accord with this finding, sodium ascorbate was found to be associated with a loss of Δψm and the release of mitochondrial cytochrome c. In addition, the loss of Δψm was nearly blocked by pretreatment of N-acetyl-l-cysteine (NAC). Therefore, this also strongly supports the role of sodium ascorbate as a prooxidant on the induction of apoptosis in B16F10 murine melanoma cells. Taken together, our findings suggest that sodium ascorbate induces apoptosis in a caspase-8–independent manner in B16F10 melanoma cell lines and that cytochrome c is a key mediator of this pathway.
Here we discovered that sodium ascorbate–induced apoptosis is mediated via the release of cytochrome c from mitochondria. However, we do not exclude the possibility of the presence of other molecules that induce the release of cytochrome c, since there are recent reports concerning several other apoptosis-mediated molecules that are released from mitochondria prior to the release of cytochrome c. These molecules include Endonuclease G, Smac/DIABLO, and HtrA2 [5, 15, 26, 28]. Endonuclease G (Endo G) is a mitochondria-specific nuclease that translocates to the nucleus during apoptosis. Once released from mitochondria, Endo G cleaves chromatin DNA into nucleosome fragments independently of caspases [15]. Smac/DIABLO and HtrA2 belong to the serine protease family and are released from mitochondria upon receiving apoptotic stimuli, and then bind to inhibitor of apoptosis (IAP) [5, 26, 28]. Therefore, these species induce atypical cell death, which is neither accompanied by a significant increase in caspase activity nor inhibited by caspase inhibitors. In the case of sodium ascorbate–induced apoptosis, we can exclude the possible involvement of Endo G, since we have confirmed caspase-3 activation after sodium ascorbate treatment, but we have not excluded the possible involvement of Smac/DIABLO or HtrA2. In addition, it has been reported that caspase-10 is another initiator of apoptosis [30], so we also did not exclude the involvement of caspase-10 in sodium ascorbate–induced apoptosis. Therefore, further investigation regarding the involvement and roles of Smac/DIABLO, HtrA2, and caspase-10 on the sodium ascorbate–induced apoptotic cell death in B16F10 melanoma is needed.
We conclude that sodium ascorbate induces the apoptosis of B16F10 murine melanoma cells through the loss of Δψm and the release of cytochrome c in a caspase-8–independent manner as a prooxidant.
Acknowledgment
This study is supported by the grant from Korea Eundan Company
Abbreviations
- NAC
N-acetyl-l-cysteine
- ROS
reactive oxygen species
- Δψm
mitochondrial membrane potential
Footnotes
Jae Seung Kang and Daeho Cho contributed equally to this work.
References
- 1.Afansev FEBS Letter. 1986;194:347. doi: 10.1016/0014-5793(86)80114-4. [DOI] [Google Scholar]
- 2.Bram Nature. 1980;284:629. doi: 10.1038/284629a0. [DOI] [PubMed] [Google Scholar]
- 3.De Anticancer Res. 1990;10:391. [PubMed] [Google Scholar]
- 4.De Anticancer Res. 1990;10:1029. [PubMed] [Google Scholar]
- 5.Du Cell. 2000;102:33. [Google Scholar]
- 6.Ferrone Immunol Today. 1995;16:487. doi: 10.1016/0167-5699(95)80033-6. [DOI] [PubMed] [Google Scholar]
- 7.Gardiner Prostaglandins Leukot Essent Fatty Acids. 1988;34:119. doi: 10.1016/0952-3278(88)90073-7. [DOI] [PubMed] [Google Scholar]
- 8.Gardiner J Nutr. 1989;119:586. doi: 10.1093/jn/119.4.586. [DOI] [PubMed] [Google Scholar]
- 9.Green Science. 1998;281:1309. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 10.Green D, Kroemer G. The central executioners of apoptosis: caspases or mitochondria? Trends Cell Biol. 1998;8:267. doi: 10.1016/s0962-8924(98)01273-2. [DOI] [PubMed] [Google Scholar]
- 11.Hahne Science. 1996;274:1363. [Google Scholar]
- 12.Huber J Immunol. 1998;148:277. [PubMed] [Google Scholar]
- 13.Krammer Nature. 2000;407:789. doi: 10.1038/35037728. [DOI] [PubMed] [Google Scholar]
- 14.Kroemer, Immunol Today. 1997;18:44. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- 15.Li Nature. 2001;412:95. [Google Scholar]
- 16.Liu Cell. 1996;86:147. [Google Scholar]
- 17.Marczewska Acta Pol Pharm. 2000;57:415. [PubMed] [Google Scholar]
- 18.Mukherji Curr Opin Oncol. 1995;7:175. doi: 10.1097/00001622-199503000-00014. [DOI] [PubMed] [Google Scholar]
- 19.Nicholson Trends Biochem Sci. 1997;22:299. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- 20.Nungester J Infect Dis. 1948;83:50. doi: 10.1093/infdis/83.1.50. [DOI] [PubMed] [Google Scholar]
- 21.Peter Curr Opin Immunol. 1998;10:545. doi: 10.1016/s0952-7915(98)80222-7. [DOI] [PubMed] [Google Scholar]
- 22.Savill Immunol Today. 1993;14:131. doi: 10.1016/0167-5699(93)90215-7. [DOI] [PubMed] [Google Scholar]
- 23.Siegel Nature. 1975;254:531. doi: 10.1038/254531a0. [DOI] [PubMed] [Google Scholar]
- 24.Stockinger Int J Occup Health Saf. 1977;46:54. [Google Scholar]
- 25.Stone Med Hypotheses. 1980;6:309. doi: 10.1016/0306-9877(80)90128-0. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki Mol Cell. 2001;8:613. doi: 10.1016/s1097-2765(01)00341-0. [DOI] [PubMed] [Google Scholar]
- 27.Thornberry Science. 1998;281:1312. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 28.Verhagen J Biol Chem. 2002;277:445. doi: 10.1074/jbc.M109891200. [DOI] [PubMed] [Google Scholar]
- 29.Victor Immunopharmacology. 2000;46:89. doi: 10.1016/S0162-3109(99)00162-9. [DOI] [PubMed] [Google Scholar]
- 30.Wang Proc Natl Acad Sci USA. 2001;98:13884. doi: 10.1073/pnas.241358198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wyllie J Pathol. 1998;142:67. [Google Scholar]
- 32.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Pen T-I, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 33.Zoratti Biochim Biophys Acta. 1995;1241:139. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]