

Abstract
The use of antineoplastic drugs for cancer treatment is frequently associated with the acquisition of a multidrug-resistant (MDR) phenotype that renders tumoural cells insensitive to antineoplastics. It remains elusive whether the acquisition of the MDR phenotype alters immunological parameters that could influence the cell sensitivity to an eventual host immune response. We report that immunisation of syngeneic mice with γ-irradiated L1210S (parental line) and L1210R (MDR phenotype) cells results in a significant rejection of subsequently implanted L1210R-based tumours, but not of the L1210S ones. Notably, L1210R tumours display a twofold reduction in vivo proliferative capacity and are less aggressive in terms of mouse survival than their sensitive counterparts. Also, analysis of surface expression of molecules involved in antigen presentation and cytokine activity revealed a slight increase in IFN-γ receptor expression, a decrease of Fas molecule, and a fourfold up-regulation of MHC class I molecules in L1210R cells. Nonetheless, both cell lines were able to induce a cytotoxic response in syngeneic mice and were equally susceptible to cytotoxicity by splenic cells. Together, these findings indicate that acquisition of drug resistance by L1210 cells is accompanied by pleiotropic changes that result in reduced tumour proliferative capacity and tumorigenicity in syngeneic mice. Hence, immunological studies of MDR tumours may assist in the design of specific therapeutic strategies that complement current chemotherapy treatments.
Keywords: Apoptosis, Cell surface molecules, Fas molecule, MDR, MHC class I, Tumour immunity
Introduction
The phenomenon of multidrug resistance (MDR) was observed when tumours treated with antineoplastic drugs developed cross-resistance to other cytotoxic agents to which they had never been exposed, effectively eliminating the possibility of treating these tumours with chemotherapy [1]. MDR phenotype is pleiotropic and frequently associated with the overexpression of efflux pumps such as P-glycoprotein (P-gp) that extrude antitumoural drugs [22]. In addition, the MDR phenotype may result from heritable changes in cancer cells that cause altered levels of specific or mutant proteins [1]. These genetic alterations can affect different aspects of cell dynamics [12, 22].
There is cumulative information suggesting a connexion between the MDR phenotype and the immunological properties of cells [11, 13, 19, 21, 23, 31, 32]. For instance, it has been described that IFN-γ up-regulates P-gp expression and activity in human peripheral blood monocyte–derived macrophages, and it may be involved in the induction of MDR in these cells [19]. Increased expression of the transport-associated protein (TAP) in MDR human cancer cell lines, and slightly higher amounts of common tumour-associated antigens on the surface of resistant cells have been also observed [11, 13, 21]. Intriguingly, while some researchers show that expression of P-gp on human MDR tumour cells did not affect their susceptibility to natural and lymphokine-activated killer cell–mediated death [23], other groups reported the resistance of MDR to natural killer–like cell-elicited cytotoxicity [32]. Consistent with this finding, the MDR phenotype appears to provide resistance to complement-mediated cytotoxicity [31]. Additionally, inconsistent results on the susceptibility of MDR cells to Fas-induced programmed cell death have been obtained [14]. For instance, Smyth et al. [26] have suggested an important protective role of P-gp on Fas-mediated apoptosis, whereas other researchers have not found such a correlation between these two factors [8, 9].
These discrepancies may be due to the different models of resistance that have been used by different researchers. Probably the acquisition of MDR phenotype induces pleiotropic changes on tumoural cells that depend on the origin and on the antineoplasic agent used to generate resistance. At this point, closer studies on the different resistance models should be done in order to adapt therapeutic strategies depending on the specific phenotype that has been induced on each type of tumoural cell.
Despite the wealth of information accrued, the alteration of immunological parameters such as tumorigenicity or surface protein expression upon the acquisition of the MDR phenotype remains elusive. We have addressed this question and report that acquisition of MDR phenotype affects the immune characteristics of the murine leukemic cell line L1210. L1210 sensitive (L1210S) and daunomycin (DNM)-resistant (L1210R) tumour cells were inoculated into syngeneic mice to monitor the onset of tumour appearance, the rate of tumour growth, and the extent of mouse survival. Immunological analysis of changes associated with the acquisition of DNM resistance are described.
Materials and methods
Mice and tumour cells
Female DBA/2 mice, 6–8 weeks old, were purchased from Jackson Laboratories (Bar Harbor, ME, USA). All mice were housed and maintained under specific pathogen-free conditions at the Animal Facilities of the University Miguel Hernández for at least 1 week before use. All animals received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 85-23, revised 1985). The L1210S, leukemic cell line of DBA/2 origin, was used as tumour model. Parental murine leukaemia L1210S cells and a DNM-resistant (L1210R) subline (~160-fold resistant to DNM) selected as previously described [6] were maintained in RPMI 1640 medium (BioWhittaker, Walkersville, MD, USA) supplemented with 10% FBS (BioWhittaker), 2 mM L-glutamine, 10 U/ml penicillin G and 10 μg/ml streptomycin sulphate (BioWhittaker).
Antibodies
Antibodies for flow cytometry and neutralisation experiments were purchased from Pharmingen (San Diego, CA, USA).
In vivo tumour model
To investigate in vivo growth characteristics of L1210S and L1210R cell lines, we injected 0.5×103 cells subcutaneously (s.c.) in the right flank of syngeneic DBA/2 mice. Tumour size (mm2) and survival (percentage) were monitored. The size of the tumours (mm2) was measured three times a week and calculated by multiplying the vertical length of the tumour by its horizontal length. To induce immunological memory, 5×105 L1210S or L1210R γ-irradiated cells (6,000 rads) were injected s.c. on the right flank of mice 1-h postirradiation. After 15 days, 0.5×103 nonirradiated cells (L1210S or L1210R) were injected on the contralateral side. Date of tumour appearance and growth rate, as well as mouse survival were monitored [33].
In vitro proliferation assays
In vitro proliferation assays were performed to compare the growth rate of L1210S and L1210R cells. The number of viable cells were counted by using trypan blue staining after plating 2×104 cells/well on a P-24 flat-bottom plate for 5 days.
Surface staining and flow cytometry
A total of 1×106 L1210S and L1210R cells were washed once with staining buffer (PBS/3% FCS), blocked with Fc block (purified antimouse CD16/CD32 monoclonal antibody), and surface-stained with the following biotin-labelled antibodies: antimouse H-2 Kd MHC class I (clone SF1-1.1), antimouse I-A/I-E MHC class II (clone 2G9), antimouse Fas (clone Jo2), antimouse IFN-γ receptor α chain (mouse CD119, clone GR20) and antimouse Fas ligand (FasL; clone MFL4). Cells were washed twice with staining buffer and incubated with PE-conjugated streptavidin (Pharmingen) (dilution 1:1,000). Cells were washed twice in staining buffer, and the analysis was performed on a flow cytometer (Beckman Coulter, Miami, FL, USA) argon-ion laser at 15 mW and 488 nm. A total of 10,000 events were collected and analysed using Expo-32 software (Beckman Coulter). When specified, cells were incubated for 24 h at 37°C with recombinant murine IFN-γ (Pharmingen).
CTL activity assay
Mice were immunised three times, 14 days apart, by i.p. injection with 1.5–2×106 γ-irradiated L1210S or L1210R cells. For challenge, 2×106 nonirradiated cells in 0.2 ml of saline, were injected i.v. 14 days after the last immunisation and 1 day before sacrificing the mice. As controls, nonimmunised mice were used. Single-cell suspensions of splenocytes from four mice per group were obtained. The splenocytes (6×106/ml) were stimulated in vitro with 8×104 L1210S-irradiated cells (or their resistant counterparts) for 5 days at 37°C, in 5% CO2 and in the presence of IL-2 (500 U/ml). For evaluation of cytotoxicity, fresh target cells, L1210S or L1210R (1×104), were mixed with effector cells at E/T ratios (effector cells to target cells) of 100:1, 30:1, 10:1 and 3:1 and incubated for 4 h. After incubation, propidium iodide (10 μg/ml) was added to each well, and the samples were analysed by flow cytometry following the procedure previously described [30].
Daunomycin accumulation assays
Steady-state intracellular accumulation assays of the fluorescent DNM in the absence or presence of Verapamil (VRP) was determined as previously described [27, 28].
Apoptosis assay
L1210S and L1210R cells were cultured in fresh medium and plated at 3×105/well on a 24-well plate. An antimouse Fas monoclonal antibody (2.5–20 μg/ml, clone Jo2, hamster IgG, λ) plus protein G (2 μg/ml) was added to the cells and incubated for 24 h. As negative control (NC), we have used a nonrelated antibody of the same isotype (purified hamster IgG2, λ) at 2.5–20 μg/ml, as specified above. After incubation, apoptosis was evaluated by using FITC-annexin V (Pharmingen) according to manufacturer’s instructions.
Statistical analysis
Data were analysed with Student’s t test. Significance was defined as p<0.05.
Results
In vitro proliferation of L1210S and L1210R cells
To study tumour development in mice induced by murine leukaemia L1210 cells, we studied the in vitro growth rate of the parental L1210S cells and that of a DNM-resistant L1210R cell subline (see “Materials and methods”). For this task, L1210S and L1210R cells (2.0×104 cells/ml) were cultured for 5 days in fresh medium, and the extent of cellular proliferation was evaluated by counting the number of viable cells using trypan blue. As illustrated in Fig. 1, the in vitro growth behaviour of L1210 cells appears to be biphasic, i.e. there is an initial slow phase followed by a fast-growing component. Note that no differences in proliferation occur between L1210S and L1210R cells in the initial phase. In contrast, L1210R cells exhibit a conspicuous ≈40% slower fast-growing step than their sensitive counterpart. Thus, acquisition of the resistant phenotype affects the in vitro proliferation rate of L1210 tumour cells.
Fig. 1.
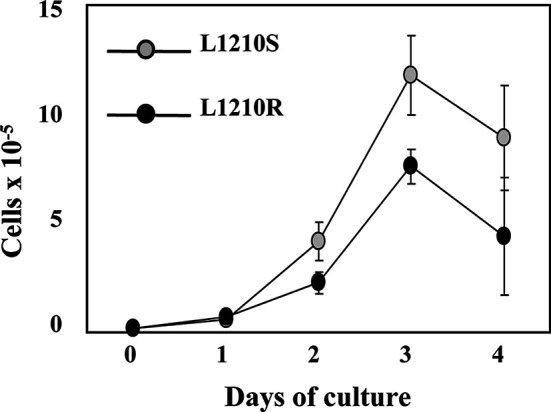
In vitro proliferative capacity of L1210S and L1210R cell lines. Cells (2×104 cells/well) were plated on a P-24 flat-bottom plate for 5 days, and the number of viable cells was counted by using trypan blue staining. Data are mean ± SEM of three separate experiments.
L1210R-based tumours grow slower in vivo than those produced by L1210S cells
To address the influence of the MDR phenotype on in vivo tumour development, we compared the tumorigenicity of L1210S and L1210R cells in syngeneic mice. For this purpose, 0.5×103 L1210S or L1210R cells were injected subcutaneously into the right flank of DBA/2 mice, and the time course of tumour development as well as mouse survival were monitored for >36 days. The onset of tumour appearance was virtually identical for both cell lines (data not shown). In contrast, a conspicuous difference in tumour growth between L1210S and L1210R cells was observed (Fig. 2a). As seen, L1210R-based tumours exhibited a remarkable 50% lower proliferation than those originated from L1210S cells. The difference in in vivo proliferation between L1210S and L1210R cells is akin to that displayed in vitro, namely while the initial slow phase was similar, the rate of tumour production that followed was different.
Fig. 2.
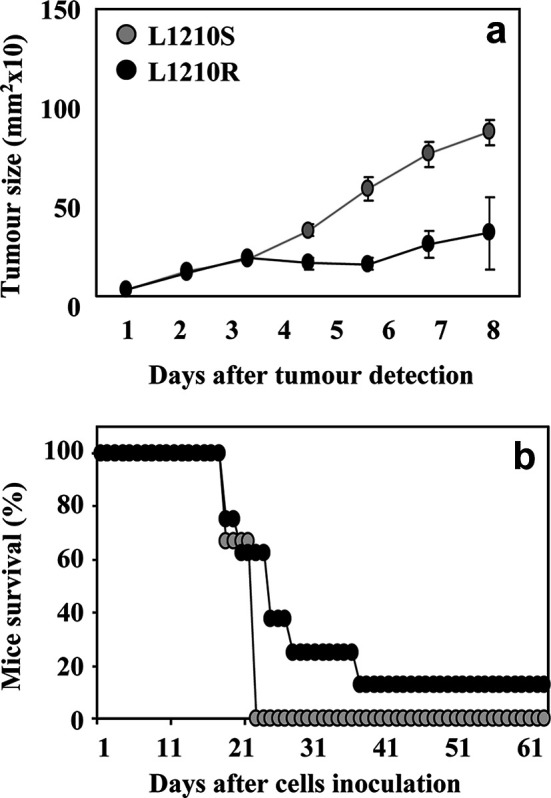
Tumorigenicity of L1210S and L1210R cells in syngeneic DBA/2 mice. Mice (eight per group) were inoculated (s.c.) in the right flank with 0.5×103 L1210S (grey circles) or L1210R (black circles) cells. Growth rate of tumours (a) and percentage of mouse survival (b) were monitorised from the day of cell inoculation (day 0).
The moderate growth capacity of L1210R-based tumours resulted in a significant prolongation of mouse survival as compared to animals expressing L1210S-based tumours (Fig. 2b). It is noteworthy that a significant 10% of the animals rejected the growth of L1210R-based tumours and survived them (Fig. 2b). These results suggest that L1210R cells are less tumorigenic in vivo than the parental sensitive cell line. The lower tumorigenicity of L1210R cells might result from both their slower growth rate and a potential higher susceptibility to the host immune system.
Induction of an immunological response by L1210S or L1210R cells affects the proliferation of L1210R-based tumours
To determine if the induction of an immune response could modulate L1210R-based tumour progress and/or tumour rejection in vivo, syngeneic mice were preimmunised with γ-inactivated cells from both L1210 sublines. For this purpose, DBA/2 mice were inoculated with 5×105 γ-irradiated L1210S or L1210R cells. Irradiated cells lost their proliferative capacity, as evidenced by the lack of cellular growth in culture (data not shown). After 2 weeks, mice were contralaterally challenged with 0.5×103 L1210S or L1210R cells, and the time course of tumour development was monitored for >36 days. The onset of tumour appearance and the tumour growth rate did not change significantly after preimmunisation with either γ-irradiated cell subline (Fig. 3a–f). As seen, L1210S and L1210R tumours were detectable 6–8 days after inoculation of viable cells (Fig. 3a, b). Analysis of the tumour growth rate revealed that, independent of the type of cells used to preimmunise the animals, L1210S-based tumours in vaccinated animals grew 2.3-fold faster (200 mm2/day) than L1210R tumours (86 mm2/day) (Fig. 3c, d). In agreement with these observations, mice inoculated with L1210S cells survived for only 21 days independent of the type of cells used for preimmunisation (Fig. 3e). In contrast, 25% of mice vaccinated with γ-irradiated L1210S and L1210R cells not only rejected the development of L1210R-based tumours but survived this challenge (Fig. 3f). Collectively, a plausible explanation of these results is that L1210R cells are more sensitive to the host immune response than the parental L1210S line, presumably because of some changes in the expression of surface proteins involved in immunity.
Fig. 3.

Tumorigenicity and immunological response of L1210S and L1210R cells in syngeneic DBA/2 mice. Mice (eight per group) were inoculated (s.c.) in the right flank with 5×105 γ-irradiated cells L1210S (Irrad-L1210S, grey circles) or L1210R (Irrad-L1210R, black circles). After 15 days, these mice were injected s.c. on the left flank with 0.5×103 L1210S (a, c, e) or L1210R (b, d, f) cells. As control (white circles) non-preimmunised mice were inoculated with 0.5×103 L1210S or L1210R cells. Percentage of tumour-free mice (a, b), growth rate of tumours (c, d) and mouse survival (e, f) were monitored from the day of viable cell inoculation (day 0).
Differential expression of surface proteins involved in immunity between L1210S and L1210R cells
The molecular basis underlying the slower growth rate and higher rejection of L1210R-based tumours in vivo could be due to the existence of alterations in surface-expressed antigens that may expose these cells or make them more sensitive to the immune system. Hence, we evaluated the expression of surface molecules implicated in cytokine activity (IFN-γR), Fas-mediated apoptosis (Fas and FasL) and antigen presentation (MHC class I, class II) on both cell sublines, by flow cytometry.
Whereas no expression of MHC class II and FasL molecules was detected in either cell line (data not shown), a slight but significant increase of IFN-γR was determined in L1210R cells (Fig. 4b). In addition, L1210R cells exhibited a significant decrease of Fas expression with respect to L1210S cells (Fig. 4c), suggesting that the resistant cells could be insensitive to Fas-induced apoptosis. To evaluate this issue, we compared the apoptosis induced by an isotype-matched control with that triggered by an anti-Fas mAb that is capable of inducing apoptotic cell death in vitro and in vivo (Fig. 5) [4, 18]. As depicted in Fig. 5a (top) and b, the anti-Fas mAb (10 μg/ml) induced a ≈4-fold increment in L1210S apoptotic death. Fas-induced apoptosis of L1210S cells remained unaltered in the concentration range of 2.5–20 μg/ml of antibody (data not shown). A similar result was observed with tymocytes (Fig. 5a [bottom] and b). In marked contrast, L1210R did not respond to Fas-mediated apoptosis up to an antibody concentration of 20 μg/ml (Fig. 5a [middle], and b), a result that is consistent with the lower surface expression of Fas exhibited by these cells.
Fig. 4.

a–c Differential expression of surface molecules on L1210S and L1210R cells. The expression levels were measured by flow cytometry using specific antibodies (black bars). As NC, a non-related antibody with the same isotype as the experimental antibodies (data not shown). A different scale has been used to represent MHC class I expression. Data are mean ± SEM of eight separate experiments. *p<0.05; **p<0.01, as compared to expression levels on L1210S parental cell line (Student’s t test).
Fig. 5.
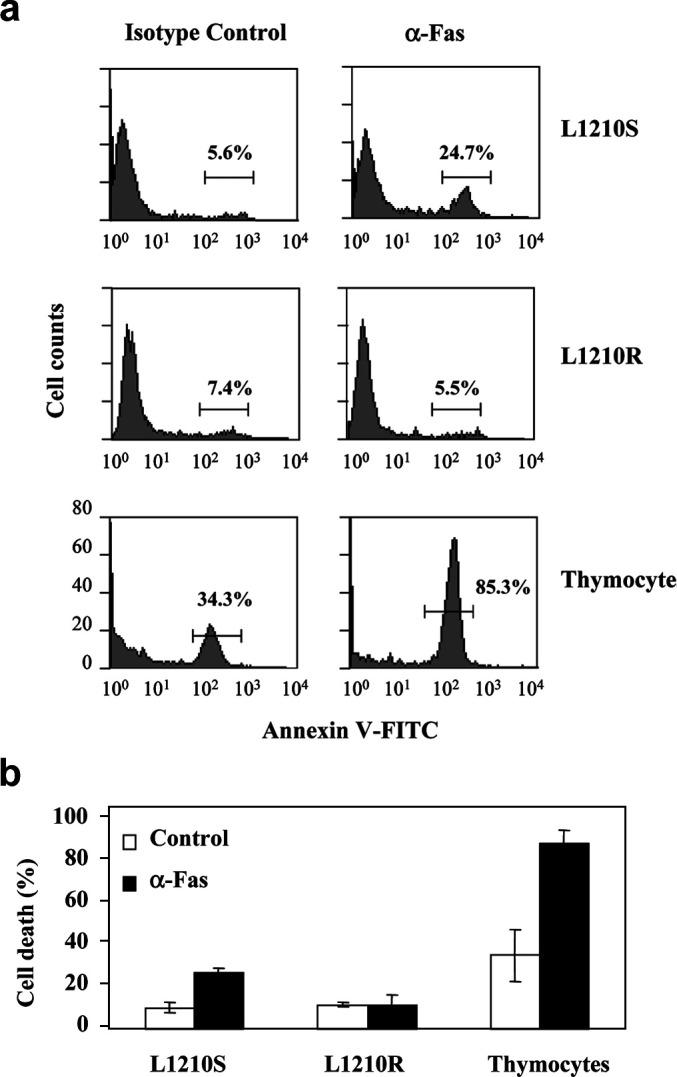
Cell death induced on L1210S and L1210R cells by an antibody specific for mouse Fas. Cells were incubated with protein G (2 μg/ml) plus anti-Fas antibody (10 μg/ml) for 24 h. As NC, protein G (2 μg/ml) plus 10 μg/ml of a non-related antibody from the same isotype as the anti-Fas antibody (hamster IgG2, λ) was used. Apoptosis was monitored with the annexin V assay. a Flow cytometry histograms of L1210S, L1210R cells and thymocytes treated with protein G plus isotype control (NC) or protein G plus anti-Fas antibody. b Bar histograms of annexin V-FITC assay results showing background death (white bars) and Fas-dependent cell death (black bars). Thymocytes were used as positive control for Fas-induced apoptotic death. Data are mean ± SEM of three separate experiments. *p<0.05; **p<0.01, as compared to NC (Student’s t test).
The most significant difference in surface antigen observed between L1210S and L1210R cells is a remarkable fourfold increase of MHC class I expression in the L1210R cells (Fig. 4a), which is involved in CTL-related cytotoxicity. No expression differences were observed in MHC class II in either subline (data not shown). Collectively, these findings demonstrate that acquisition of the MDR phenotype by L1210 cells is associated with significant changes in the expression of surface proteins directly involved in immune response, specially MHC class I.
L1210R cells display similar susceptibility to lysis by cytotoxic T lymphocytes to that of their sensitive counterpart
Based on the data of MHC class I expression on resistant cells, we next assessed whether this characteristic of L1210R cells was responsible for an increased recognition and elimination of the tumour cells by the host immune response that would explain the lower tumorigenicity of L1210R cells related to the parental subline. For this purpose, we investigated whether both cell lines could have a different capacity to induce tumour-specific CTLs as well as different susceptibility as target cells in cytotoxic assays. As illustrated in Fig. 6, mice immunised with L1210S or L1210R were capable of generating tumour-specific CTLs. Furthermore, we observed that CTLs from mice immunised with the parental cell line lysed both L1210S and L1210R. Similarly, mice immunised with L1210R cells were also able to lyse both sensitive and resistant cells. These results indicate similar sensitivity to CTL-mediated killing of both parental cells and their isogenic sublines with the acquired MDR phenotype. Thus, despite the differential expression of MHC class I and IFN-γR, our data demonstrate that both cell lines are capable of inducing CTL activity and are equally susceptible to in vitro lysis by CTLs.
Fig. 6.
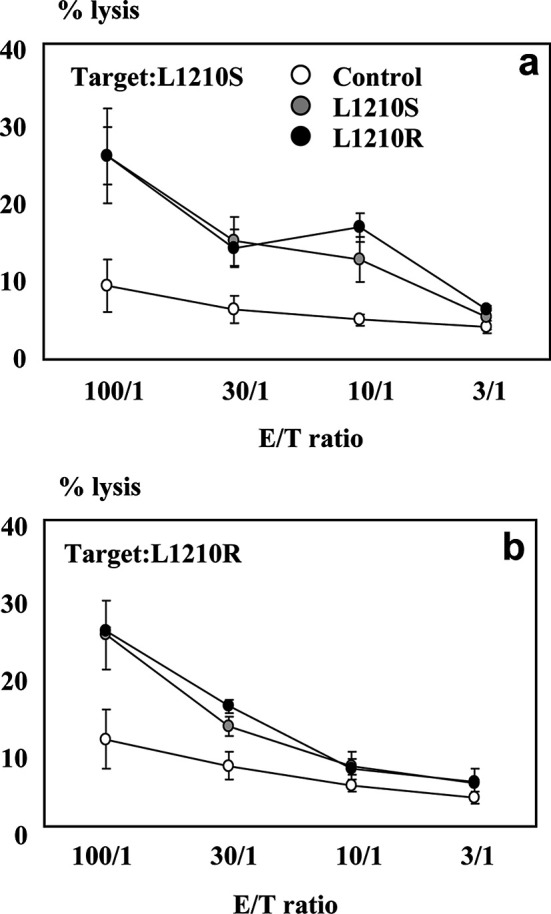
Cytotoxic activity of L1210S- or L1210R-immunised mice. a Specific lysis of L1210S cells (target cells) by splenic cells from control, L1210S- or L1210R-immunised mice. b Specific lysis of L1210R cells (target cells) by CTLs from control, L1210S- or L1210R-immunised mice. Data are mean ± SEM of four separate experiments.
Discussion
A better understanding of the MDR phenomenon requires the characterisation of all cellular changes associated with this phenotype. Numerous in vitro studies using drug-resistant, MDR-transfected or chemosensitiser-treated cells have shown the up-regulation of extrusion pumps like P-gp on these cells and its role in different physiological processes [3, 18, 22]. However, few studies have addressed whether acquisition of the pleiotropic MDR phenotype can modify their susceptibility to factors other than antineoplastic drugs, such as the capacity for tumour surveillance by the host immune system.
Here, we have studied sensitive and resistant cells of the leukaemic cell line L1210 in its phenotypic and functional immunological aspects. A salient contribution of our work is that L1210R cells give rise to tumours that grow much slower than their L1210S counterparts (Fig. 2c). Furthermore, preimmunisation with either γ-irradiated L1210S or L1210R cells drastically inhibited L1210R tumour progression and in a significant 25% subset of animals led to complete tumour rejection and disappearance (Fig. 3b). These findings imply that L1210R cells have lower in vivo proliferative capacity and are more easily defeated by the immune system than the parental drug-sensitive line. Accordingly, the acquisition of DNM resistance by the leukaemic L1210 cell line is associated with changes in the expression of surface antigens that could make resistant cells more vulnerable to a host immune response.
To address this issue, we performed in vitro experiments aimed at correlating the in vivo findings with changes in both the proliferation capacity of L1210 cells upon acquiring the MDR phenotype, and the expression of surface molecules involved in immune system recognition. Thus we noticed that L1210R cells proliferated 40% slower than L1210S cells when cultured in vitro in fresh medium (Fig. 1). Although this characteristic could significantly contribute to the distinct in vivo proliferation capabilities between both cell lines, it appears inadequate to account for the high rejection percentage of L1210R-based tumours in vaccinated mice, implying the occurrence of additional immune mechanisms. In this regard, we analysed whether an increased susceptibility of L1210R cells to apoptotic stimuli could be related to the lower tumour progression observed in L1210R-based tumours in comparison with those induced by L1210S cells. L1210R cells displayed a down-regulation of Fas molecule with respect to their parental counterpart, and exhibited a complete insensitivity to Fas-induced programmed cell death (Fig. 5). Our findings are in agreement with results reported by others showing that acquisition of MDR in human myeloma (8226) and T-cell leukaemia (CEM) is accompanied by a decrease in the number of Fas surface sites and a desensitisation to Fas-induced apoptosis [7, 15–17]. However, such observations are not compatible with the involvement of Fas mediating a signalling pathway to explain the differences in tumour progression between L1210R- and L1210S-based tumours. Nonetheless, caution should be exerted, taking into account that L1210R cells may have a higher susceptibility than L1210S cells to enter into apoptosis by mechanisms other than the interaction of Fas with its ligand FasL. In fact, Castro-Galache et al. [6] have recently reported that L1210R cells are more sensitive to apoptosis induction by histone deacetylase inhibitors than the parental L1210S cell line. In addition, we observed that L1210R are more sensitive to stress by nutrient deprivation, especially when growing at a high cellular density (data not shown). Thus, while acquisition of the MDR phenotype insensitises the tumour cells against Fas-induced cell death, it may concomitantly prime these cells to other cell death-inducing pathways.
Interestingly, L1210R cells display a substantial up-regulation of MHC class I, a molecule which plays a key role in CTL-mediated cytotoxicity. The higher expression of MHC class I should increase the efficiency of L1210R cells in tumour-associated or tumour-specific antigen presentation, thus favouring its detection by the tumour-specific CTLs [24, 29]. These findings provide a molecular explanation for previous observations describing a higher immunogenicity of L1210R cells related to an increment in the amount or density of tumour-associated antigens on the cell surface [11, 21]. Our results (Fig. 6) show that the enhanced expression of MHC class I in L1210R cells did not make any difference in the capacity of these cells to induce tumour-specific cytotoxic cells or in their sensitivity to lysis in vitro by the effector cells. These results are in agreement with Scheper et al. [23] who have shown that the selection of RPMI-8226 myeloma cells for resistance to doxorubicin or mitoxantrone did not alter the sensitivity to in vitro lysis by LAK or NK. Additionally, data from Shtil et al. [25] demonstrate similar sensitivity to CTL-mediated killing of MPC11 parental cells and their isogenic sublines with acquired MDR. In our system, differential contribution of NK cells and CTLs to tumour cytotoxicity could account for the similar results observed in L1210S and L1210R cells. A plausible explanation is the presence of a “compensatory mechanism of lysis” exerted by NK cells and CTLs. At the first stages of tumour development, innate immunity would defend the organism from proliferation of transformed cells; in particular, NK cells would recognise tumour cells with a deficient expression of class I molecules. The decreased expression of MHC I molecules in L1210 sensitive cells would make them sensitive targets of NK cells. In contrast, the higher expression of MHC class I on L1210R cells would protect them from the initial attack by NK cells, but would increase their susceptibility to specific immunity, i.e. recognition and killing by CD8+ T cells. This coordinated mechanism of cytotoxicity would be consistent with the similar lytic activity obtained from cytotoxic splenic cells against L1210S and L1210R cells. Taken together, these data demonstrate a host-induced immune response by L1210S and L1210R cells, and suggest that lysis of tumour cells by NK or CTLs may contribute to, but it does not appear to determine, the lower tumorigenicity of resistant vs sensitive cells. Further studies are necessary to uncover the molecular mechanisms underlying the reduction in in vivo tumorigenicity of MDR cells.
In summary, our findings lend support to previous observations that the generation of immunogenicity was characteristic of tumour resistance [10]. Indeed, Fichtner et al. [10] found that drug-resistant P388/Mitox and P388/Vinc developed immunogenicity that was absent in the parental cell line P388. Furthermore, vaccination with lethally irradiated drug-resistant cells resulted in a substantial rejection of viable tumour cells of the same line. Moreover, Azuma et al. [2] demonstrated CTL generation against the MDR cell line P388/ADR but not against the parental cells. Although additional experimental evidence is required, our results along with those of Fichtner et al. [10] suggest that acquisition of drug resistance is concomitantly associated with increased immunogenicity.
Conclusion
In conclusion, we reported that tumours from resistant cells grow at a slower rate in vivo, and are more sensitive to preimmunisation with either γ-irradiated cells. The slower growth rate may be explained in terms of a higher susceptibility to spontaneous apoptotic death. The higher rejection of L1210R-based tumours as compared with those of the parental, sensitive line might be due to significant changes in the expression of tumour-specific or tumour-associated antigens presented in the context of MHC class I molecules that show higher expression levels on the resistant cells in comparison to the sensitive cell line. These facts would make these cells more noticeable to a host immune response. However, a slower metabolic rate of resistant tumours and/or defects on angiogenesis cannot be fully ruled out [16]. It is tempting to propose that adjuvants or agents that stimulate and potentiate the immune response against resistant tumour-presenting antigens, alone or in combination with antineoplastic drugs, may decrease the devastating effects of the MDR phenotype. Thus, more studies toward a precise characterisation of the immunological characteristics of resistant tumours should be performed for efficient therapy design.
Acknowledgements
We thank Dr Manuel Sánchez and Dr Rosa Planells-Cases for critical reading of this manuscript, Dr Miguel Saceda and Dr Gonzalo Rubio for helpful suggestions and discussions, and Dr Agustín Beltrán de Heredia-Rueda and Dr José A. Pérez de Gracia for helpful assistance with γ-irradiation of cells and in the animal facilities.This work has been supported by the Comisión Interministerial de Ciencia y Tecnología (CICYT) and the European Commission, grants SAF-2000-0142 and 1FD97-0662-C02-01, and the Instituto de Salud Carlos III grant FISSS-01/0038-02 and 01/0038-01 (to J.A.F. and A.F-M.). Elena Martín-Orozco was the recipient of a contract from the Spanish Ministry of Science and Technology.
References
- 1.Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. 1999;39:361. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 2.Azuma E, Masuda S, Qi J, Kumamoto T, Hirayama M, Nagai M, Hiratake S, Umemoto M, Komada Y, Sakurai M. Cytotoxic T-lymphocytes recognizing P-glycoprotein in murine multidrug-resistant leukemias. Eur J Haematol. 1997;59(1):14. doi: 10.1111/j.1600-0609.1997.tb00954.x. [DOI] [PubMed] [Google Scholar]
- 3.Bellamy WT. P-glycoproteins and multidrug resistance. Annu Rev Pharmacol Toxicol. 1996;36:161. doi: 10.1146/annurev.pa.36.040196.001113. [DOI] [PubMed] [Google Scholar]
- 4.Bradley M, Zeytun A, Rafi-Janajreh A, Nagarkatti PS, Nagarkatti M. Role of spontaneous and interleukin-2-induced natural killer cell activity in the cytotoxicity and rejection of Fas+ and Fas− tumor cells. Blood. 1998;92(11):4248. [PubMed] [Google Scholar]
- 5.Cao X, Chen G, He L, Zhan W, Yu Y, Wang J. Involvement of MHC class I molecule and ICAM-I in the enhancement of adhesion and cytotoxic susceptibility to immune effector cells of tumor cells transfected with the interleukin (IL)-2, IL-4 or IL-6 gene. J Cancer Res Clin Oncol. 1997;123:602. doi: 10.1007/s004320050112. [DOI] [PubMed] [Google Scholar]
- 6.Castro-Galache MD, Ferragut JA, Barbera VM, Martin-Orozco E, Gonzalez-Ros JM, Garcia-Morales P, Saceda M. Susceptibility of multidrug resistance tumor cells to apoptosis induction by histone deacetylase inhibitors. Int J Cancer. 2003;104(5):579. doi: 10.1002/ijc.10998. [DOI] [PubMed] [Google Scholar]
- 7.Costello RT, Mallet F, Gaugler B, Sainty D, Arnoulet C, Gastaut JA, Olive D. Human acute myeloid leukemia CD34+/CD38− progenitor cells have decreased sensitivity to chemotherapy and Fas-induced apoptosis, reduced immunogenicity, and impaired dendritic cell transformation capacities. Cancer Res. 2000;60:4403. [PubMed] [Google Scholar]
- 8.Cullen K, Davey R, Davey M. The drug resistance proteins, multidrug resistance-associated protein and P-glycoprotein, do not confer resistance to Fas-induced cell death. Cytometry. 2001;43(3):189. doi: 10.1002/1097-0320(20010301)43:3<189::aid-cyto1048>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 9.Cullen KV, Davey RA, Davey MW. Drug resistance does not correlate with resistance to Fas-mediated apoptosis. Leuk Res. 2001;25(1):69. doi: 10.1016/s0145-2126(00)00085-0. [DOI] [PubMed] [Google Scholar]
- 10.Fichtner I, Stein U, Hoffmann J, Winterfeld G, Pfeil D, Hentschel M. Characterization of four drug-resistant P388 sublines: resistance/sensitivity in vivo, resistance and proliferation markers, immunogenicity. Anticancer Res. 1994;14(5A):1995. [PubMed] [Google Scholar]
- 11.Fuji H, Murakami MJ. Differential tumour immunogenicity of DBA/2 mouse lymphoma L1210 and its sublines, III: control of host resistance to drug-resistant L1210 sublines by H-2-linked and non-H-2-linked genes. J Natl Cancer Inst. 1983;70(1):119. [PubMed] [Google Scholar]
- 12.Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 13.Izquierdo MA, Neefjes JJ, Mathari AE, Flens MJ, Scheffer GL, Scheper RJ. Overexpression of the ABC transporter TAP in multidrug-resistant human cancer cell lines. Br J Cancer. 1996;74(12):1961. doi: 10.1038/bjc.1996.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 15.Labroille G, Dumain P, Lacombe F, Belloc F. Flow cytometric evaluation of fas expression in relation to response and resistance to anthracyclines in leukemic cells. Cytometry. 2000;39(3):195. [PubMed] [Google Scholar]
- 16.Landowski TH, Gleason-Guzman MC, Dalton WS. Selection for drug resistance results in resistance to Fas-mediated apoptosis. Blood. 1997;89(6):1854. [PubMed] [Google Scholar]
- 17.Laurent G, Jaffrézou JP. Signaling pathways activated by daunorubicin. Blood. 2001;98:913. doi: 10.1182/blood.v98.4.913. [DOI] [PubMed] [Google Scholar]
- 18.Ogasawara J, Watanabe-Fukunage R, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y, Itoh N, Suda T, Nagata S. Lethal effect of the anti-Fas antibody in mice. Nature. 1993;364:806. doi: 10.1038/364806a0. [DOI] [PubMed] [Google Scholar]
- 19.Puddu P, Fais S, Luciani F, Gherardi G, Dupuis ML, Romagnoli G, Ramoni C, Cianfriglia S, Gessani S. Interferon-gamma up-regulates expression and activity of P-glycoprotein in human peripheral blood monocyte-derived macrophages. Lab Invest. 1999;79(10):1299. [PubMed] [Google Scholar]
- 20.Qin Z, Blankesistein T. CD4+ T cell-mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFNg receptor expression by nonhematopoietic cells. Immunity. 2000;12:677. doi: 10.1016/S1074-7613(00)80218-6. [DOI] [PubMed] [Google Scholar]
- 21.Rapp L, Fuji H. Differential antigenic expression of the DBA/2 lymphoma L1210 and its sublines: cross-reactivity with C3H mammary tumours as defined by syngeneic monoclonal antibodies. Cancer Res. 1983;43(6):2592. [PubMed] [Google Scholar]
- 22.Roninson IB. The role of the MDR1 (P-glycoprotein) gene in multidrug resistance in vitro and in vivo. Biochem Pharmacol. 1992;43:95. doi: 10.1016/0006-2952(92)90666-7. [DOI] [PubMed] [Google Scholar]
- 23.Scheper RJ, Dalton WS, Grogan TM, Schlosser A, Bellamy WT, Taylor CW, Scuderi P, Spier C. Altered expression of P-glycoprotein and cellular adhesion molecules on human multi-drug-resistant tumor cells does not affect their susceptibility to NK-and LAK-mediated cytotoxicity. Int J Cancer. 1991;48(4):562. doi: 10.1002/ijc.2910480414. [DOI] [PubMed] [Google Scholar]
- 24.Seliger B, Harders C, Wollscheid U, Staege MS, Reske-Kunz AB, Huber C. Suppression of MHC class I antigens in oncogenic transformants: association with decreased recognition by cytotoxic T lymphocytes. Exp Hematol. 1996;24(11):1275. [PubMed] [Google Scholar]
- 25.Shtil AA, Turner JG, Durfee J, Dalton WS, Hua Y. Cytokine-based tumor cell vaccine is equally effective against parental and isogenic multidrug-resistant myeloma cells: the role of cytotoxic T lymphocytes. Blood. 1999;93(6):1831. [PubMed] [Google Scholar]
- 26.Smyth MJ, Krasovkis E, Sutton VR, Johnstone RW. The drug efflux protein, P-glycoprotein, additionally protects drug-resistant tumour cells from multiple forms of caspase-dependent apoptosis. Proc Natl Acad Sci U S A. 1998;95(12):7024. doi: 10.1073/pnas.95.12.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soto F, Canaves JM, Gonzalez-Ros JM, Ferragut JA. Rapid kinetics of the interaction between daunomycin and drug-sensitive or drug-resistant P388 leukemia cells. FEBS Lett. 1992;301:119. doi: 10.1016/0014-5793(92)80223-4. [DOI] [PubMed] [Google Scholar]
- 28.Soto F, Planells-Cases R, Canaves JM, Ferrer-Montiel AV, Aleu J, Gamarro F, Castanys S, Gonzalez-Ros JM, Ferragut JA. Possible coexistence of two independent mechanisms contributing to anthracycline resistance in leukemia P388 cells. Eur J Cancer. 1993;29A(15):2144. doi: 10.1016/0959-8049(93)90050-p. [DOI] [PubMed] [Google Scholar]
- 29.VandenDriessche T, Bakkus M, Toussaint-Demylle D, Thielemans K, Verschueren H, De Baetselier P. Tumorigenicity of mouse T lymphoma cells is controlled by the level of major histocompatibility complex class I H-2Kk antigens. Clin Exp Metastasis. 1994;12(1):73. doi: 10.1007/BF01784336. [DOI] [PubMed] [Google Scholar]
- 30.Wang YY, Zheng XX. A flow cytometry-based assay for quantitative analysis of cellular proliferation and cytotoxicity in vitro. J Immunol Methods. 2002;268:179. doi: 10.1016/s0022-1759(02)00190-4. [DOI] [PubMed] [Google Scholar]
- 31.Weisburg JH, Curcio M, Caron PC, Raghu G, Mechetner EB, Roepe PD, Scheinberg DA. The multidrug resistance phenotype confers immunological resistance. J Exp Med. 1996;183(6):2699. doi: 10.1084/jem.183.6.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woods G, Lund LA, Naik M, Ling V, Ochi A. Resistance of multidrug-resistant lines to natural killer-like cell-mediated cytotoxicity. FASEB J. 1988;2(12):2791. doi: 10.1096/fasebj.2.12.3044904. [DOI] [PubMed] [Google Scholar]
- 33.Zhihai Q, Blankestein T. CD4+ T cell-mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFNγ receptor expression by nonhematopoietic cells. Immunity. 2000;12:677. doi: 10.1016/S1074-7613(00)80218-6. [DOI] [PubMed] [Google Scholar]