

Abstract
The therapeutic unconjugated anti-CD20 Mab rituximab is used for the treatment of B-non-Hodgkin’s lymphomas. We have studied the direct biological effects, signalling and gene expression profiles induced by rituximab in two human B-lymphoma cell lines, DHL4 and BJAB, using microarray, quantitative PCR and gel shift analysis. Rituximab alone inhibited thymidine uptake and induced homotypic adhesion in DHL4 only, but not BJAB. Analysis of Affymetrix microchips carrying probes for about 10,000 human cDNAs, allowed us to identify 16 genes in DHL4 and 12 in BJAB induced by rituximab at 4 h. Eleven and seven of these genes were specific for DHL4 and BJAB, respectively; whereas the remaining five were up-regulated in both cell lines. Mean induction ranged from 2- to 16-fold. Real time PCR analysis allowed us to confirm up-regulation of all genes identified, except one in BJAB. Time course of induction of eight genes was studied, showing peak induction in most cases at 4 h. The up-regulation of 5/5 genes was also observed with the F(ab′)2 fragment of rituximab. Analysis of three further B-cell lymphoma lines showed that gene induction is not restricted to BJAB and DHL4. Finally, we show that rituximab alone can induce AP1 activation in both cell lines and provide evidence that the ERK1/2 pathway is involved in the rituximab-mediated up-regulation of gene expression. These data demontrate that rituximab alone has direct signalling capacity in different B-lymphoma lines, inducing distinct but overlapping sets of genes which may play a role in the biological and/or therapeutic effect of the antibody.
Keywords: AP1, CD20, Lymphoma, Microarray, Rituximab
Introduction
The monoclonal antibody rituximab is a chimeric anti-CD20 IgG1κ antibody used successfully in the treatment of B-non-Hodgkin’s lymphomas (B-NHLs) and more recently in antibody-mediated autoimmune diseases [6, 10, 14, 24, 46]. Rituximab activates efficiently both complement and antibody-dependent cellular cytotoxicity (ADCC) [8, 21, 39], and these immune-mediated mechanisms are thought to be mainly responsible for the rapid depletion of normal B cells in vivo and for the therapeutic activity of the antibody against lymphoma cells [3, 5, 13]. However, other factors may play a role, as suggested by the direct effects on normal or neoplastic B cells of anti-CD20 antibodies, including rituximab, in terms of apoptosis and/or inhibition of proliferation. Indeed the murine anti-CD20 MAb B1, as well as rituximab, has been shown to block normal B-cell proliferation in vitro, following stimulation with polyclonal B-cell mitogens [21, 23, 50], or to inhibit proliferation of some lymphoma cell lines in vitro [41, 48]. Furthermore, B1 and to a weaker extent rituximab have been reported to induce apoptosis, although in the case of rituximab, cross-linking appears to be required [4, 17, 33, 35, 43]. Recent evidence however indicates that the apoptosis induced by anti-CD20 antibodies is partial and characterised by membrane and mitochondrial permeability changes but not DNA fragmentation [4, 53], raising questions about the nature of the signalling pathway induced by the antibody in B-lymphoma cells. Interestingly, recent evidence suggests that the therapeutic activity of B1 in a xenograft lymphoma model does not require complement activation or NK cells but rather may depend on its signalling effects [7]. The signalling capacity of other Mabs has also been reported to contribute significantly to their therapeutic activity in vivo, suggesting that activation of CDC and ADCC may not be sufficient for the therapeutic efficacy of unconjugated Mabs [51].
The direct functional effects of anti-CD20 MAbs on neoplastic and/or normal B cells imply the triggering of a signalling cascade by the CD20 molecule, although the actual signalling pathway(s) leading to these biological responses have not been fully defined: CD20 is a highly hydrophobic molecule carrying four membrane-spanning domains and is thought to form tetramers that directly or indirectly control Ca++ fluxes across the plasma membrane [49]. Antibody binding to CD20 leads to redistribution of the molecule to insoluble membrane rafts [38], resulting in association with the PAG adaptor protein and src family kinases [11, 12, 37, 42]. The role of this redistribution into membrane rafts in CD20-mediated signalling and in the biological response of cells to anti-CD20 antibodies is still unclear [8, 12, 44]. CD20 has also been reported to be associated with MAPK pathway activation [33, 35].
We have studied here the signalling pathway induced by rituximab in lymphoma cells, using two different cell lines as targets and microarray gene chip analysis. We identify two different but overlapping sets of genes induced by rituximab, which may play a role in the biological effects and/or therapeutic activity of the antibody.
Materials and methods
Cell culture and reagents
The DHL4 and WSU-NHL follicular lymphoma, and BJAB and EsIII Burkitt’s lymphoma cell lines have been described elsewhere [21, 22]. The Raji cell line was a kind gift of Prof. D.H. Crawford, Edinburgh University, Scotland. All cell lines were grown in RPMI 1640 medium (Seromed, Berlin, Germany) supplemented with 2 mM glutamine (Life Technologies, Paisley, Scotland), 10% fetal calf serum (Hyclone, Logan, UT, USA) and 100 U/ml penicillin/streptomycin (complete medium). They were maintained in exponential growth in a 5% CO2 incubator.
Thymidine uptake
A total of 104 cells/well were plated in triplicates in flat-bottomed 96-well plates (Falcon, Le-Pont-de-Chaix, France) in 200 μl complete medium in presence or absence of 1–100 μg/ml of the antibodies rituximab (a kind gift of Roche Italia), daclizumab (Zenapax, humanised anti-hIL-2Rβ; Roche, provided by Dr A. Rambaldi, Ospedali Riuniti, Bergamo, Italy), campath-1H (humanised anti-CD52 antibody, a kind gift of Schering, Rome, Italy), rituximab F(ab′)2 fragment (a kind gift of Prof. E.S. Vitetta, University of Texas, Dallas, TX, USA) and in some cases antihuman IgG1 (Fc-specific MAb, clone HP6001; Sigma) diluted 1:500. After 48 h, cells were pulsed for 16 h with 0.5 μCi [3H]-thymidine per well (Amersham Pharmacia Biotech, Little Chalfont, Bucks, UK) and collected in a cell harvester (Tomtec, Hamden, CT, USA).
Apoptosis assay
Cells were plated at 8×104/ml in presence or absence of 10 μg/ml rituximab. Cells were collected at 48 or 96 h, and fixed in 70% ethanol. Apoptosis was measured using the in situ cell death detection assay kit (Roche Diagnostics, Monza, Italy) following the manufacturer’s instructions. Analysis was performed on a FACstarPlus cytometer (Becton Dickinson).
Homotypic adhesion
Cells were plated in triplicates at 2.5×103 cells/well in flat-bottomed 96-well plates in presence or absence of 10 μg/ml rituximab. Homotypic adhesion was evaluated 24 h later by microscopic examination, and by counting the number of aggregates of >10 cells in at least five different randomly selected fields/condition.
RNA preparations for microarrays and PCR
Exponentially growing DHL4 or BJAB cells were plated at 10×106 /ml for microarray experiments and at 1×106/ml for PCR assays, in 6-well plates. Rituximab, daclizumab or campath-1H were added at 10 μg/ml. In some experiments the inhibitors PD98059 and PD169316 (Calbiochem) were added at 10 μM and 30 μM 10 min before the addition of rituximab. Cells were collected at different times, centrifuged and lysed in guanidinium isothiocyanate solution. Total RNA was purified by standard cesium chloride gradient centrifugation, as described previously [20]. RNA was precipitated in 100 mM NaCl and 70% ethanol, air-dried, resuspended at 5 μg/ml in RNase-free water. RNA quantity and quality were verified in all cases by running an aliquot in a standard 1% agarose/formaldehyde gel.
Microarrays
The cRNA ‘targets’ were generated as described by the Affymetrix Expression Analysis Gene Chip Technical Manual, protocol P/N 900218 rev. 2, P/N 70021 rev. 3. Briefly, double-stranded cDNA was synthesized using the SuperScript Choice System (Life Technologies, Paisley, UK) and a primer containing poly(dT) and a T7 RNA polymerase promoter sequence (MWG). In vitro transcription using double-stranded cDNA as a template in the presence of biotinylated UTP and CTP was carried out using BioArray High Yield RNA Transcript Labeling Kit (Enzo Diagnostic, Farmingdale, NY, USA). Biotinylated cRNA was purified, fragmented and hybridized to the Affymetrix HG-U95av2 chips (Affymetrix, Santa Clara, CA, USA), containing 12,625 probe sets specific for mostly full-length genes. The cRNA was detected with streptavidin-phycoerythrin (Molecular Probes, Eugene, OR, USA), and analysis was completed using a Hewlett-Packard Gene Array Scanner (Affymetrix). Furthermore the 20x eukaryotic hybridization control kit was included, as recommended by the manufacturer (Affymetrix Data Analysis Fundamentals manual). The signal for each probe set was quantified using Micro Array Suite 5.0 software (Affymetrix). The recommended internal controls and quality checks were performed: briefly, average background and noise were verified to be comparable between different experiments; the percentage of probes scored as present were similar in treated and untreated samples (28.4% and 32% for DHL4, and 33.2% and 26.8% for BJAB, respectively); the ratios of 3’ and 5’ probe sets for GAPDH and actin were below 2; finally the scaling factors were comparable between different arrays. The default parameters were used for the statistical algorithm and for probe set scaling using Microarray Suite 5.0 (with a target intensity of 100). The data were then filtered so that the absolute value of the fold change was equal or more than 2. Additionally, genes that were scored as absent in experimental and baseline files were removed. The statistical significance in the comparison analyses was calculated using the Wilcoxon signed rank test as described in the Affymetrix Data Analysis Fundamentals manual. The default cutoff p value of 0.0025 was used. The sorting for robust change (≥ twofold) was chosen to minimise false positives.
Annotations about individual genes were obtained in the Net Affx Analysis Center (http://www.affymetrix.com) [32]) and the NCBI site (http://www.ncbi.nlm.nih.gov; OMIM and PubMed).
Real-time PCR
Total RNA (4 μg) was reverse transcribed in a final volume of 80 μl (5 mM Mg2Cl, 10 mM Tris-HCl, pH 8.3, 50 mM KCl, 1 mM dNTPs, 0.5 U/μl RNase inhibitor, 5 U/μl M-MuLV Reverse Transcriptase [Roche Diagnostics, Monza, Italy], 2.5 μM random hexamers) for 45 min at 42°C, followed by 5 min at 95°C. Oligonucleotides were designed using the Primer Express 1.5 software (Applied Biosystems). They were as follows:
| DUSP2 | 5′-GGAGGCCATAGGCTTCATTGA-3′ |
| 5′-AGGTATGCCAGACAGATGGTGG-3′ | |
| RGS2 | 5′-AAATCACCACAGAGCCTCATGC-3′ |
| 5′-TATGGCAGGTCACAGTCCTTCC-3′ | |
| NR4A1 | 5′-TGCCAATCTCCTCACTTCCCT-3′ |
| 5′-CCAGCATCTTCCTTCCCAAAG-3′ | |
| DDIT3 | 5′-AACCAGCAGAGGTCACAAGCAC-3′ |
| 5′-TCCTGGTTCTCCCTTGGTCTTC-3′ | |
| ID3 | 5′-TGCTGGACGACATGAACCACT-3′ |
| 5′-ATGACGCGCTGTAGGATTTCC-3′ | |
| CD83 | 5′AGAGAGGATGGAGACACCCCA-3′ |
| 5′-TGATAGTGCTGTCCCCTGAGGT-3′ | |
| TIEG | 5′-GCTTCCGGGAACACCTGATT-3′ |
| 5′-GGAGTCAAACAAAATGCTGGGA-3′ | |
| ZFP36 | 5′-TTCGCCCACTGCAACCTC-3′ |
| 5′CATAGCTCAGTCTTGTAGCGCG-3′ | |
| JUNB | 5′-CAGGCTCGGTTTCAGGAGTTT-3′ |
| 5′GATGTGCACTAAAATGGAACAGC-3′ | |
| ETR101 | 5′-GCCAAAGTCAGCCGCAAA-3′ |
| 5′-TCTTCTTCCTTTTCTTCCAGACG-3′ | |
| TGOLN2 | 5′-CACGGGTAGCGAGAAGGATG-3′ |
| 5′-CTGCCATTTCCAGAACCGTT-3′ | |
| VEGF | 5′- TGGATGTCTATCAGCGCAGC-3′ |
| 5′-TGTCCACCAGGGTCTCGATT-3′ | |
| SGK | 5′- GGTGGCAATTCTCATCGCTT-3′ |
| 5′-TCGTTCAGACCCATCCTCCT-3′ | |
| INSIG1 | 5′-GCGCTGCATAGCAGTTTTTG-3′ |
| 5′-TCCAATTTAGCACTGGCGTG-3′ | |
| FOS | 5′-TCAACGCGCAGGACTTCTG-3′ |
| 5′-TGAAGTTGGCACTGGAGACG-3′ | |
| MAPK6 | 5′-GAAGAGCATGCCAGGCTTTT-3′ |
| 5′-ATACTTGAGCCCCCGTAGCA-3′ | |
| ZPF36L1 | 5′- TGCAGGAAGGAAGGCTGAAA-3′ |
| 5′- TCCATCTAGCTTCCCCTTCCTC-3′ | |
| DSIPI | 5′-AGGGTGCTTGGCTTAATCCC-3′ |
| 5′-TCCTCGTTTCTGAAGCAGGG-3′ | |
| DDX11 | 5′-TTTTGGAGGCTGGCAAGATT-3′ |
| 5′-TCCCAGTGCCAGTTGGACTC-3′ | |
| ADM | 5′-GCATCCGAGTCAAGCGCTAC-3′ |
| 5′-GGAGGCCCTGGAAGTTGTTC-3′ | |
| β-ACTIN | 5′-CCCAAGGCCAACGCGAGAAGAT-3′ |
| 5′-GTCCCGGCCAGCCAGGTCCAG-3′ |
A total of 2 μl reverse transcriptase product was amplified in triplicate in a 30 μl reaction using the Sybr Green Master Mix (Applied Biosystems) and 300 nM of each oligonucleotide. Amplification reactions were carried out in 96-well optical plates and GeneAmp 5700 Sequence Detection System (Applied Biosystems), according to the manufacturers’ instructions. To normalise for differences in cDNA contents between different samples, all samples were normalised by amplification of the β-actin gene. The calculations of the fold difference in gene expression and normalisations were carried out according to the manufacturer’s instructions (Applied Biosystems, User Bulletin No. 2): Fold change in gene expression was calculated according to the formula: 2−(ΔCT test−ΔCT control), where ΔCT is the mean cycles for the test gene—mean cycle time for β-actin for the same sample. Standard curves were generated for each gene analysed in test experiments and were found to differ from the theoretical value by less than 5% in all cases, justifying the use of the above formula for all amplifications. Furthermore, controls were performed with RNA preparations amplified in absence of reverse transcriptase to verify the absence of DNA contamination.
Statistical analysis was performed using Prism 4.0 (Graph Pad software, San Diego, CA, USA) and performing Student’s t tests.
Electrophoretic mobility shift assays (EMSA)
A total of 10×106 exponentially growing DHL4 cells was resuspended in 1 ml fresh medium in presence of 10 μg/ml rituximab, daclizumab or phorbol myristic acetate (PMA, 20 ng/ml; Sigma). After 3 h, cells were collected, washed two times in x1 PBS and lysed in 300 μl lysis buffer (25 mM HEPES, pH 7.8, 50mM KCl, 0.2% NP40, 0.1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml pepstatin A, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM sodium orthovanadate) at 4°C for 5 min. The nuclei were centrifuged at 3,000 rpm for 5 min at 4°C and extracted in 40 μl extraction buffer (25 mM HEPES, pH 7.8, 500 mM KCl, 10% glycerol, 0.1 mM dithiothreitol, and the phosphatase and protease inhibitors as above). Extracted nuclei were centrifuged at 13,000 rpm for 10 min and supernatant aliquoted and stored at −70°C. Protein content was measured using the BioRad protein assay (BioRad) and serial dilutions of BSA.
The annealed AP1 consensus oligonucleotides (dGTGTGATGACTCAGGTTTCC) were labelled with 32P-dCTP and Klenow enzyme according to standard procedures. The labelled AP1 probe was purified on a 1 ml Sephadex G-50 column (Amersham Pharmacia Biotech) equilibrated in Tris-EDTA buffer containing 0.1% SDS. The binding reaction was carried out for 15 min at room temperature using 5 μg nuclear extract and 50,000 cpm probe in 40 mM Tris-HCl, pH 7.5, 120 mM KCl, 4 mM ethylenediaminetetraacetic acid (EDTA), 1 mM DTT, 8% Ficoll 400 and 1 μg polydIdC (Amersham Pharmacia Biotech). In some cases, 1 μl anti-c-fos (clone 4) or anti-jun antibodies (clone D; Santa Cruz, Heidelberg, Germany) or a 50-fold excess cold competitor DNA were added to the binding reaction. Binding reactions were separated in a 5% acrylamide gel in x0.5 Tris-borate-EDTA gel according to standard procedures. After running about 2 h at 150 V, the gels were dried and exposed to X-AR5 films (Kodak).
Results
Rituximab inhibits thymidine uptake and induces homotypic adhesion in the DHL4 but not BJAB cell line
To investigate the signalling pathways triggered by rituximab, we chose two cell lines, DHL4 and BJAB. DHL4 had been reported previously to respond to several anti-CD20 antibodies, in terms of proliferation and/or apoptosis in vitro [17, 41]. Both cell lines express high and comparable levels of CD20 (210,000 and 170,000 molecules/cell, respectively, as quantified by direct immunofluorescence using calibrated beads, data not shown). As shown in Fig. 1a (solid circles), rituximab inhibited thymidine uptake in the DHL4 cell line by 30–60% in a dose-dependent manner (p<0.001, with 10 μg/ml rituximab in three experiments). By contrast, BJAB cells (Fig. 1a, open circles) were unresponsive to up to 100 μg/ml rituximab in the same experimental conditions (p>0.05). That the inhibition of thymidine uptake was specific was shown by the lack of effect of other two humanised IgG1 antibodies daclizumab (anti-IL2-Rβ) and campath-1H (anti-CD52) (Fig.1b). The latter binds to DHL4 cells, which are CD52-positive (data not shown).
Fig. 1.

Rituximab inhibits thymidine uptake by DHL4 but not BJAB cells. a Exponentially growing DHL4 (solid circles) or BJAB lymphoma cells (open circles) were plated in presence or absence of increasing concentrations of rituximab and [3H]-thymidine uptake was measured at 64 h. b DHL4 cells were cultured in presence or absence of 10 μg/ml rituximab, daclizumab or campath-1H. c DHL4 cells were plated in presence or absence of 10 μg/ml rituximab and/or monoclonal antihuman IgG1 (1:500). d DHL4 cells were plated in presence or absence of 10 μg/ml rituximab, the rituximab F(ab′)2 fragment or campath-1H antibody. [3H]-Thymidine uptake was measured at 64 h in all cases. All results shown are the mean and standard deviation of triplicate or quadruplicate wells and are representative of three independent experiments.
Since some authors have suggested that cross-linking of anti-CD20 antibodies may increase their biological effects [17, 33, 43], we wanted to investigate this point further. To carry out cross-linking selectively, we used a monoclonal antihuman IgG1 Fc antibody (clone HP6001). This clone was shown by indirect immunofluorescence to bind to rituximab specifically but not to the sIgG of DHL4 cells (data not shown) and was therefore used in cross-linking experiments. As shown in Fig. 1c, antihuman IgG antibody alone had no effect on thymidine uptake by DHL4, nor did it significantly affect the inhibition induced by rituximab (p>0.05). These data suggest that rituximab cross-linking is not required to inhibit thymidine uptake in DHL4 cells.
We then determined whether the F(ab′)2 fragment of rituximab was effective at inhibiting thymidine uptake. Strong inhibition, to levels equivalent to those observed with the whole antibody, was observed also with F(ab′)2 (Fig. 1d). These data demonstrate that the effect is dependent on CD20 signalling.
Since the decrease in thymidine uptake can be due to either inhibition of proliferation or induction of apoptosis or both, we measured apoptosis at different times after rituximab exposure by TUNEL assay. As shown in Table 1, we could not detect significant apoptosis induced by rituximab. These data are consistent with previous data using the same cell line and in absence of cross-linking [21, 41], suggesting that rituximab inhibits proliferation of these cells.
Table 1.
Apoptosis assay (TUNEL)
| Time |
Mean percentage apoptotic cells (SD) | |
|---|---|---|
| Control | + Rituximab | |
| 48 h | 3.7 (2) | 6.1 (1.6) |
| 96 h | 5.0 (1.1) | 6.6 (1.8) |
During these experiments, we observed that rituximab alone also induced homotypic adhesion of DHL4 cells, but not BJAB, leading to formation of large cell aggregates (Fig. 2). This effect took at least 8–12 h to become observable and was best measured at 24 h. Like thymidine uptake, homotypic adhesion took place also in presence of the F(ab′)2 fragment but not in presence of control campath-1H antibody (data not shown), suggesting a role for CD20 signalling in this response. Altogether these data show that rituximab binding to CD20 induces different biological responses in different lymphoma lines, leading to homotypic adhesion and inhibition of thymidine uptake in the DHL4 but not BJAB cell line.
Fig. 2.

Rituximab induces homotypic adhesion of DHL4 but not BJAB cells. Exponentially growing DHL4 or BJAB cells were plated in presence or absence of 10 μg/ml rituximab or control antibody daclizumab. Cells were examined microscopically 24 h later. a DHL4 + daclizumab, b DHL4 + rituximab, c BJAB + daclizumab, d BJAB + rituximab. The results are representative of at least five experiments.
Pattern of genes induced by rituximab in DHL4 and BJAB
To try to dissect the molecular events that follow rituximab binding to lymphoma cells, we have investigated the pattern of genes whose expression is induced or repressed by the antibody. DHL4 and BJAB cells were treated with 10 μg/ml rituximab or with the same dose of control antibody daclizumab. Cells were collected at 4 h in two separate experiments with each cell line and additionally at 8 h in one experiment with DHL4 and RNA extracted. RNA quality and quantity were verified in all cases on standard agarose/formaldehyde gels (data not shown). The biotinylated cRNA ‘targets’ were generated and used to hybridise Affymetrix gene chips carrying 12,625 human probe sets representing about 10,000 mostly full-length genes (HG-U95A chips). The results of paired samples were analysed using the MAS 5.0 software. The quality of the probes and hybridisation was verified as described in ‘Materials and methods’ and was found to be within the expected values. We opted for an analysis of robust changes, i.e. of probe sets induced or repressed at least twofold in all 3 DHL4 experiments or in both BJAB experiments and with a p value for change below 0.0025, as recommended. The results are shown in Tables 2 and 3. A total of 16 genes in DHL4 and 12 genes in BJAB were found to be reproducibly induced in these conditions, with mean p values for change in all cases less than 0.001 (generally <0.0001) (Tables 2 and 3). No gene was consistently repressed by ≥twofold in either cell line.
Table 2.
Results of microarray analysis following rituximab stimulation (DHL4)
| Gene | GenBank accession number | Fold increase | Mean fold change (SD) | Mean p value for changea | Mean signal (SD) | Functionb | ||
|---|---|---|---|---|---|---|---|---|
| Experiment 1 (4 h) | Experiment 2 (4 h) | Experiment 3 (8 h) | ||||||
| RGS2 (GOS8) | L13463 | 8.6 | 3.7 | 4.0 | 5.4 (2.7) | <0.0001 | 1,425 (752) | ST |
| DUSP2 (PAC1) | NM_004418 | 11.3 | 2.6 | 5.3 | 6.4 (4.4) | <0.0001 | 1,240 (691) | ST |
| ETR101 | NM_004907 | 3.7 | 6.1 | 3.7 | 4.5 (1.3) | <0.0001 | 1,343 (543) | ST |
|
NR4A1 (TR3) 2 probe setsc |
L13740 | 9.2 | 7.5 | 2.3 | 4.7 (3) | <0.0001 | 368 (164) | TF |
| 4.0 | 3.5 | 1.7 | ||||||
| ZFP36 | NM_003407 | 2.8 | 3.7 | 3.3 | 3.3 (0.5) | <0.0001 | 502 (231) | TF |
| MAPK6 (Erk3) | NM_002748 | 2.5 | 2.1 | 2.0 | 2.2 (0.2) | <0.0001 | 240 (37) | ST |
| JunB | NM_002229 | 4.9 | 3.0 | 2.6 | 3.5 (1.2) | <0.0001 | 306 (56) | TF |
| FOS | NM_005252 | 29.9 | 2.5 | 14.9 | 15.8 (13.7) | 0.0003 | 47 (11) | TF |
| DDIT3 (GADD153)2 probe setsc | NM_004083 | 5.3 3.7 | 24.3 5.3 | 11.3 5.7 | 9.3 (7.8) | <0.0001 | 731 (563) | TF |
| ID3 | NM_002167 | 2.8 | 3.7 | 2.8 | 3.1 (0.5) | <0.0001 | 987 (326) | TF |
| TIEG | NM_005655 | 3.7 | 2.3 | 3.0 | 3.0 (0.7) | <0.0001 | 212 (72) | TF |
| ZFP36L1 (Erf1) | NM_004926 | 2.5 | 4.0 | 2.3 | 2.9 (0.9) | 0.0008 | 386 (82) | TF |
| DSIPI | NM_004089 | 12.1 | 4.3 | 3.7 | 6.7 (4.7) | <0.0001 | 213 (67) | TF |
| VEGF | NM_003376 | 2.1 | 3.0 | 2.0 | 2.4 (0.6) | <0.0001 | 320 (185) | SP |
| CD83 (HB15) | NM_004233 | 2.6 | 2.5 | 2.3 | 2.5 (0.2) | <0.0001 | 430 (73) | MP |
| INSIG1 | NM_198336 | 2.0 | 2.6 | 2.1 | 2.3 (0.3) | <0.0001 | 703 (256) | MP |
ap Values for change were obtained as described in the Affymetrix Data Analysis Fundamentals manual (http://www.affymetrix.com)
b ST signal transduction protein, TF transcription factor/DNA-RNA regulator, MP membrane, SP secreted protein
cFor DDIT3 and NR4A1, the results obtained with two different probe sets are shown. Mean fold induction and mean signals in this case refer to both probe sets
Table 3.
Results of microarray analysis following rituximab stimulation (BJAB)
| Gene | GenBank accession number | Fold increase | Mean fold change (SD) | Mean p value for changea | Mean signal (SD) | Functionb | |
|---|---|---|---|---|---|---|---|
| Experiment 1 (4 h) | Experiment 2 (4 h) | ||||||
| RGS2 (GOS8) | L13463 | 4.6 | 2.6 | 3.6 (1.0) | <0.0001 | 1879 (659) | ST |
| DUSP2 (PAC1) | NM_004418 | 5.7 | 5.7 | 5.7(0.0) | <0.0001 | 1252 (303) | ST |
| ETR101 | NM_004907 | 2.3 | 2.6 | 2.5 (0.2) | <0.0001 | 2056 (247) | ST |
|
NR4A1 (TR3) 2 probe setsc |
L13740 | 4.6 | 5.7 | 4.2 (1.2) | <0.0001 | 253 (193) | TF |
| 3.0 | 3.5 | ||||||
| ZFP36 | NM_003407 | 3.2 | 3.7 | 3.4 (0.2) | <0.0001 | 322 (47) | TF |
| CCL3 (MIP 1α) | D90144 | 11.3 | 4.3 | 7.8 (3.5) | <0.0001 | 3786 (1832) | SP |
| CCL4 (MIP 1β) | NM_002984 | 12.1 | 2.6 | 7.4 (4.8) | <0.0001 | 1129 (914) | SP |
| DDX11 (CHLR1) | U75968 | 2.1 | 2.0 | 2.0 (0.1) | 0.00011 | 232 (63) | TF |
| TGOLN2 (TGN48) | NM_006464 | 13.9 | 3.2 | 8.5 (5.3) | 0.00011 | 101 (27) | MP |
| RGS1 (BL34) | NM_002922 | 8.0 | 2.0 | 5.0 (3.0) | 0.0005 | 384 (148) | ST |
| SGK | AJ000512 | 6.5 | 2.6 | 4.5 (2.0) | <0.0001 | 385 (4) | ST |
| ADM | NM_001124 | 3.0 | 2.3 | 2.6 (0.3) | <0.0001 | 219 (30) | SP |
ap Values for change were obtained as described in the Affymetrix Data Analysis Fundamentals manual (http://www.affymetrix.com)
b ST signal transduction protein, TF transcription factor/DNA-RNA regulator, MP membrane, SP secreted protein
cFor DDIT3 and NR4A1, the results obtained with two different probe sets are shown. Mean fold induction and mean signals in this case refer to both probe sets
Interestingly, 5 genes (RGS2, DUSP2, ETR101, NR4A1 and ZFP36) were up-regulated in both DHL4 and BJAB (indicated in bold in Tables 2 and 3), whereas another 11 genes were up-regulated specifically in DHL4, and 7 only in BJAB. Of note is that in some cases two different probe sets for the same gene were found induced (NR4A1 and DDIT3), strengthening the validity of the data obtained (Tables 2 and 3). Mean fold induction ranged from two to nearly sixteen. Tables 2 and 3 also report the mean signals for rituximab-treated samples. These varied from a mean signal of 47 (FOS in DHL4) to one of 3786 (CCL3 in BJAB) (Tables 2 and 3). Thus rituximab has direct signalling capacity in both BJAB and DHL4, as demonstrated by the distinct although overlapping sets of genes induced in these two cell lines.
The genes in Tables 2 and 3 have been organised by function. The vast majority are transcription factors/RNA regulators (TF; ten different genes) or signal transduction molecules (ST: kinases, phosphatases, G protein inhibitors; six genes). In addition seven membrane or secreted proteins (MP/SP) were found to be induced.
We then searched for further information on all genes identified, and the results are summarised in Table 4. The transcription factors belonged to several different families, including bZIP (DDIT3), zinc fingers (ZFP36, ZPF36L1, TIEG), steroid receptor family (NR4A1), jun/fos family (JunB, FOS) and inhibitory members of the HLH family (ID3) (Table 4, second column). Among signal transduction molecules, three of the genes induced are regulators of G protein signalling: RGS1, RGS2 [26] and DUSP2 (the last is a phosphatase that inhibits MAPK signalling [54]) (Table 4). Annotations about their biological functions showed that 12 of the 16 genes induced in DHL4 and 7 of the 12 up-regulated in BJAB are involved in the control of cell growth and/or apoptosis (Table 4). Also of particular interest from their functional point of view is the induction of vascular endothelial growth factor (VEGF) in DHL4 and adrenomedullin (ADM), both secreted proteins that promote angiogenesis [16], and induction of CCL3 (MIP-1α) and CCL4 (MIP-1β) in BJAB, which are C-C chemokines that attract neutrophils and NK cells.
Table 4.
Functions and expression of genes induced by rituximab
| Gene | Molecular function | Biological function | Expression |
|---|---|---|---|
| BJAB+DHL4 | |||
| RGS2 | Regulator of G protein signalling Gq inh. (GTPase) | Growth control oncogenesis | Immediate-early, mitogen-induced |
| DUSP2 | Dual-specificity protein phosphatase, inhibitor of MAPK | p53 dep. apoptosis anti-proliferative | Induced by MEK/ERK, by mitogens mostly haematopoietic |
| ETR101 | Putative TF, weak homology to jun family | Not known | Immediate-early, PMA + stress-induced |
| NR4A1 | TF, steroid receptor family | Proliferation proapoptotic | Ca-dependent, mitogen + PMA-induced |
| ZFP36 | Zinc finger protein, regulator of TNF-α+GM-CSF mRNA (destabilisation) | Inflammation | Mitogen-induced |
| DHL4 | |||
| MAPK6 | Ser/thr kinase, erk1 related | Proliferation | PMA + mitogen-induced |
| JunB | TF, AP1 family | Tumour suppressor | NFκB-induced |
| FOS | bZIP TF, component of AP-1 | Proliferation differentiation | Immediate-early, mitogen-induced |
| DDIT3 | bZIP TF C/EBP inhibitor | Proapoptotic | Stress-induced gene induced by MAPK+AP1 |
| ID3 | HLH TF inhibitor | B-cell proliferation development | Mitogen-induced through ERK pathway |
| TIEG | Zinc finger TF | Proapoptotic antiproliferative | Immediate-early, TGF-β1 + mitogen-induced |
| ZFP36L1 | Zinc finger TF | Proliferation development | PMA+EGF-induced |
| VEGF | Growth factor | Angiogenesis proliferation | Hypoxia- and GF-induced (MAPK pathway) |
| CD83 | Membrane protein, adhesion receptor of SIGLEC family | Immune response | Activated B cells + DCs |
| INSIG1 | ER protein | Cholesterol homeostasis proliferation | Insulin + proliferation-induced |
| DSIPI | Leucine zipper protein, trascriptional regulator | Anti-inflammatory immunosuppressive | IL-10–stimulated macrophages |
| BJAB | |||
| TGOLN2 | Trans-golgi network glycoprotein | Secretion | Ubiquitous |
| CCL3 | C-C chemokine | Chemotaxis | Immune cells, activated B cells |
| CCL4 | C-C chemokine | Chemotaxis | Immune cells, activated B cells |
| RGS1 | Regulator of G protein signalling GTPase activator | Growth control immune response | Immediate-early gene B cells, monocytes |
| SGK | Ser/thr kinase | Apoptosis Na+ transport | Serum/glucocorticoid-induced, ubiquitous |
| ADM | Soluble peptide | Angiogenesis, cell survival | Hypoxia- and activation-induced in T cells and epith. |
| DDX11 | DNA helicase | Cellular growth and division | Ubiquitous |
Interestingly, the vast majority of induced genes (17/23) are known to be up-regulated by PMA, mitogens or MAPK signalling in different cell types including B cells [1, 2, 15, 19, 25–27, 29, 30, 45, 47]. In addition, three have been shown to be stress/hypoxia-induced genes (DDIT3, ETR101, ADM) (Table 4, fourth column) [28].
Characterisation of gene induction using quantitative PCR analysis
To verify directly that gene chip analysis reliably identified rituximab-induced genes in DHL4, we tested all induced genes by quantitative PCR. With a view to validating both the biological and technical reproducibility of gene regulation, the RNA used for these analyses was not the same as that used for gene chip analysis, but was derived from independent stimulation experiments with rituximab using both DHL4 and BJAB. All samples were normalised for β-actin expression and at least two separate experiments were performed for each gene. The results are presented graphically in Fig. 3. All 16 tested genes identified in DHL4 were confirmed to be significantly induced using real-time PCR techniques, with fold induction ranging from 1.7- to 15-fold and p values below 0.01 in all cases (Fig. 3a, two to seven experiments performed for each gene). In BJAB, 11 of the 12 genes were found to be significantly induced (two to five experiments performed for each gene, p<0.05). Only one, RGS1, was not confirmed to be up-regulated in two independent PCR experiments (Fig.3b). Thus we can deduce that gene chip analysis reliably identified gene induction in over 95% of cases analysed.
Fig. 3.

Real-time PCR analysis of genes induced by rituximab. Exponentially growing DHL4 (a) or BJAB (b) were plated in presence or absence of 10 μg/ml rituximab, and total RNA was extracted after 4 h. Fold induction of the indicated genes by rituximab over control was determined by real-time PCR using specific oligonucleotides (striped bars) and are compared with the fold inductions obtained by chip analysis (black bars). The results of PCR analysis are the mean and standard deviation of two to seven independent experiments.
Further characterisation of selected genes was carried out. The choice of the genes for further work was based on their potentially interesting biological activity, on the fact that they were induced in both cell lines or at relatively high levels. Thus, using three genes in DHL4 (NR4A1, DUSP2 and RGS2) and two specific for BJAB (CCL3 and CCL4), we first determined whether induction was dependent upon binding of the antibody to CD20. For this purpose, cells were treated with either rituximab whole IgG or its F(ab′)2 fragment for 4 h and RNA was analysed by real-time PCR. As shown in Fig. 4, all five tested genes were up-regulated to similar levels by both rituximab and F(ab′)2.
Fig. 4.

Both rituximab whole antibody and F(ab′)2 fragment can up-regulate gene expression. Exponentially growing DHL4 (a) or BJAB (b) cells were plated in presence of 10 μg/ml rituximab or its F(ab′)2 fragment. Total RNA was extracted after 4 h. Fold induction of the indicated genes by either rituximab or F(ab′)2 fragment over control was determined by real-time PCR using specific oligonucleotides. The results shown are the mean and standard deviation obtained in two independent experiments.
A time course of five genes in DHL4 and seven genes in BJAB was then performed by real-time PCR to determine whether induction was transient or sustained. The results of analysis at 1, 4 and 24 h for DHL4, and additionally at 48 h for BJAB, are shown in Fig.5a, b. They demonstrate that most genes are induced with a peak expression at 4 h. One gene, ZFP36, peaked at 24 h (Fig.5b). Several genes, including DUSP2, CCL3 and ETR101, still have sustained up-regulation at 24 h (2.5- to 10-fold over control), whereas expression of the other genes was more transient (Fig 5a, b).
Fig. 5.

Time course of gene expression after exposure to rituximab. Exponentially growing DHL4 (a) or BJAB (b) cells were plated in presence or absence of 10 μg/ml rituximab antibody, and total RNA was extracted at 1, 4, 24 or 48 h. Induction of the indicated genes by rituximab relative to control antibody was determined by real-time PCR using specific oligonucleotides. The results are the mean and standard deviation of triplicate samples.
To investigate the pattern of gene induction in a wider range of B-lymphoma cell lines, we have stimulated the WSU-NHL, ESIII and RAJI cell lines with rituximab in the same conditions as those used for DHL4 and BJAB. RNA was extracted at 4 h and analysed by quantitative PCR. A subset of genes was tested on these cells representative of the three patterns observed, namely three of the genes that had been found induced in both DHL4 and BJAB (NR4A1, RGS2 and DUSP2), four genes induced in DHL4 only (ERK3, ID3, FOS and JunB) and two genes induced specifically in BJAB (CCL3 and CCL4). Quantitative PCR was carried out as before with normalisation with β-actin. The results are shown in Fig. 6 and demonstrate that gene induction is not restricted to DHL4 and BJAB, and that different patterns of gene expression are observed in different cell lines. Thus NR4A1 and FOS were induced in all three cell lines, whereas RGS2, DUSP2, ID3 and CCL3 were induced only in WSU-NHL and ESIII. RAJI showed in general a more restricted set of gene inductions, since of the eight genes tested, only NR4A1 and FOS were found to be induced at least twofold (Fig. 6). Interestingly, the three cell lines were more similar to BJAB than DHL4 in terms of biological response, in that they did not show an inhibition of cell proliferation in response to rituximab or homotypic adhesion (data not shown).
Fig. 6.

Pattern of gene induction in B-lymphoma cell lines. Exponentially growing WSU-NHL (black bars), ESIII (thin-striped bars) and Raji (thick-striped bars) B-lymphoma cells were plated in presence or absence of 10 μg/ml rituximab antibody, and total RNA was extracted at 4 h. Induction of the indicated genes by rituximab relative to control antibody was determined by real-time PCR using specific oligonucleotides. The results are the mean and standard deviation of triplicate samples.
Rituximab activates the AP1 transcription factor
Since several of the genes induced by rituximab in DHL4 were known to be up-regulated by PMA, a known activator of the MAPK pathway and of the AP1 transcription factor, or were functionally related to the MAPK pathway, we investigated whether rituximab was able to activate AP1 in DHL4 and BJAB cells. As shown in Fig. 7a (lanes 3 and 6), electrophoretic mobility shift assays using a labelled AP1 consensus sequence, demonstrated that rituximab indeed activates AP1 in these cells. Although AP1 activation by rituximab was weaker than that obtained with PMA (Fig. 7a, lane 2), it was clearly specific and was not observed in the presence of control daclizumab antibody (lane 4). The specificity of the retarded band was verified by supershift with anti-jun (Fig. 7b, lane 3) or anti-fos antibodies (lane 4) and by competition with a specific (lane 5) but not with a nonspecific competitor (lane 6). These data demonstrate that rituximab activates the AP1 transcription factor in both DHL4 and BJAB cell lines. This activation is likely to be responsible for the induction of at least some of the genes identified by microarray technology.
Fig. 7.
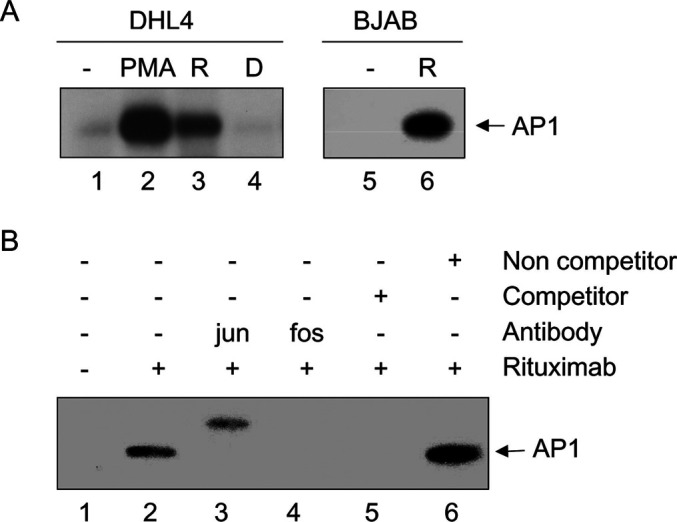
Rituximab activates AP1 in both cell lines. Exponentially growing DHL4 or BJAB cells were incubated for 4 h in presence or absence of 10 μg/ml rituximab or daclizumab, or in presence of 20 ng/ml PMA. Nuclear extracts were prepared and used in electrophoretic mobility shift assays with a labelled AP1 probe. a The retarded bands obtained with unstimulated DHL4 (lane 1), or after stimulation with PMA (lane 2), rituximab (R, lane 2) or daclizumab (D, lane 4) or with unstimulated BJAB (lane 5) or BJAB stimulated with rituximab (lane 6) are shown. The results are representative of at least three independent experiments. In b, EMSA was performed using DHL4 extracts in absence (lanes 1–2) or presence of antibodies against the fos (lane 3) and jun proteins (lane 4), or in presence of excess specific (lane 5) or nonspecific unlabelled (cold) AP1 probe (lane 6). The results are representative of at least three independent experiments.
Rituximab signals through the MEK1/MEK2 kinase pathway
AP1 activity is regulated by the MAPK-signalling cascades, which are composed of three major pathways, the MEK1/2-ERK1/2, p38 and JNK/SAPK pathways. We have therefore investigated the effect of two different inhibitors, available for the MEK1/2 and p38 pathways, to try to dissect the role of these pathways in rituximab-induced gene expression. DHL4 cells were pretreated with different doses of the inhibitor PD98059, a specific inhibitor of MEK1/2, or PD169316, a specific inhibitor of p38, and then exposed to the standard dose of rituximab. Cells were collected after 4 h, and RNA extracted for analysis by real-time PCR of the two most induced genes. As shown in Fig. 8, induction of NR4A1 and DUSP2 was inhibited by 70–80% in presence of 30 μM of the MEK1/2 inhibitor but not the p38 inhibitor. These data suggest that rituximab signalling induces activation of the MEK1/2-ERK1/2 pathway.
Fig. 8.

Rituximab induces gene expression through the MEK1/MEK2-ERK1 pathway. Exponentially growing DHL4 cells were preincubated for 10 min in absence (black bars) or presence of 10 μM (light grey bars) or 30 μM (dark grey bars) PD98059 (MEK1/2 inhibitor) or 10 μM (thin-striped bars) or 30 μM (thick-striped bars) PD169316 (p38 inhibitor), followed by 4 h in presence or absence of 10 μg/ml rituximab. Total RNA was extracted, and expression of the indicated genes was analysed by real-time PCR. The results are the mean and standard deviation of three independent experiments.
Altogether the data presented allow us to draw a likely signalling pathway triggered by rituximab in DHL4 cells, which includes MAPK pathway activation through MEK1/2-ERK1/2, AP1 activation and induction of at least some of the genes identified.
Discussion
In this report we have investigated the direct effects of rituximab in several B-lymphoma cell lines. We studied their biological response, gene expression pattern and signalling pathway following stimulation with rituximab. Overall, the data show that rituximab alone has direct signalling activity on B-lymphoma lines and that different cell lines can show different although overlapping responses. Indeed rituximab alone, in absence of further cross-linking, inhibited proliferation and induced homotypic adhesion in the follicular lymphoma line DHL4 but not in the Burkitt’s lymphoma line BJAB. We could not detect apoptosis induced by rituximab alone. These results are in agreement with previous data obtained with DHL4 and rituximab in vitro [41, 48]. Some authors have reported variable degrees of apoptosis induced by different anti-CD20 antibodies, including rituximab, in DHL4 cells, although in most cases this effect required antibody cross-linking [17, 33, 43] or seemed to involve a peculiar type of apoptosis independent from caspase activation and involving little or no DNA degradation [4]. We could not detect any additional effect of antibody cross-linking on apoptosis or thymidine uptake over the growth inhibition observed with rituximab alone. This may have been due to the cross-linking method that we have used, which employed a highly specific anti-human IgG1 monoclonal antibody which perhaps did not lead to the extensive cross-links obtained with chemical or other methods.
To further define the signalling events triggered by rituximab, we investigated the early gene pattern and signalling pathway induced by rituximab in DHL4 and BJAB by gene chip analysis. We have thus identified for the first time a set of 16 genes in DHL4 and 12 in BJAB reproducibly induced by rituximab alone. Eleven and seven genes were specific for DHL4 and BJAB, respectively, whereas the other five were induced in both lines. Quantitative PCR analysis confirmed all genes induced, except one in BJAB (RGS1), demonstrating over 95% correspondence with the results obtained by microarray. Up-regulation of these genes was specific for CD20, since they were not induced by an irrelevant humanised IgG1 antibody or by the anti-CD52 antibody campath-1H. Furthermore the F(ab′)2 fragment of rituximab also induced expression of five out of five genes tested, suggesting that gene up-regulation was the result of direct triggering of the CD20 molecule and was independent of the Fc portion binding to Fc receptors on the B cells.
Several of the genes identified are of functional interest. For example 12 out of the 16 genes induced by rituximab in DHL4 cells are known to be involved in the control of cell growth and/or apoptosis in different cell types, nine of which were specifically induced in DHL4. Thus, one or more of these genes may be involved in the inhibition of proliferation observed in DHL4 cells in presence of the antibody. Similarly, one or more of the genes up-regulated by rituximab in DHL4 cells may be involved in inducing homotypic adhesion in these cells. More work will be required to investigate these points which are beyond the scope of this article. The direct effects of rituximab analysed here are of particular interest in view of the recent finding that the therapeutic activity of the B1 antibody does not appear to require CDC or NK cells [7], suggesting that inhibition of cell growth or sensitisation to apoptosis in some lymphoma cells may indeed contribute to the efficacy of anti-CD20 antibodies.
We have analysed induction of a set of nine genes in by quantitative PCR analysis in three other B-lymphoma cell lines. These data showed that, although each cell has a specific pattern of gene induction, some genes, such as RGS2, DUSP2, ID3 and FOS, are induced in 4/5 lines and one (NR4A1) in 5/5 lines. Thus rituximab is likely to induce a common early signalling cascade leading to the induction of immediate-early genes.
Gene expression of several secreted molecules, in particular VEGF, ADM, and the chemokines CCL3 and CCL4, have been shown here for the first time to be up-regulated by rituximab in DHL4 and BJAB cells, respectively. Induction of these molecules in vivo may have functional consequences on tumour growth and/or antibody therapeutic activity. Indeed up-regulation of the angiogenic factors VEGF and ADM may increase tumour vascularisation, favouring tumour growth [16]. CCL3 and CCL4 expression on the other hand may attract immune cells and facilitate rituximab mediated ADCC and tumour regression [9, 52]. In vivo models are being set up to test these novel hypotheses.
The molecular pathway leading to gene up-regulation was also further characterised. We showed here that rituximab activates the AP1 transcription complex in both DHL4 and BJAB. This is in accordance with the fact that many of the genes up-regulated by rituximab are known to be induced by PMA or other AP1 activators [1, 2, 25, 26, 29, 30, 34, 45, 47]. Rituximab activates AP1 in both DHL4 and BJAB suggesting that differences in the composition of the AP1 factor or of the genes it induces downstream are responsible for the diverse biological response of these cells to rituximab. Activation of AP1 by rituximab had already been suggested previously [33], but in the latter case, cross-linking of the antibody by binding to plastic was found to be required. That rituximab can activate AP1 was further confirmed by finer analysis of the MAPK cascades. We have shown here that induction of two test genes (NR4A1 and DUSP2) was inhibited by the MEK1/2 inhibitor PD98059 and not by the p38 inhibitor PD169316, suggesting involvement of the MEK1/2-ERK1/2 pathway in the response of DHL4 to rituximab [31]. These data also add strong evidence that at least some of the genes induced by rituximab (including NR4A1 and DUSP2) are up-regulated through AP1 activation. The MEK/ERK pathway does not however appear to be involved in signalling homotypic adhesion since inhibitor PD98059 did not significantly inhibit clump formation in response to rituximab (Golay J, unpublished observations). Interestingly the genes induced by rituximab included a subset (at least eight genes, such as CCL3 and CCL4) also induced by BCR stimulation of B cells [18, 19, 30]. The MEK/ERK pathway is indeed known to be activated following an immunogenic or tolerogenic signal through the BCR and is responsible for induction of a subset of genes induced by BCR signalling [19, 40]. These data altogether confirm on a molecular and genome-wide basis the observations of a common signalling pathway between sIg and CD20 [33, 36]. It is worth noting however that the response of DHL4 cells to rituximab was relatively weak, with induction of about 0.13% of the probe sets analysed, whereas other signals such BCR stimulation in B cells or PDGF/EGF in fibroblasts up-regulate 0.3–2% of genes at early time points [15, 19]. Direct comparisons are however difficult due to the different assay conditions. Furthermore, we have chosen to look for robust changes in gene expression (at least twofold change in replicate experiments). Nonetheless, the results obtained suggest that rituximab induces a rather restricted and specific signalling pathway(s) in B-lymphoma cells.
To conclude, the data presented offer for the first time a genome-wide analysis of the gene expression profiles induced by rituximab in different human B-lymphoma cells lines, genes which could play a role in the therapeutic activity of rituximab in vivo. Work is in progress to further investigate this point.
Acknowledgements
We thank Prof. E.S. Vitetta (Cancer Immunobiology Cancer, University of Texas Southwestern Medical Center, Dallas, TX, USA) for her generous gift of rituximab F(ab′)2 fragment.This work was supported by the Italian Ministry for University and Research (projects FIRB no. RBAU01 J2ER and no. RBAU01H8SX), the Italian Ministry of Health (ICS PERF00001), the Associazione Italiana Ricerca sul Cancro (AIRC to J.G. and M.I.), Roche Italia (Monza, Italy) and the Istituto Superiore di Sanità (ISS Rome, project 30D.41) and the Associazione Italiana contro le Leucemie (AIL)-sezione Paolo Belli Bergamo.
References
- 1.Bain G, Cravatt CB, Loomans C, Alberola-Ila J, Hedrick SM, Murre C. Regulation of the helix-loop-helix proteins, E2A and Id3, by the Ras-ERK MAPK cascade. Nature Immunol. 2001;2:165–171. doi: 10.1038/84273. [DOI] [PubMed] [Google Scholar]
- 2.Bustin SA, Nie XF, Barnard RC, JKumar V, Pascall JC, Brown KD, Leigh IM, Williams NS, McKay IA. Cloning and characterisation of ERF-1, a human member of the Tis11 family of early-response genes. DNA Cell Biol. 1994;13:449–459. doi: 10.1089/dna.1994.13.449. [DOI] [PubMed] [Google Scholar]
- 3.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor Fc gammaIIIa gene. Blood. 2002;99:754–758. doi: 10.1182/blood.V99.3.754. [DOI] [PubMed] [Google Scholar]
- 4.Chan HT, Hughes D, French RR, Tutt AL, Walshe CA, Teeling JL, Glennie MJ, Cragg MS. CD20-induced lymphoma cell death is independent of both caspases and its redistribution into triton X-100 insoluble membrane rafts. Cancer Res. 2003;63:5480–5489. [PubMed] [Google Scholar]
- 5.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nature Med. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 6.Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H, Bouabdallah R, Morel P, Van den BNeste E, Salles G, Gaulard P, Reyes F, Gisselbrecht C. CHOP chemotherapy plus rituximab compared to chop alone in elderly patients with diffuse large B cell lymphoma. New Engl J Med. 2002;346:235–242. doi: 10.1056/NEJMoa011795. [DOI] [PubMed] [Google Scholar]
- 7.Cragg MS, Glennie M. Antibody specific controls in vivo effector mechanisms of anti-CD20 reagents. Blood. 2003;103:1238–1243. doi: 10.1182/blood-2003-06-2031. [DOI] [PubMed] [Google Scholar]
- 8.Cragg MS, Morgan SM, Chan HT, Morgan BP, Filatov AV, Johnson PW, French RR, Glennie MJ. Complement-mediated lysis by anti-CD20 mAb correlates with segregation into lipid rafts. Blood. 2003;101:1045–1052. doi: 10.1182/blood-2002-06-1761. [DOI] [PubMed] [Google Scholar]
- 9.Crittenden M, Gough M, Harrington K, Olivier K, Thompson J, Vile RG. Expression of inflammatory chemokines combined with local tumor destruction enhances tumor regression and long-term immunity. Cancer Res. 2003;63:5505–5512. [PubMed] [Google Scholar]
- 10.Czuczman MS, Fallon A, Mohr A, Stewart C, Bernstein ZP, McCarthy P, Skipper M, Brown K, Miller K, Wentling D, Klippenstein D, Loud P, Rock MK, Benyunes M, Grillo-Lopez AJ, Bernstein SH. Rituximab in combination with CHOP or fludarabine in low-grade lymphoma. Semin Oncol. 2002;29:36–40. doi: 10.1053/sonc.2002.30766. [DOI] [PubMed] [Google Scholar]
- 11.Deans JP, Schieven GL, Shu GL, Valentine MA, Gilliland LA, Aruffo A, Clark EA, Ledbetter JA. Association of tyrosine and serine kinases with the B cell specific surface antigen CD20. J Immunol. 1993;151:4494–4504. [PubMed] [Google Scholar]
- 12.Deans JP, Li H, Polyak MJ. CD20-mediated apoptosis: signalling through lipid rafts. Immunology. 2002;107:176–182. doi: 10.1046/j.1365-2567.2002.01495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Gaetano N, Cittera E, Nota R, Vecchi A, Grieco V, Scanziani E, Botto M, Introna M, Golay J. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol. 2003;171:1581–1587. doi: 10.4049/jimmunol.171.3.1581. [DOI] [PubMed] [Google Scholar]
- 14.Edwards JC, Leandro MJ, Cambridge G. B-lymphocyte depletion therapy in rheumatoid arthritis and other autoimmune disorders. Biochem Soc Trans. 2002;30:824–828. doi: 10.1042/BST0300824. [DOI] [PubMed] [Google Scholar]
- 15.Fambrough D, McClure K, Kazlauskas A, Lander ES. Diverse signalling pathways activated by growth factor receptors induce broadly overlapping, rather than independent, sets of genes. Cell. 1999;97:727–741. doi: 10.1016/S0092-8674(00)80785-0. [DOI] [PubMed] [Google Scholar]
- 16.Fernandez-Sauze S, Delfino C, Mabrouk K, Dussert C, Chinot O, Martin PM, Grisoli F, Ouafik L, Boudouresque F. Effects of adrenomedullin on endothelial cells in the multistep process of angiogenesis: involvement of CRLR/RAMP2 and CRLR/RAMP3 receptors. Int J Cancer. 2004;108:797–804. doi: 10.1002/ijc.11663. [DOI] [PubMed] [Google Scholar]
- 17.Ghetie M-A, Bright H, Vitetta E. Homodimers but not monomers of Rituxan (chimeric anti-CD20) induce apoptosis in human B-lymphoma cells and synergise with a chemotherapeutic agent and an immunotoxin. Blood. 2001;97:1392–1398. doi: 10.1182/blood.V97.5.1392. [DOI] [PubMed] [Google Scholar]
- 18.Glynne R, Akkaraju S, Healy JI, Rayner J, Goodnow CC, Mack DH. How self-tolerance and the immunosuppressive drug FK506 prevent B-cell mitogenesis. Nature. 2000;403:672–676. doi: 10.1038/35001102. [DOI] [PubMed] [Google Scholar]
- 19.Glynne R, Ghandour G, Rayner J, Mack DH, Goodnow CC. B-lymphocyte quiescence, tolerance and activation as viewed by global gene expression profiling on microarrays. Immunol Rev. 2000;176:216–246. doi: 10.1034/j.1600-065x.2000.00614.x. [DOI] [PubMed] [Google Scholar]
- 20.Golay J, Capucci A, Arsura M, Castellano M, Rizzo V, Introna M. Expression of c-myb and B-myb, but not A-myb, correlates with proliferation in human hematopoietic cells. Blood. 1991;77:149–158. [PubMed] [Google Scholar]
- 21.Golay J, Zaffaroni L, Vaccari T, Lazzari M, Borleri GM, Bernasconi S, Tedesco F, Rambaldi A, Introna M. Biologic response of B lymphoma cells to anti-CD20 monoclonal antibody rituximab in vitro: CD55 and CD59 regulate complement-mediated cell lysis. Blood. 2000;95:3900–3908. [PubMed] [Google Scholar]
- 22.Golay J, Gramigna R, Facchinetti V, Capello D, Gaidano G, Introna M. Acquired immunodeficiency syndrome-associated lymphomas are efficiently lysed through complement-dependent cytotoxicity and antibody-dependent cellular cytotoxicity by rituximab. Br J Haematol. 2002;119:923–929. doi: 10.1046/j.1365-2141.2002.03935.x. [DOI] [PubMed] [Google Scholar]
- 23.Golay JT, Clark EA, Beverley PC. The CD20 (Bp35) antigen is involved in activation of B cells from the G0 to the G1 phase of the cell cycle. J Immunol. 1985;135:3795–3801. [PubMed] [Google Scholar]
- 24.Grillo-Lopez AJ, White CA, Varns C, Shen D, Wei A, McClure A, Dallaire BK. Overview of the clinical development of rituximab: first monoclonal antibody approved for the treatment of lymphoma. Sem Oncol. 1999;26:66–73. [PubMed] [Google Scholar]
- 25.Grumont RJ, Rasko JE, Strasser A, Gerondakis S. Activation of the mitogen-activated ptotein kinase pathway induces transcription of the PAC-1 phosphatase gene. Mol Cell Biol. 1996;16:2913–2921. doi: 10.1128/mcb.16.6.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heximer SP, Watson N, Linder ME, Blumer KJ, Hepler JR. RGS2/GOS8 is a selective inhibitor of Gq-alpha function. Proc Natl Acad Sci U S A. 1997;94:14389–14393. doi: 10.1073/pnas.94.26.14389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iyer VR, Eisen MB, Ross DT, Schuler G, Moore T, Lee JC, Trent JM, Staudt LM, Hudson J, Jr, Boguski MS, Lashkari D, Shalon D, Botstein D, Brown PO. The transcriptional program in the response of human fibroblasts to serum. Science. 1999;283:83–87. doi: 10.1126/science.283.5398.83. [DOI] [PubMed] [Google Scholar]
- 28.Kawahara K, Oyadomari S, Gotoh T, Kohsaka S, Nakayama H, Mori M. Induction of CHOP and apoptosis by nitric oxide in p53-deficient microglial cells. FEBS lett. 2001;506:135–139. doi: 10.1016/S0014-5793(01)02898-8. [DOI] [PubMed] [Google Scholar]
- 29.Kozlow EJ, Wilson GL, Fox CH, Kehrl JH. Substractive cDNA cloning of a novel member of the Ig gene superfamily exzpressed at high levels in activated B lymphocytes. Blood. 1993;81:454–461. [PubMed] [Google Scholar]
- 30.Krzysiek R, Lefevre EA, Zou W, Foussat A, Bernard J, Portier A, Galanaud P, Richard Y. Antigen receptor engagement selectively induces macrophage inflammatory protein-1 alpha (MIP-1 alpha) and MIP-1 beta chemokine production in human B cells. J Immunol. 1999;162:4455–4463. [PubMed] [Google Scholar]
- 31.Kummer JL, Rao PK, Heidenreich KA. Apoptosis induced by withdrawal of trophic factors is mediated by p38 mitogen-activated protein kinase. J Biol Chem. 1997;272:20490–20494. doi: 10.1074/jbc.272.33.20490. [DOI] [PubMed] [Google Scholar]
- 32.Liu G, Loraine AE, Shigeta R, Cline M, Cheng J, Valmeekam V, Sun S, Kulp D, Siani-Rose MA. NetAffx: Affymetrix probesets and annotations. Nuclic Acids Res. 2003;31:82–86. doi: 10.1093/nar/gkg121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mathas S, Rickers A, Bommert K, Doerken B, Mapara MY. Anti-CD20- and B-cell receptor-mediated apoptosis:evidence for shared intracellular signalling pathways. Cancer Res. 2000;60:7170–7176. [PubMed] [Google Scholar]
- 34.Nomiyama H, Hieshima K, Hirokawa K, Hattori T, Takatsuki K, Miura R. Characterization of cytokine LD78 gene promoters: positive and negative transcriptional factors bind to a negative regulatory element common to LD78, interleukin-3, and granulocyte-macrophage colony-stimulating factor gene promoters. Mol Cell Biol. 1993;13:2787–2801. doi: 10.1128/mcb.13.5.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pedersen IM, Buhl AM, Klausen P, Geisler CH, Jurlander J. The chimeric anti-CD20 antibody rituximab induces apoptosis in B-cell chronic lymphocytic leukemia cells through a p38 mitogen activated protein-kinase-dependent mechanism. Blood. 2002;99:1314–1319. doi: 10.1182/blood.V99.4.1314. [DOI] [PubMed] [Google Scholar]
- 36.Petrie RJ, Deans JP. Colocalization of the B cell receptor and CD20 followed by activation-dependent dissociation in distinct lipid rafts. J Immunol. 2002;169:2886–2891. doi: 10.4049/jimmunol.169.6.2886. [DOI] [PubMed] [Google Scholar]
- 37.Polyak MJ, Deans JP. Alanine-170 and proline-172 are critical determinants for extracellular CD20 epitopes; heterogeneity in the fine specificity of CD20 monoclonal antibodies is defined by additional requirements imposed by both amino acid sequence and quaternary structure. Blood. 2002;99:3256–3262. doi: 10.1182/blood.V99.9.3256. [DOI] [PubMed] [Google Scholar]
- 38.Polyak MJ, Tailor SH, Deans JP. Identification of a cytoplasmic region of CD20 required for its redistribution to a detergent-insoluble membrane compartment. J Immunol. 1998;161:3242–3248. [PubMed] [Google Scholar]
- 39.Reff ME, Carner C, Chambers KS, Chinn PC, Leonard JE, Raab R, Newman RA, Hanna N, Anderson DR. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994;83:435–445. [PubMed] [Google Scholar]
- 40.Richards JD, Dave SH, Chou CH, Mamchak AA, DeFranco AL. Inhibition of the MEK/ERK signalling pathway blocks a subset of B cell responses to antigen. J Immunol. 2001;166:3855–3864. doi: 10.4049/jimmunol.166.6.3855. [DOI] [PubMed] [Google Scholar]
- 41.Rose AL, Smith BE, Maloney DG. Glucocorticoids and rituximab in vitro: synergistic direct anti-proliferative and apoptotic effects. Blood. 2002;100:1765–1773. [PubMed] [Google Scholar]
- 42.Semac I, Palomba C, Kulangara K, Klages N, van Echten-Deckert G, Borisch B, Hoessli DC. Anti-Cd20 therapeutic antibody rituximab modifies the functional organisation of rafts/microdomains of B lymphoma cells. Cancer Res. 2003;63:534–540. [PubMed] [Google Scholar]
- 43.Shan D, Ledbetter JA, Press OW. Apoptosis of malignant human B cells by ligation of CD20 with monoclonal antibodies. Blood. 1998;91:1644–1652. [PubMed] [Google Scholar]
- 44.Shan D, Ledbetter JA, Press OW. Signalling events involved in anti-CD20-induced apoptosis of malignant human B cells. Cancer Immunol Immunother. 2000;48:673–683. doi: 10.1007/s002620050016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shimizu N, Ohta M, Fujiwara C, Sagara J, Mochizuki N, Oda T, Utiyama H. Expression of a novel immediate early gene during TPA-induced macrophage differentiation of HL-60 cells. J Biol Chem. 1991;266:12157–12161. [PubMed] [Google Scholar]
- 46.Silverman GJ, Weisman S. Rituximab therapy and autoimmune disorders: prospects for anti-B cell therapy. Arthritis Rheum. 2003;48:1484–1492. doi: 10.1002/art.10947. [DOI] [PubMed] [Google Scholar]
- 47.Tachibana I, Imoto M, Adjei PN, Gores GJ, Subramaniam M, Spelsberg TC, Urrutia R. Overexpression of the TGF-beta-regulated zinc finger encoding gene TIEG, induces apoptosis in pancreatic epithelial cells. J Clin Invest. 1997;99:2365–2374. doi: 10.1172/JCI119418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taji H, Kagami Y, Okada Y, Andou M, Nishi Y, Saito H, Seto M. Growth inhibition of CD20-positive B lymphoma cell lines by IDEC-C2B8 anti-CD20 monoclonal antibody. Jpn J Cancer Res. 1998;89:748–756. doi: 10.1111/j.1349-7006.1998.tb03280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tedder TF, Engel P. CD20: a regulator of cell-cycle progression of B lymphocytes. Immunol Today. 1994;15:450–454. doi: 10.1016/0167-5699(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 50.Tedder TF, Boyd AW, Freedman AS, Nadler LM, Schlossman SF. The B cell surface molecule B1 is functioanally linked with B cell activation and differentiation. J Immunol. 1985;135:973–979. [PubMed] [Google Scholar]
- 51.Tutt AL, French RR, Illidge TM, Honeychurch J, McBride HM, Penfold CA, Fearon DT, Parkhouse RM, Klaus GG, Glennie MJ. Monoclonal antibody therapy of B cell lymphoma: signalling activity on tumor cells appears more important than recruitment of effectors. J Immunol. 1998;161:3176–3185. [PubMed] [Google Scholar]
- 52.Uekusa Y, Yu WG, Mukai T, Gao P, Yamaguchi N, Murai M, Matsushima K, Obika S, Imanishi T, Higashibata Y, Nomura S, Kitamura Y, Fujiwara H, Hamaoka T. A pivotal role for CC chemokine receptor 5 in T-cell migration to tumor sites induced by interleukin 12 treatment in tumor-bearing mice. Cancer Res. 2002;62:3751–3758. [PubMed] [Google Scholar]
- 53.van der Kolk LE, Evers LM, Omene C, Lens SM, Lederman S, van Lier RA, van Oers MH, Eldering E. CD20-induced B cell death can bypass mitochondria and caspase activation. Leukemia. 2002;16:1735–1744. doi: 10.1038/sj.leu.2402559. [DOI] [PubMed] [Google Scholar]
- 54.Ward Y, Gupta S, Jensen P, Wartmann M, Davis RJ, Kelly K. Control of MAP kinase activation by the mitogen-induced threonine/tyrosine phosphatase PAC1. Nature. 1994;367:651–654. doi: 10.1038/367651a0. [DOI] [PubMed] [Google Scholar]