

Abstract
Chronic myelogenous leukemia (CML) is characterized by a t(9;22) translocation resulting in expression of BCR-ABL fusion oncoproteins which are unique to the leukemic cells, necessary for oncogenesis, and potentially immunogenic. We have previously shown that human dendritic cells transduced with an adeno-associated virus vector encoding the fusion region of the b3a2 splice variant (p210b3a2) of the BCR-ABL oncoprotein elicit specific T-cell responses in vitro. Two cytotoxic T lymphocyte (CTL) clones generated in this fashion displayed restriction with previously unreported HLA alleles. The first, T1/B9, was CD4+ and restricted by DRB5*0101 (autologous) or DRB1*1101 (allogeneic). The minimum cytotoxic epitope (MCE) binding to DRB5*0101 for this clone was identified as FKQSSKALQ, overlapping the p210b3a2 fusion point (boldface). The MCE of DRB1*1101 for this clone differed from DRB5*0101, but also included the fusion point. The clonality of CTL T1/B9 was verified by analyses of TCRα/β chain usage and DNA sequence analyses. To our knowledge, this is the first description of a single clone recognizing both DRB5*0101 and DRB1*1101. The other CTL clone, T1/33, was CD8+ and recognized HLA-B*3501 or B*3503 complexed with an MCE, RPVASDFEP, derived from the c-abl sequence in proximity to the p210b3a2 fusion point. K562 cells transfected with plasmids encoding HLA-DRA + B5*0101, B*3501, or B*3503 but not controls expressing DRA + DRB1*1501 were lysed by cognate CTL clones, confirming that DRB5*0101 and B*3501/3 could present p210b3a2 joining region epitopes via endogenous processing. The identification of three additional HLA alleles (DRB5*0101, B*3501, and B*3503) presenting the p210b3a2 fusion-region antigen will broaden the application of vaccine strategies for targeting CML cells. The findings of single CTL clones cross-recognizing autologous (DRB5*0101 or B*3501) and allogeneic (DRB1*1101 or B*3503) HLA alleles presenting BCR-ABL fusion-region epitopes implies the potential separation of graft-versus-leukemia from graft-versus-host effects.
Keywords: Human, CTL, T-cell receptor, MHC, Antigen presentation
Introduction
Many human cancers, including leukemia, lymphomas, and certain solid tumors are associated with chromosomal translocations that result in the expression of novel, potentially immunogenic, fusion proteins (reviewed in [1, 13, 42]). For example, chronic myelogenous leukemia (CML) is a malignant disorder characterized by clonal proliferation of cells that possess a reciprocal chromosomal translocation leading to the fusion of the BCR gene on chromosome 22 and the Abelson (c-abl) oncogene on chromosome 9. The BCR-ABL fusion gene described in 1984 was the first example of a fusion oncogene in a human malignancy. It is now accepted that generation of the BCR-ABL fusion gene is critical to the pathogenesis of CML, and results in the unregulated expression of the c-abl tyrosine kinase [7]. Furthermore, the BCR-ABL fusion protein is present only in the leukemic cells, and results in formation of unique targets to which an antileukemic immune response might be generated. Thus, the BCR-ABL fusion protein is an attractive target for the development of CML-targeted immunotherapy. Indeed, several studies have demonstrated that CML-specific T cells could be generated both in individuals with the disease and in healthy donors, adding further interest to this approach.
One strategy for induction of specific immune responses focuses upon the use of dendritic cells (DCs), the most potent antigen-presenting cells (APCs) currently known. DCs process antigens and present them as peptides to effector T cells in an antigen-specific, MHC-restricted fashion. Recently, recombinant viral vectors have been employed to introduce specific antigens into DC for efficient processing and presentation. This strategy permits the expression of large, multiepitope proteins which can be subsequently processed by DC into appropriately sized peptides for presentation to T cells in the context of MHC molecules. In this regard, we have previously shown that primary human DCs could be successfully transduced with recombinant adeno-associated virus (rAAV2) in vitro. rAAV-based vectors possess several advantages for introduction of antigens into DC, including lack of toxicity, and removal of virus-encoded genes that could interfere with antigen processing/presentation. To assess whether this strategy might be effective for the generation of an anti-CML immune response, we transduced DCs with an rAAV2 vector encoding a truncated p210b3a2 protein containing the fusion region, and generated CTL lines from peripheral blood lymphocytes from healthy donors in vitro [39]. To assess MHC restriction, CTL clones generated using this system were screened against a panel of 14 HLA divergent B cell lines pulsed with the p210b3a2 fusion-region peptide. Findings were subsequently confirmed using two approaches: (1) anti-HLA monoclonal antibody cytotoxicity blocking experiments, and (2) introduction of specific HLA alleles into either p210b3a2 peptide pulsed, or cell lines endogenously expressing p210b3a2 in chromium release assays (CRAs). Using these strategies, we describe three new HLA alleles (DRB5*0101, B*3501, and B*3503) that present fusion-region epitopes of the p210b3a2 oncoprotein. Here we distinguish between epitopes of the fusion point and fusion region. In the latter instance, the epitope may be in proximity to, but does not necessarily include, the fusion point. K562 cells, a leukemic cell line expressing p210b3a2, engineered to express HLA-DRA + DRB5*0101, B*3501, or B*3503 but not DRA + DRB1*1501 were lysed by cognate CTL clones, indicating these peptide-MHC complexes generated via the endogenous pathway could be recognized. These findings widen the application of vaccine strategies for targeting CML cells by broadening the potential immune-responsive population. We also identified CTL clones that cross-reacted both to autologous and to allogeneic HLA alleles complexed with p210b3a2 fusion-region–derived peptides, which provides evidence for the potential separation of graft-versus-leukemia (GVL) from graft-versus-host-disease (GVHD) effects.
Materials and methods
Cells, viruses, and vectors
The adenovirus-transformed human embryonic kidney cell line 293, the p210b3a2 expressing CML cell line K562, the human B cell line Ramos, and COS-1 were obtained from the American Type Culture Collection (ATCC, Manassas, VA). The HLA reference cell lines KOSE and FH3 were obtained from the International Histocompatibility Working Group (IHWG, Seattle, Washington). Epstein-Barr-virus–infected peripheral blood mononuclear cells isolated from healthy donors were used to derive immortalized B cell lines. Blood was obtained using an institutionally reviewed and approved protocol. A total of 11 B cell lines were generated in this study, and their HLA alleles were typed in the City of Hope HLA laboratory. Eight B cell lines with HLA divergent but non-redundant alleles were selected to demonstrate restriction elements for CTL clones (Table 1). The HLA alleles of other B cell lines, FH3, KOSE, and Ramos, as determined elsewhere, are also listed in Table 1. COS-1 and 293 were propagated in DMEM; other cells were maintained in RPMI 1640. Cells were maintained in media containing 10% FCS and 2 mM l-glutamine at 37°C in humidified, 5% CO2. All cell lines were monitored and proven to be mycoplasma free. An rAAV2 vector encoding a truncated p210b3a2 protein including the fusion region has been previously described [39].
Table 1.
HLA types of the cells used in this study
| Cell line | HLA-I | HLA-II | ||||||
|---|---|---|---|---|---|---|---|---|
| A* | B* | Cw* | DR | DQ | DP | |||
| B1 | 0301 | 0702 | 0401 | B1*0101 | B5*0101 | A1*0101 | B1*0501 | B1*0101 |
| B1 | 3501 | 0702 | 1501 | 0102/3 | B1*0602 | 0401 | ||
| B2 | 0301 | 0702 | 0304 | B1*1501 | B5*0101 | A1*0102 | B1*0301 | B1*0401 |
| B2 | 4001 | 0702 | 1602 | 0202 | 0505 | 0602 | 0402 | |
| B4 | 1101 | 4001 | 0303 | B1*0801 | B3*0301 | A1*0103 | B1*0301 | B1*0202 |
| B4 | 0702 | 1201 | 0503 | 0601 | 0501 | |||
| B5 | 0301 | 0702 | 0702 | B1*0701 | B3*0202 | A1*0104 | B1*0202 | B1*0301 |
| B5 | 3201 | 1401 | 0802 | 1401 | B4*0101/3 | 0201 | 0503 | 1001 |
| B6 | 0301 | 3501 | 0401 | B1*0403 | B3*0101 | A1*0103 | B1*0301 | B1*0301 |
| B6 | 1101 | 47? | 0602 | 1301 | B4*0103 | 0303 | 0603 | 1501 |
| B7 | 0201 | 0702 | 0501 | B1*0401 | B3*0202 | A1*0103 | B1*0301 | B1*0401 |
| B7 | 4402 | 0702 | 1301 | B4*0103 | 0303 | 0603 | ||
| B9 | 0201 | 4402 | 07? | B1*1101 | B3*0202 | A1*0502 | B1*0301 | B1*0401 |
| B9 | 4901 | 0505 | ||||||
| B10 | 0101 | 0801 | 0303 | B1*0301 | B3*0101 | B1*0201 | B1*0401 | |
| B10 | 0201 | 1501 | 0701 | 1301 | 0603 | |||
| FH3 | 3301 | 1402 | 0401 | B1*1101 | ||||
| FH3 | 3101 | 3502 | 0802 | 1104 | ||||
| KOSE | 0201 | 3503 | 1203 | B1*1302 | ||||
| KOSE | 1401 | |||||||
| Ramos | 0301 | 4402 | 1601 | B1*0701 | B4*0101/3 | B1*0202 | ||
| Ramos | 5101 | |||||||
Plasmids and peptides
Plasmid pRSVgptDRB1*1501 was kindly provided by Dr Eric Long (NIH, Bethesda, MD). Plasmid pCWRSPNDRA expressing HLA-DRA was constructed in our laboratory [5, 39]. pNSB*3501 was obtained from the IHWG (contributed by Dr Osam Mazda, Kyoto, Japan), and pcDNA3-B*3503 was kindly provided by Dr Pierre van der Bruggen (Ludwig Institute for Cancer Research, Brussels, Belgium). HLA expression plasmids also contained a selection cassette that conferred resistance to G418. A 25-residue peptide b3a2 (IVHSA TGFKQ SS-K-AL QRPVA SDFEP, where “-K-” represents the new amino acid at the fusion point of p210b3a2), truncated variants, and a negative control peptide b3b4 (IVHSA TGFKQ SSNLY CTLEV DSFGY) from the BCR gene were synthesized by the peptide Core Facility at the City of Hope.
DNA Transfection
The 293 cells were transfected with HLA-DR encoding plasmids using calcium phosphate coprecipitation per the manufacturer’s protocol (CellPhect, Amersham Pharmacia, Piscataway, NJ). COS-1 cells were transfected with HLA-B encoding plasmids, and K562 cells with HLA-B and HLA-DR encoding plasmids using Lipofectin (GibcoBRL, Grand Island, NY) as per manufacturer’s instructions. Transfected cells were selected with media containing 400 μg/ml of active G418 for 2 weeks. G418-resistant cells were monitored by FACS, and cells expressing the appropriate HLA allele were sorted at the City of Hope FACS Core Facility.
Chromium release assays (CRA) and antibody-blocking analysis
CRAs were performed as previously described [39]. Briefly, human B-cell targets were pulsed with indicated peptides (specific epitope or control) and labeled with 100-μCi 51Cr (Amersham Biosciences, Piscataway, NJ) per 106 cells for 1.5 h. Labeled targets were seeded in triplicate into 96-well V-bottom plates in the presence of lymphocytes at indicated effector/target (E/T) ratios. After incubating at 37°C for 4 h, released supernatant 51Cr was counted as experimental release (Re) in a TopCount.NXT(Packard Instruments, Meriden, CT). Spontaneous 51Cr release (Rs) was determined by incubating targets with media alone, and maximal 51Cr release (Rm) was obtained by incubating targets with media containing 1% SDS. Specific lysis was calculated as 100% × (Re − Rs) / (Rm − Rs). Blocking experiments were performed with or without addition of monoclonal antibody at 10 μg/ml to the target preparation and to the CRA cultures. Monoclonal anti-HLA-A, B, DR, DQ, or DP antibodies recognize an intralocus determinant on HLA molecules (Lab Vision, Fremont, CA).
Analysis of TCRα and β chain usage and their sequences
Total RNA from clone T1/B9 was extracted, subjected to RT-PCR using the First Choice RLM-RACE kit (Ambion, Austin, TX), and amplified fragments were subcloned via TA cloning (Invitrogen, Carlsbad, CA), following manufacturer’s protocols. Ten colonies with inserts of each amplified-TCRα or β chain were sequenced in the City of Hope Sequencing Core Facility. Sequences were analyzed using the Entrez Nucleotide Database. TCRs were designated according to nomenclature outlined in the WHO-IUIS subcommittee on TCR designation [19], and published reports for joining (J) segments [3, 21].
Results
Generation of p210b3a2 fusion-region–specific CTL cell lines and clones
We induced anti-BCR-ABL T-cell responses using human primary DCs transduced with a recombinant AAV2 vector encoding a truncated p210b3a2 protein fragment containing the BCR-ABL fusion region. Several truncated-p210b3a2 protein-specific T cell lines were generated via this system [39]. Human B cell lines pulsed with a 25 amino acid peptide, b3a2, which included the p210b3a2 fusion point, were used as targets in CRAs to screen T cell lines for BCR-ABL fusion-region–specific CTLs (Fig. 1). Several T cell lines displayed BCR-ABL antigen-specific cytotoxicity. Two CTL lines from donor 1 demonstrated restriction with previously unreported HLA alleles, and were cloned and studied further (Fig. 1). The first CTL clone, T1/B9 (Fig. 1A), was CD4+ and of the Th1 functional phenotype [39], and the second, T1/33 (Fig. 1B), was CD8+.
Fig. 1A, B.

Cytotoxicity profiles of p210BCR-ABL specific CTL clones against a panel of targets with divergent HLA alleles. A. Cytotoxicity profile of CD4 CTL clone T1/B9 (DRB5*0101). Cytolysis of the B9 lymphoblastoid line and FH3 indicated that HLA-DRB1*1101 is an allogeneic restriction element for T1/B9. Cytolysis of peptide-pulsed B9 could be specifically blocked with an anti-HLA-DR monoclonal antibody (striped bar, b3a2/anti-HLA-DR). B. Cytotoxicity profile of CTL clone T1/33 (B*3501). T1/33 recognizes autologous HLA-B*3501 and allogeneic B*3503. A panel of human B cell lines with divergent HLA alleles (from B1 to KOSE), COS-1 cells expressing HLA-B*3501 or B*3503, or mock-transfected COS-1 not expressing HLA (Mock) were pulsed with the peptide b3a2 (open bars), or the control peptide b3b4 (closed bars) and tested against CTL clone T1/B9 (A) or T1/33 (B) in standard 4-h CRAs at serial E/T ratios. The data shown were picked from a single E/T ratio of 20, derived from at least three experiments and expressed as mean values ± SE
Identification of new HLA-restriction elements for presentation of the p210b3a2 fusion-region epitopes to the two CTL clones
We analyzed HLA-restriction elements of the p210b3a2 fusion-region–specific CTL clones using a panel of HLA divergent B cell lines (Table 1) pulsed with the b3a2 peptide as targets in CRA. The autologous restriction element for the CD4 CTL clone T1/B9 was previously defined as HLA-DRB5*0101 [39]. Interestingly, the DRB5*0101 negative B cell line, B9 (DRB1*1101), could also present the BCR-ABL antigen to T1/B9. In contrast, B cell lines with other HLA alleles, DRB3*0202 (B5 and B7), or DPB1*0401 (B7) (Fig. 1A; Table 1) could not present the BCR-ABL antigen in the same assay, suggesting that DRB1*1101 was an allogeneic restriction element for the T1/B9 clone. b3a2 peptide-pulsed FH3 cells, a DRB1*1101 positive reference cell line, were also recognized and lysed by T1/B9, confirming this hypothesis. The restricted recognition of the T1/B9 clone to the HLA-DR, but not DQ, locus on allogeneic target B9 was further confirmed by monoclonal-antibody-blocking experiments. An anti-HLA-DR monoclonal antibody specifically blocked T1/B9-mediated cytolysis of b3a2 peptide-pulsed B9 cells (Target B9+b3a2/anti-HLA-DR in Fig. 1A), while anti-HLA-A, B, DQ, or DP monoclonals had no effect (data not shown). Of note is that the DRB1*1101 locus has been reported to be capable of presenting a p210b3a2 fusion-region peptide [2, 33], but there are no previous reports of a single CTL clone that recognizes both DRA + DRB5*0101 and DRA + DRB1*1101. Further data supporting this hypothesis is presented below.
The inability of cell line B2 (A*0301, B*0702, and Cw*0702, Table 1), or FH3 (Cw*0401) to present the p210b3a2 fusion-region epitope to the CD8+ CTL clone T1/33 (Fig. 1B) suggested that HLA-B*3501 was the autologous restriction element (in italics in Table 1) for this antigen. It has been reported that CTL clones restricted by B*3501 can also recognize allogeneic B*3502 [20], B*3503 [25, 20], B*5101 [43], and also an HLA-A1 locus presenting the same or different antigenic epitopes derived from infectious organisms or non-BCR-ABL tumor antigens [24]. To determine whether the HLA-B*3501-restricted clone T1/33 could recognize BCR-ABL presented by other HLA alleles, BCR-ABL peptide-pulsed FH3 (B*3502) and KOSE (B*3503) cell lines were used as targets in CRAs. The data demonstrated that in addition to HLA-B*3501, HLA-B*3503 but not HLA-B*3502 could present the p210b3a2 fusion-region epitope to clone T1/33 (Fig. 1B). Cell lines B10 (HLA-A1), Ramos (B*5101), and other B cell lines with divergent HLA alleles did not serve as targets for T1/33 (data not shown), indicating that clone T1/33 does not recognize these allorestriction elements for the presentation of the BCR-ABL peptide b3a2. To confirm that HLA-B*3501 and B*3503 truly functioned as antigen-presenting molecules for the p210b3a2 fusion region, we transfected COS-1 cells with individual plasmids expressing these HLA genes. COS-1 cells expressing HLA-B*3501 or B*3503 and pulsed with b3a2 were specifically lysed by clone T1/33, confirming that these molecules can indeed present the p210b3a2 fusion-region epitope to T cells (Fig. 1B).
It is possible that the apparent recognition of an allogeneic and an autologous MHC by a single T-cell clone could also result from the expression of a dual receptor [32] or lack of true clonality (Fig. 2). To verify clonality of our CTLs, the TCR gene cDNA of the T1/B9 was cloned following RT-PCR amplification, and TCRα and β chain usage and sequence analyses were determined. As depicted in Fig. 2, the DNA sequences of TCRα or β chains from 10 different bacterial colonies derived from T1/B9 TCR gene cDNA inserts were identical, confirming that T1/B9 was truly clonal (Genbank accession numbers AF542062 and AY157620), and that the cross-recognition of both autologous and allogeneic alleles was mediated by a single receptor.
Fig. 2.

CDR3 sequences of TCRα and β chains from the CD4 CTL clone T1/B9. Total RNA from T1/B9 was extracted and subjected to RT-PCR and cloned as described under “Materials and methods.” Ten colonies with inserts of each amplified-TCRα or β chain were sequenced and the consensus sequence is shown. The CDR3 and adjacent sequences, the last three residues of the V region and the first three residues from the conserved phenylalanine in the J region, are depicted. The J segment residues contributed to CDR3 are underlined. The nomenclature used is described in the text
Identification of the minimum cytotoxic epitopes (MCEs) for the p210b3a2 fusion region for the newly defined HLA-restriction elements
MHC class I molecules bind peptides of 8–11 amino acids with both amino and carboxyl termini tightly fixed in its groove (Fig. 3). In contrast, class II ligands, consisting of 9–25 residues, are not fixed but extend out at the ends of the groove. The shortest required amino acid sequence necessary for binding to MHC molecules and for recognition by CTL is termed the MCE. To determine the MCEs derived from the p210b3a2 fusion region that bind to these newly identified HLA-restriction elements, a panel of short overlapping peptides (Fig. 3A) were synthesized which spanned the p210b3a2 fusion region. B cell lines—B1, B9, or KOSE—were pulsed with varying concentrations of these peptides, and used as targets for the T-cell clones T1/B9 or T1/33 in CRAs (Fig. 3B and C).
Fig. 3A–C.

Identification of the MCEs of the p210b3a2 fusion region for newly identified HLA-restriction elements. A panel of short peptides corresponding to the p210b3a2 fusion region (A) were tested in CRAs with different effector target pairs. The bolded K corresponds to the new amino acid inserted at the BCR-ABL fusion point. KOSE or B cell lines, B1 or B9, were pulsed with varying concentrations of synthetic peptides, and used as targets for T1/B9 or T1/33 CTL at serial E/T ratios in CRAs. Data with 5-μM peptides are shown as mean values ± SE from at least three experiments (B). Striped bars indicate CTL clone T1/B9(DRB5*0101) vs target B9 (allogeneic HLA-DRB1*1101) pulsed with different peptides indicated on the Y-axis, open bars clone T1/33(HLA-B*3501) vs B1 (autologous HLA-B*3501) and gray bars T1/33(HLA-B*3501) vs KOSE (allogeneic HLA-B*3503) pulsed with indicated peptides. Peptide avidity profiles (C) of T1/B9(DRB5*0101) vs B9(HLA-DRB1*1101) + A17S (triangle), T1/33(HLA-B*3501) vs B1(HLA-B*3501) + R9P (diamond), and T1/33(HLA-B*3501) vs KOSE(HLA-B*3503) + R9P (square) were compared over a wide range of peptide concentrations
Using a similar panel of peptides, we have previously shown that FKQSSKALQ (peptide F9Q), which includes the p210b3a2 fusion point (boldface K), was the MCE for presentation by the autologous HLA class II molecule (DRA + DRB5*0101) to CD4 CTL clone T1/B9 [39]. In contrast, only A17S (ATGFKQSSKALQRPVAS) and b3a2 were recognized by T1/B9 using allogeneic B9 (DRB1*1101) targets. These results suggest that the binding motif of the p210b3a2 fusion region for the allogeneic DRB1*1101 was different from that of the autologous DRB5*0101. The MCE for DRB1*1101 might be either A17S, or possibly a shorter motif in A17S, but must include the p210b3a2 fusion point because peptides I15L (IVHSATGFKQSSKAL) and Q16P (QSSKALQRPVASDFEP) that contain carboxyl or amino terminal truncations of A17S were not recognized in this assay (Fig. 3B). B9 pulsed with previously described peptides K11 V (KQSSKALQRPV), F11P (FKQSSKALQRP), T10L (TGFKQSSKAL), F10R (FKQSSKALQR), and K9R (KQSSKALQR) consisting of amino or carboxyl terminal truncations of the A17S sequence [39] were not recognized by CTL T1/B9 (data not shown). Further experiments to define the MCE presented by allogeneic DRB1*1101 to T1/B9 were not pursued since it is not necessary for epitope peptides to be processed into MCEs to bind MHC class II molecules.
For the CD8+ CTL clone T1/33 (B*3501 restricted), B1 (B*3501) cells were pulsed with a variety of peptides shorter than b3a2, and tested in CRAs. R9P (RPVASDFEP) but not P8P (PVASDFEP) elicited a CTL response (Fig. 3), suggesting that R9P is the MCE from the p210b3a2 fusion region binding to B*3501 and recognized by clone T1/33. KOSE, a HLA-B*3503 cell line, was pulsed with R9P or P8P, and assayed in a similar fashion. T1/33 exhibited similar cytolytic recognition patterns, suggesting that R9P also functions as the MCE for binding to B*3503. Of note is that the R9P is derived from the c-abl sequence in proximity to, but not inclusive of, the fusion point of both b3a2 and b2a2 variants, So R9P might be considered a tumor-associated antigen.
To compare the avidity of CTL clones T1/B9 (CD4) or T1/33 (CD8) for recognition of these epitopes following presentation by autologous or allogeneic HLA alleles, B9, B1, or KOSE were pulsed with varying concentrations of peptides A17S or R9P and used as targets in CRAs (Fig. 3C). The avidity of T1/33 (B*3501, CD8) versus target B1(B*3501) + R9P or KOSE(B*3503) + R9P is similar; a peptide concentration between 0.5 μM and 5.0 μM results in half maximal lysis. In contrast, the concentration of A17S necessary to pulse B9 (DRB1*1101) to get half maximum lysis by T1/B9 (DRB5*0101) was about 3 μM, a value about three-fold lower than that necessary for 50% lysis of A17S-pulsed B1(DRB5*0101) (about 10 μM) [39]. However, the avidity as determined by this assay is not very sensitive, and we conclude that the avidity of these two HLA alleles for A17S were of the same order of magnitude. Of note is that avidity as measured with EBV-immortalized B cell lines with intact antigen-processing pathways is generally 10–100 times lower than that measured by TAP-deficient cell lines, such as T2 [11].
Presentation of endogenously processed p210b3a2 fusion-region epitopes by newly defined HLA-restriction elements
In the above experiments, p210b3a2 fusion-region peptides were used to test CTL clones T1/B9 (CD4) and T1/33 (CD8) for restriction elements and MCEs. Although these two clones were generated after priming and stimulation with rAAV-transduced, mature DCs, we wished to further confirm whether these HLA class I restriction elements B*3501 and B*3503 could present endogenously processed epitopes of the p210b3a2 protein. K562, a leukemic cell line that endogenously expresses the p210b3a2 oncoprotein, was transfected with expression plasmids encoding the HLA genes B*3501 or B*3503, and tested for cytolysis with T1/33 (CD8) in CRAs. In Fig. 4, K562 cells expressing B*3501 or B*3503 but not a control HLA allele were lysed by T1/33. Similar experiments have been performed using the autologous restriction element HLA-DRB5*0101 for T1/B9 [39]. Therefore, naturally expressed p210b3a2 can be processed endogenously to epitopes that bind to DRA + B5*0101, B*3501, or B*3503 and are recognized by CTLs (Fig. 4).
Fig. 4.
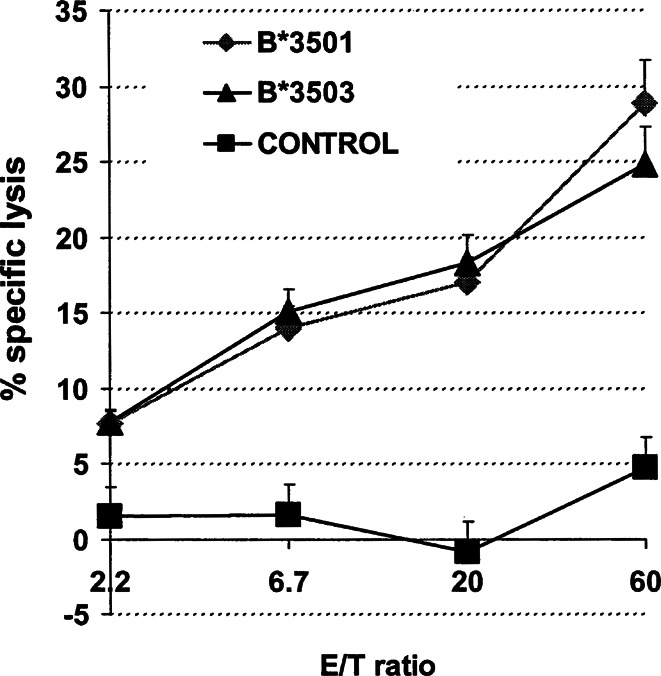
Cytotoxicity profile of clone T1/33 against K562 cells expressing various MHC alleles. K562, a p210b3a2 expressing CML cell line lacking HLA I and II expression, was transfected with genes encoding DRA + DRB1*1501 (negative control, square), B*3501 (diamond), or B*3503 (triangle). HLA-B or DR expression was analyzed by FACS, and bright cells were sorted and used as targets for T1/33 in CRA. Data shown as mean values ± SE from at least three experiments
Discussion
We constructed an rAAV2 vector encoding a truncated protein containing the p210b3a2 fusion region of the BCR-ABL oncogene. Primary DCs transduced with this vector were able to prime lymphocytes from healthy donors, and generated BCR-ABL–specific CTL lines in vitro [39]. A panel of HLA-defined B cell lines was subsequently used to identify HLA-restriction elements for these antigen-specific T cell lines. In the screening process, we used a 25-residue b3a2 peptide that spanned the fusion region. Because most MHC-binding motifs are 8–11 residues in length [34], this 25-residue peptide should contain all HLA-I MCEs, most, if not all, class II MCEs that cover the fusion point, and some MCEs outside the fusion point. Accumulating evidence indicates that synthetic peptides used to load APCs can be efficiently trimmed to MHC-binding epitopes by ubiquitous endoplasmic [10, 23] and/or serum proteases [12], supporting this approach for screening for antigen-specific cytolytic responses.
We identified HLA-B*3501, B*3503, and DRB5*0101 as new HLA restriction elements for p210b3a2 fusion-region epitopes. An HLA-DRB1*1501 restricted, CD4+ T cell line with proliferative but not cytotoxic responses specific for a p210b3a2 fusion-region peptide had previously been described [41]. A CTL line was later shown to demonstrate cytotoxic and proliferative responses to a p210b2a2 fusion-region peptide in a DRB5*0101-restricted fashion [40]. HLA-B*3501 and B*3503 molecules are expressed by approximately 19% of the Caucasian population; and DRB1*1501 by up to 20% [26]. DRB5*0101 most commonly associates with DRB1*1501, but can also associate with DRB1*0101, 1502, and 1602 [36]. Taken together, these findings broaden the BCR-ABL immune-responsive population, potentially widening the application of immunotherapy for CML.
Using serially truncated peptides in cytotoxicity assays, we identified RPVASDFEP as the MCE for HLA-B*3501 recognized by the CD8+ CTL clone T1/33. This epitope was derived from c-abl sequences in close proximity to the fusion point of both b3a2 and b2a2 splicing variants. RPVASDFEP was also the MCE for allogeneic B*3503 recognized by T1/33 (Fig. 3). At the molecular level, B*3503 differs from B*3501 only at position 116 (S to F) for the floor of the peptide-binding groove [20]. However, not all peptides binding to B*3501 can bind to B*3503 [25]. We previously identified an MCE of the DRA + B5*0101 complex recognized by the CD4+ CTL clone T1/B9 as FKQSSKALQ, which includes the p210b3a2 fusion point [39]. CTL clone T1/B9 also recognized DRB1*1101 (allogeneic) presenting different epitopes. In contrast to B*3501 and B*3503, there is a large sequence divergence between these MHC II alleles, and information comparing their three-dimensional structures is not available. It is possible that they may form a similar structure when complexed with a specific epitope. Our findings expand the potential motifs for B*3501. Peptide Q16P (Fig. 3), as well as several longer peptides containing the MCE, were able to sensitize targets to T1/33 (CD8) (data not shown) further confirming that exogenously loaded peptides can be trimmed to antigenic epitopes efficiently.
Identification of RPVASDFEP, which does not contain the BCR-ABL fusion point, as the MCE for B*3501 raised several issues. BCR and ABL proteins are normal cellular constituents, and peptides derived from them might not be highly immunogenic because of self-tolerance. However, self-tolerance may be quantitative rather than absolute [27]. Compared with native BCR and ABL proteins, BCR-ABL oncoproteins are both over and aberrantly expressed in CML cells. In normal cells, only one allele of ABL or BCR is expressed, whereas the BCR-ABL protein is produced in addition to the ABL and BCR products in leukemic cells [4]. Also, the c-abl protein is normally found in both the nucleus and the cytoplasm, while BCR-ABL is exclusively cytoplasmic [45]. Thus, the overexpression, structural alteration, and delocalization of the hybrid BCR-ABL protein might modify processing and presentation of epitopes at the cell surface. Therefore, BCR-ABL sequences outside of the fusion region in CML cells could also be considered as tumor-associated antigens [27, 35]. Potential binding motifs derived from the entire BCR-ABL sequence have been tested for binding to HLA-A1, A2, A3, A11, B7, B8, B27, and B44. Several antigenic peptides were identified and CTLs specific for these peptides were generated from healthy HLA-A2 or B7 donors and one individual with CML. Demonstration of specific immune responses against p210BCR-ABL, but not against fusion-domain peptides, in interferon-treated CML patients in long-term remission provides further support for this contention [30]. Our data provides additional support to the hypothesis that the immune response to normal BCR and c-abl proteins, and to the BCR-ABL oncogene may be different. The CTL clone T1/33 proliferated specifically after incubation with autologous DCs transduced with an rAAV vector encoding truncated BCR-ABL, but not against analogous DCs transduced with a control rAAV vector encoding human placental alkaline phosphatase (data not shown). Furthermore, antigen-specific cytotoxicity was only detected against BCR-ABL peptide-pulsed human B or COS-1 cells expressing HLA-B35 (Figs. 1 and 3), suggesting that the RPVASDFEP epitope might not be readily generated from the native c-abl protein in these APCs. These findings might also reflect quantitative differences in the amounts of BCR-ABL protein in leukemic cells compared with c-abl protein in normal cells.
Numerous clinical studies support the hypothesis that host antitumor cellular immune responses play a major role in the control of CML. For example, patients who develop GVHD following bone marrow transplantation (BMT) for CML experienced a lower rate of relapse [44]. In contrast, those undergoing allogeneic BMT for CML using T-cell-depleted grafts experienced a much higher incidence of relapse [15], implying that T-cells are important in disease control via a mechanism termed GVL. The observation that donor lymphocyte infusions can induce remissions in a high percentage of patients with relapsed CML provided further support for this contention. Several studies suggest that the T-cell population responsible for GVL may be distinct from alloreactive T cells causing GVHD [9, 38], and identification of these antitumor T cells may lead to novel adoptive immunotherapy for cancer [14]. It is generally accepted that alloreactivity causing GVHD results from cross-reactive recognition of foreign MHC complexes by self-MHC-restricted T cells rather than by a particular population of T cells [37]. T-cell recognition of allogeneic MHC is often peptide dependent or specific. Recently, several self-MHC-restricted CTLs clones have been shown to specifically recognize allogeneic MHC combined with antigenic peptides derived from commonly expressed proteins [17, 18], overexpressed tumor-associated antigens [24], tumor-specific antigens [25], or infectious organisms [20, 43]. At the clonal level, donor lymphocytes that cross-recognize commonly expressed antigens may cause GVHD as well as GVL, while others that cross-recognize tumor-associated or specific antigens might cause GVL only. The characterization of CTL clone T1/B9 is the first report of allorestriction between DRB5*0101 and DRB1*1101 and allogeneic recognition of epitopes derived from the BCR-ABL oncoprotein; findings which might be useful in a novel immunotherapy for CML with CTLs restricted by non-self MHC [14, 38]. Clone T1/33 recognizes self-HLA-B*3501 and foreign HLA-B*3503 complexes with the BCR-ABL–derived peptide, which might contribute to GVL particularly with donor lymphocyte infusions. Because most patients receive BMT from donors who, despite serological HLA matching, are genetically mismatched, alloreactivity is often of high avidity. Allorestricted antigen recognition of these T-cell precursors will result in their faster activation and proliferation, with subsequent clonal expansion [28].
An ever-growing number of MHC alleles capable of binding peptides corresponding to the junctional regions of the most common BRC-ABL oncoproteins has become evident [22]. However, until recently, studies failed to provide definitive proof of processing of the BCR-ABL hybrid oncoprotein into appropriate antigenic peptides; for example, elution of BCR-ABL junctional peptides from HLA molecules on CML cells [31]. Several BCR-ABL–specific CTL cell lines (some polyclonal) have been generated using peptide-pulsed APCs in vitro, but reports are conflicting as to whether these CTLs can lyse CML cells expressing endogenous BCR-ABL proteins [6, 29, 30, 46]. However, progress is being made. For example, in a clinically relevant murine model, protective immune responses resulting in prolonged survival have been generated after immunization with a p210b3a2 fusion-region–specific peptide [16]. Recently, the first direct evidence of leukemic cells presenting HLA-associated peptides derived from the p210b3a2 fusion region has been reported using mass spectrometry with nanospray ionization [8]. Our T-lymphocyte lines generated after priming and stimulation with rAAV-transduced, mature DCs have shown specific responses to autologous DCs expressing truncated p210b3a2 endogenously [39]. When the restriction elements and MCEs were precisely defined for clones T1/B9 and T1/33, we demonstrated that K562, a p210b3a2 expressing CML cell line, was specifically lysed after introduction of the appropriate HLA genes, supporting the conclusion that native BCR-ABL can be processed, presented, and recognized by our T-cell clones (Fig. 4). Characterization of the precise epitopes generated by intracellular degradation of p210b3a2 would, at least theoretically, pave the way to an immunotherapeutic approach of CML.
In summary, we describe three previously unreported HLA alleles (DRB5*0101, B*3501, and B*3503) that were capable of presenting p210b3a2 fusion epitopes. In addition, we confirmed the ability of previously described HLA alleles, such as DR11, to present the p210b3a2 fusion region. These findings have the potential for increasing the utility of CML immunotherapy. For example, DRB5*0101 associates not only with DRB1*1501, but also with other alleles including DRB1*0101, 1502, and 1602 [36]. In addition, HLA-B*3501 and B*3503 are expressed by approximately 19% of the Caucasian population [26]. Overall, these findings expand the population base that can generate immune responses to the BCR-ABL fusion region and thus broaden the potential application of immunotherapy for CML.
Acknowledgements
We thank Drs Eric O. Long, Pierre van der Bruggen, Osam Mazda, and Gerald Nepom for generously providing plasmids or cell lines; and James Bolen, Lucy Brown, and Claudio Spalla of the City of Hope FACS Core Facility for help with FACS analyses. This work was supported in part by Grants IR01CA75186, 5P01 CA30206, and CA33572 from the National Institutes of Health and the National Cancer Institute. TCRα and β chain gene cDNA sequences of variable region and partial joint region reported in this paper have been deposited in the GenBank database, accession numbers AF542062 and AY157620.
References
- 1.Bergsagel Oncogene. 2001;20:5611. doi: 10.1038/sj.onc.1204641. [DOI] [PubMed] [Google Scholar]
- 2.Bocchia Blood. 1996;87:3587. [PubMed] [Google Scholar]
- 3.Breiteneder Immunogenetics. 1995;42:53. doi: 10.1007/BF00164987. [DOI] [PubMed] [Google Scholar]
- 4.Buzyn Eur J Immunol. 1997;27:2066. doi: 10.1002/eji.1830270834. [DOI] [PubMed] [Google Scholar]
- 5.Chatterjee Curr Top Microbiol Immunol. 1996;218:61. doi: 10.1007/978-3-642-80207-2_5. [DOI] [PubMed] [Google Scholar]
- 6.Chen J Immunother. 1998;21:257. doi: 10.1097/00002371-199807000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Clark Leuk Lymphoma. 2001;42:871. doi: 10.3109/10428190109097706. [DOI] [PubMed] [Google Scholar]
- 8.Clark Blood. 2001;98:2887. doi: 10.1182/blood.V98.10.2887. [DOI] [PubMed] [Google Scholar]
- 9.Datta Bone Marrow Transplant. 1994;14:517. [PubMed] [Google Scholar]
- 10.Day Proc Natl Acad Sci USA. 1997;94:8064. doi: 10.1073/pnas.94.15.8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diamond Blood. 1997;90:1751. [PubMed] [Google Scholar]
- 12.Eberl Mol Immunol. 1999;36:103. doi: 10.1016/S0161-5890(99)00023-1. [DOI] [PubMed] [Google Scholar]
- 13.Elliott Cell Mol Life Sci. 2002;59:373. doi: 10.1007/s00018-002-8429-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao Blood. 1999;94:2999. [PubMed] [Google Scholar]
- 15.Hale Bone Marrow Transplant. 1994;13:597. [PubMed] [Google Scholar]
- 16.He Cancer Immunol Immunother. 2001;50:31. doi: 10.1007/PL00006680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heemskerk Proc Natl Acad Sci USA. 2001;98:6806. doi: 10.1073/pnas.111162298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kageyama J Immunol. 2001;166:3028. doi: 10.4049/jimmunol.166.5.3028. [DOI] [PubMed] [Google Scholar]
- 19.Kazatchkine Immunogenetics. 1995;42:451. [Google Scholar]
- 20.Khanna Eur J Immunol. 1999;29:1587. doi: 10.1002/(SICI)1521-4141(199905)29:05<1587::AID-IMMU1587>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 21.Koop Genomics. 1994;19:478. [Google Scholar]
- 22.Leeksma OC, Kessler JH, Huijbers IJ, Ten Bosch GJ, Melief CJ. BCR-ABL directed immunotherapy: a virtual reality? Leuk Lymphoma. 2000;38:175. doi: 10.3109/10428190009060331. [DOI] [PubMed] [Google Scholar]
- 23.Levitt Eur J Immunol. 2001;31:1181. doi: 10.1002/1521-4141(200104)31:4<1181::AID-IMMU1181>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 24.Luiten Tissue Antigens. 2000;56:77. doi: 10.1034/j.1399-0039.2000.560110.x. [DOI] [PubMed] [Google Scholar]
- 25.Mandruzzato J Immunol. 2000;164:4130. doi: 10.4049/jimmunol.164.8.4130. [DOI] [PubMed] [Google Scholar]
- 26.Marsh SG, Parham P, Barber LD (2000) The HLA factsbook. Academic Press, London
- 27.Morgan Immunol. 1998;160:643. [PubMed] [Google Scholar]
- 28.Munz J Immunol. 1999;162:25. [Google Scholar]
- 29.Nieda Blood. 1998;91:977. [PubMed] [Google Scholar]
- 30.Oka Leukemia. 1998;12:155. doi: 10.1038/sj.leu.2400919. [DOI] [PubMed] [Google Scholar]
- 31.Papadopoulos Blood. 1997;90:4938. [PubMed] [Google Scholar]
- 32.Padovan Science. 1993;262:422. doi: 10.1126/science.8211163. [DOI] [PubMed] [Google Scholar]
- 33.Pawelec Blood. 1996;88:2118. [PubMed] [Google Scholar]
- 34.Rammensee Immunogenetics. 1995;41:178. doi: 10.1007/BF00172063. [DOI] [PubMed] [Google Scholar]
- 35.Rongcun J Immunol. 1999;163:1037. [PubMed] [Google Scholar]
- 36.Schreuder Hum Immunol. 1991;32:141. doi: 10.1016/0198-8859(91)90111-L. [DOI] [PubMed] [Google Scholar]
- 37.Sherman Annu Rev Immunol. 1993;11:385. doi: 10.1146/annurev.immunol.11.1.385. [DOI] [PubMed] [Google Scholar]
- 38.Stauss Immunol Today. 1999;20:180. doi: 10.1016/s0167-5699(99)01443-7. [DOI] [PubMed] [Google Scholar]
- 39.Sun Cancer Res. 2002;62:3175. [PubMed] [Google Scholar]
- 40.Ten Leukemia. 1995;9:1344. [Google Scholar]
- 41.Ten Blood. 1999;94:1038. [PubMed] [Google Scholar]
- 42.Tomescu Trends Mol Med. 2001;7:554. doi: 10.1016/S1471-4914(01)02244-4. [DOI] [PubMed] [Google Scholar]
- 43.Tomiyama Eur J Immunol. 2000;30:2521. doi: 10.1002/1521-4141(200009)30:9<2521::AID-IMMU2521>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 44.Weiden N Engl J Med. 1979;300:1068. doi: 10.1056/NEJM197905103001902. [DOI] [PubMed] [Google Scholar]
- 45.Wetzler J Clin Invest. 1993;92:1925. doi: 10.1172/JCI116786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zorn Transplantation. 2001;71:1131. [Google Scholar]