

Abstract
Advanced metastatic melanoma is incurable by standard treatments, but occasionally responds to immunotherapy. Recent trials using dendritic cells (DC) as a cellular adjuvant have concentrated on defined peptides as the source of antigens, and rely on foreign proteins as a source of help to generate a cell-mediated immune response. This approach limits patient accrual, because currently defined, non-mutated epitopes are restricted by a small number of human leucocyte antigens. It also fails to take advantage of mutated epitopes peculiar to the patient's own tumour, and of CD4+ T lymphocytes as potential effectors of anti-tumour immunity. We therefore sought to determine whether a fully autologous DC vaccine is feasible, and of therapeutic benefit. Patients with American Joint Cancer Committee stage IV melanoma were treated with a fully autologous immunotherapy consisting of monocyte-derived DC, matured after culture with irradiated tumour cells. Of 19 patients enrolled into the trial, sufficient tumour was available to make treatments for 17. Of these, 12 received a complete priming phase of six cycles of either 0.9×106 or 5×106 DC/intradermal injection, at 2-weekly intervals. Where possible, treatment continued with the lower dose at 6-weekly intervals. The remaining five patients could not complete priming, due to progressive disease. Three of the 12 patients who completed priming have durable complete responses (average duration 35 months+), three had partial responses, and the remaining six had progressive disease (WHO criteria). Disease regression was not correlated with dose or with the development of delayed type hypersensitivity responses to intradermal challenge with irradiated, autologous tumour. However, plasma S-100B levels prior to the commencement of treatment correlated with objective clinical response (P=0.05) and survival (log rank P<0.001). The treatment had minimal side-effects and was well tolerated by all patients. Mature, monocyte-derived DC preparations exposed to appropriate tumour antigen sources can be reliably produced for patients with advanced metastatic melanoma, and in a subset of those patients with lower volume disease their repeated administration results in durable complete responses.
Keywords: Dendritic cells, Melanoma, Immunotherapy, Cytotoxic T lymphocytes, S-100B
Introduction
Regional therapies, surgery and radiotherapy have little to offer outside palliation in late stage disseminated melanoma. The search for systemic treatments is logical; however, current chemotherapies are unsatisfactory due to the poor durability of response, accompanied by unacceptable toxicity profiles in patients with a limited lifespan. Systemic cytokine therapies, such as IL-2, interferons and IL-12, similarly cause severe side-effects.
In contrast, immunotherapies are usually well tolerated, but those in current use against metastatic melanoma rarely induce complete regression in patients with visceral disease [2, 27, 30]. In active immunotherapy regimes, this may be due to insufficient stimulation of cell-mediated anti-tumour responses. Dendritic cells (DC) are the most potent antigen-presenting cells for the stimulation of T lymphocytes, and their use as a cellular adjuvant in experimental cancer therapy is engaging many trials [1, 22]. An important common outcome of these trials has been the excellent tolerability of DC-based vaccines. Outstanding differences between studies have been the sources of potential tumour antigens, and the kinds of DC used.
Many studies have used peptide epitopes identified in common lineage or marker proteins of malignancies. Such peptides have practical advantages because of their availability as "off-the-shelf" reagents and their utility in the laboratory procedures with which immunological responses are measured. High densities of epitope on the presenting DC can also be achieved, which may be important in determining the effectiveness of presentation [17]. Indeed, the use of multiple antigenic peptides pulsed onto CD34+-derived DC has had excellent clinical efficacy in HLA-A2+ melanoma patients [2].
However, the choice of synthetic peptides as the source of antigen has the clinical disadvantages of epitope restriction to specific human leucocyte antigen (HLA) types and the possible non-expression of the antigen by some clones of the malignancy, which then escape the immune response to that epitope [35]. Furthermore, opportunities will be lost for recognising patient-specific, tumour antigens [12, 19] generated by some of the very large number (>10,000) of random mutations which arise in most malignancies [8] over an extended but variable pre-malignant period. Finally, there is also little opportunity for the discovery of novel, conserved tumour antigens associated with clinical responses in patients undergoing peptide/DC treatments. Therefore, the case for autologous tumour as the best representative source of the full, unique spectrum of tumour-associated antigens for each individual patient is compelling. Because patients do not need to be selected on the basis of their tissue type, it also provides the potential for the discovery of a much broader spectrum of conserved epitopes covering most HLAs. A major practical issue potentially limiting the applicability of autologous tumour-based vaccines is the availability of sufficient tumour tissue. Clearly, this issue is related to those of dose and treatment regimen. It was therefore important to consider these questions in the formulation of a trial aimed at treating patients with a wide variation in disease burdens.
Tumour burden has been widely recognised as a determining factor in the outcome of immunotherapy [18, 26], but the precise measurement of tumour volume by radiological methods is technically challenging. The S-100B protein is a member of a highly conserved family of calcium-binding proteins originally isolated from brain tissues, but also expressed by some tumours, notably malignant melanoma. The concentration of S-100B in the blood of patients with malignant melanoma correlates with the stage of disease [9] and number of metastases [16], while pre-treatment levels are of prognostic value for survival [15]. In the current study, we measured plasma S100-B to determine whether it could predict patients' response to therapy.
Aside from the choice of antigen source, other differences between immunotherapy trials using DC include their source (blood derived, or cultured from CD34+ or monocyte precursors), state of maturation, mode of injection, and dosing regimen. The objective of our study was to evaluate the safety and clinical effectiveness of two dose levels of an intradermally delivered, fully autologous, matured monocyte-derived DC therapy for American Joint Cancer Committee (AJCC) stage IV metastatic melanoma, employing irradiated autologous tumour cells as the source of antigen.
The effect of the tumour cells on the DC was examined, to ensure that their in vitro stimulatory capacity was not impaired. Since the treatment resulted in a high proportion of objective clinical responses, we were able to examine the relevance of DC maturation, and the prognostic value of pre-treatment plasma S-100B, to clinical outcome.
Patients and methods
Selection criteria
Patients considered for the trial had multiple, distant, melanoma metastases defining stage IV disease, and a good performance status (<2 on the Eastern Cooperative Oncology Group scale). Exclusion criteria included: cerebral involvement, any significant non-malignant disease, such as autoimmune disease, prior use of immunotherapy or chemotherapy within the previous 8 weeks, positive HIV, hepatitis B or C serology indicating active infection, and pregnancy. All eligible patients were accepted into the trial. They were allocated to their treatment group (dose level), and are numbered in the text, according to their order of enrolment. Ten of the 19 patients had previous therapy apart from surgery—chemotherapy, chemo-immunotherapy or radiotherapy (Table 1).
Table 1.
Patient treatment and response characteristics
| Patient no. | Sex | Age on entry | Previous therapy | No. of injections | Tumour source for vaccine | Organs involved on entry | Response | Survival post vaccine 1 (months) | DTH response (mm) |
|---|---|---|---|---|---|---|---|---|---|
| 1a | F | 53 | S | 14 | S-C, chest wall | LN, skin, S-C | PR (skin) | 19.5 | 0 |
| 2a | M | 60 | S | 9 | S-C, abdomen | Skin, S-C, LN, lung, mediastinum | PR (skin) | 8 | NT |
| 3a | M | 50 | S | 3c | Axilla | S-C, LN, adrenal, kidney | PD | 1 | 0 |
| 4a | F | 37 | S, C, CI | 10 | Head of pancreas | LN, pancreas | PD | 10 | 0 |
| 5a | M | 39 | S | 36d | Retroperitoneal node | LN, kidney, adrenal, mediastinum, pleuro-pericardium, soft tissue | CR | 55+f | <1 |
| 6b | F | 47 | S, I, R | 6 | Axilla | Lung, LN, S-C, skin | PD | 3.5 | 0 |
| 7 | M | 49 | S, R | 0c | S-C, scapula | Skin, lung, liver | PD | – | NT |
| 8b | F | 51 | S, C | 3c | Shin nodules | Skin, LN, lung, bone | PD | 3 | NT |
| 9b | M | 67 | Nil | 1c | Axilla | Skin, LN, liver, lung, adrenal, mediastinum | PD | 1 | NT |
| 10b | F | 64 | S, C, CI, R | 5c | S-C, scapula | Skin, lung, mediastinum | PD | 5.5 | 7.5 |
| 11b | M | 21 | Nil | 19 | Small bowel | LN, mesentery, lung, soft tissue | PR (LN) | 21 | 5.8 |
| 12 | M | 49 | S, R | 0c | Nodule, cubital fossa | Lung, adrenal | PD | – | NT |
| 13a | M | 66 | S | 6 | Lung | Lung | PD | 5 | 4 |
| 14b | F | 24 | S | 30d | Small bowel | Mesenteric nodes | CR (+ bonee) | 44+f | 41 |
| 15b | M | 49 | S | 28d | Sternal lesion | Liver, lung, sternum | PD | 38 | 7 |
| 16a | M | 27 | S, I, R | 3c | S-C, mid back | LN, bowel | PD | 10 | 3 |
| 17a | M | 31 | S, I | 6 | Bone | S-C, LN, soft tissue | PD | 6.5 | 0 |
| 18a | F | 46 | S, H, I | 6 | Axilla | Skin, LN, liver, pancreas, adrenal, mesenteric nodes | PD | 7.5 | 0 |
| 19a | F | 34 | S, H | 16d | Lung | Lung | CR | 33+f | NT |
C, Chemotherapy; CI, chemo-immunotherapy; H, hormone therapy; I, Immunotherapy; LN, lymph node; R, radiotherapy; S, surgery; S-C, subcutaneous; Skin, multiple skin metastases; DTH, average delayed type hypersensitivity response to intradermal irradiated autologous melanoma injection; PD, progressive disease; PR, partial response; CR, complete response; NT, not tested
aLow dose
bHigh dose
cDid not complete initial course
dContinues on maintenance therapy
eBone metastasis developed during course of trial
fPatient remains alive, with no evidence of disease
The research ethics committees of both the clinical (Mater Hospital) and scientific (Queensland Institute of Medical Research) institutions approved the study protocol. All patients gave written, informed consent prior to screening and at the time of enrolment. The study spanned 2 1/2 years, from November 1997 to May 2000.
Clinical measurements
Immediately before the first vaccination, after the fourth vaccination and 2 weeks after completion of the six priming vaccinations, each patient underwent magnetic resonance imaging (MRI) of the brain, computed tomography (CT) of the thorax, abdomen and pelvis and a technetium bone scan to estimate the extent of malignant disease. A full blood count and a serum biochemical profile, including C-reactive protein, were performed every 2 weeks.
Standard (World Health Organisation) definitions of complete response (CR), partial response (PR) and progressive disease (PD) were used [25]. CR is the disappearance of all macroscopic disease, determined by two observations not less than 4 weeks apart. PR is defined as a 50% or more decrease in total tumour load of the lesions that have been measured to determine the effect of therapy, by two observations not less than 4 weeks apart, with no progressive or new lesions. We included any mixed response or stable disease under progressive disease (PD).
Vaccine production
For each patient, autologous melanoma cells were mechanically disaggregated from fresh surgical specimens under sterile conditions, using crossed scalpels. The use of enzymes was avoided to limit the possibility of non-tumour-specific immune responses at injection sites. A fraction of the tumour cell suspension (<10%) was cultured to provide a cell line (successful in 14/17 patients from whom tumour cells were obtained). The remainder was cryopreserved in 10% dimethyl sulphoxide (Sigma-Aldrich, Sydney)/30% autologous plasma (AP, from heparinised blood) after being rendered replicatively inactive by gamma irradiation (150 Gy, 60Co source) at 4°C. DC were cultured from blood monocyte precursors [4, 28, 29, 31]. Briefly, mononuclear cells were isolated over Ficoll-Paque Plus (Amersham Pharmacia Biotech Pty Ltd, Sydney) from peripheral blood derived from venesection (400 ml, low dose) or leukapheresis (high dose), and the plastic-adherent fraction cultured in 1% autologous plasma/RPMI-1640 (CSL, Parkville, Australia) ("medium") supplemented with 1,000 U/ml each of interleukin-4 and granulocyte/macrophage-colony stimulating factor (kind gifts from Schering-Plough). The resulting "immature" DC were cryopreserved on day 6. Prior to each treatment administration, thawed DC were cultured in cytokine supplemented medium with irradiated autologous tumour cells (in the ratio of one tumour cell to four DC). After 6 h, monocyte-derived conditioned medium (MCM, a 24-h supernatant from autologous monocytes plated onto Petri dishes pre-coated with 10 mg/ml human immunoglobulin [29]) was added (20% of final volume) to induce DC maturation. Two and a half days later, the cells were harvested, washed twice, resuspended in 0.2 ml of 1% plasma/phosphate-buffered saline (0.2 g/litre KCl, 0.2 g/litre KH2PO4, 8 g/litre NaCl, 1.15 g/litre Na2HPO4; Sigma-Aldrich, Sydney) and held at 4°C for injection within 2 h.
Vaccine administration
All trial participants were treated as outpatients. Injections were given intradermally, at a single site, into abdominal skin. Two priming dosage regimens were performed: six fortnightly injections at either low dose [on average 0.9 (±0.4) ×106 cells] or high dose [5 (±3) ×106 cells]. Following this priming phase, where possible, patients continued treatment (at 106 cells/dose) at 6-weekly intervals.
Immunological assessments
Delayed-type hypersensitivity (DTH) reaction to irradiated tumour cell challenge
Where sufficient tumour biopsy cells were available, 0.5×106 irradiated, cryopreserved, autologous tumour cells (thawed, washed and resuspended as for vaccine) were injected intradermally into the volar aspect of a non-affected arm at the same time as vaccination. The DTH response was measured as the maximum induration diameter between 24 and 48 h.
Peripheral blood samples were taken just prior to vaccination for the in vitro assessment of anti-tumour cell-mediated immunity. These results are the subject of a separate report (in preparation).
In vitro assessments of DC phenotype and allostimulatory activity
Vaccines were assessed for phenotype by flow cytometric analysis using pre-titred, fluorescently labelled monoclonal antibodies against CD14 (PE-Cy5 labelled clone RMO52) and CD83 (PE labelled clone HB15a; both from Beckman-Coulter). A minimum of 1,000 cells distinguished from lymphocytes by their high forward/orthogonal scatter were analysed, using isotype-matched control antibodies to determine thresholds for positivity.
The effect of co-culture with tumour cells upon the ability of DC to stimulate allogeneic responses was assessed. Peripheral blood mononuclear cells from three healthy donors (50,000 cells/U-bottom well) were cultured with graded numbers of stimulator populations (different melanoma patient DC preparations at 1:1,000–1:30 stimulator to responder ratios), in triplicate, for 6 days at 37°C in 10% pooled human serum/RPMI 1640 (CSL, Australia). They were pulsed for the final 18 h with 1 μCi/well 3H-thymidine (Amersham), before harvesting to measure their proliferation as the amount of incorporated label. The following hyperbolic equation was fitted to the data using multiple function non-linear regression (SigmaStat V.2):
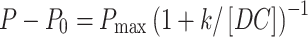 |
1 |
where P= proliferation of responder cells (cpm), P 0= background proliferation, in the absence of stimulator cells, P max= theoretical maximum proliferation, k= dose ratio of DC/responders stimulating a half-maximal response, and [DC]= dose ratio of DC/responder cells added. The value k was used as a measure of DC function, with lower values signifying higher allostimulatory capacity. To test for the significance of the differences detected +/− tumour, a three-way analysis of variance was performed (SigmaStat V.2).
Histopathology
Punch biopsies were performed selectively on DTH challenge sites, vaccine sites and regressing nodules. Sections from these biopsies were stained with haematoxylin and eosin, and antibodies developed with immunoperoxidase including S-100 for the confirmation of melanoma cells, CD3, CD4 and CD45 RO for the presence of T cells, CD68 for macrophages and CD20 for B cells.
Plasma S-100B measurements
Heparinised plasma samples taken prior to the first vaccination were stored at −70°C and analysed blind using the Sangtec 100 immunoluminometric assay (Immunodiagnostics, Sydney, Australia). Receiver operator characteristic (ROC) analysis [23, 34] was used to assess the ability of pre-treatment plasma S-100B levels to predict the clinical response. The optimum threshold that maximised sensitivity and specificity was used to categorise patients into low and high S-100B groups. A Kaplan-Meier survival analysis was then used to compare survival in these two groups, and the significance of the difference was determined with the log rank test.
Results
Vaccine characteristics
Preliminary experiments (not shown) indicated that the allostimulatory potential of healthy donor DC preparations grown in 1% autologous plasma/RPMI-1640, and matured with monocyte-derived condition medium, was optimal over a range of IL-4 concentrations (100–10,000 U/ml), and that MCM concentrations of >5% were adequate to yield high levels of CD83+ cells. However, MCM preparations from different melanoma patients varied significantly in their capacity to mature healthy donor DC (data not shown). The average percentage of CD83+ cells in vaccine preparations for different trial patients ranged from 34% to 80% (mean 57%).
Accompanying the uncultured, disaggregated suspensions of autologous melanoma cells used as the source of antigen were variable amounts of infiltrating leucocytes, as well as stromal cells and necrotic tumour cells. Since these could have a variable effect on the antigen-presenting function of the DC, we tested the allostimulatory capacity of the latter in both the presence and the absence of tumour cells. As shown in Table 2, small differences between the two DC populations were seen in three of five melanoma patients tested. In two cases (B, C) an overall enhancement of allostimulation was seen in the presence of tumour cells (P<0.02), in one case inhibition was seen (D, P< 0.001) and in the others no significant difference could be detected (P>0.05).
Table 2.
Effect of irradiated, autologous melanoma cells upon the allostimulatory capacity of DC
| Stimulating DC | ± tumour | Responding mononuclear cells | ||
|---|---|---|---|---|
| a | b | c | ||
| A | + | 6±3a | 3±1 | 9±2 |
| − | 3±1 | 3±1 | 7±2 | |
| B | + | 7±4 | 5±2 | 6±3 |
| − | 13±7 | 15±5 | 11±5 | |
| C | + | 10±3 | 6±2 | >50b |
| − | 24±6 | 11±3 | >50b | |
| D | + | 18±4 | 21±4 | 21±4 |
| − | 7±2 | 8±1 | 10±2 | |
| E | + | 17±4 | 17±5 | 26±6 |
| − | 15±3 | 17±5 | 29±6 | |
a103×k, where k is the ratio of DC to mononuclear cells needed to achieve half-maximal allostimulation ± standard error (Eq. 1). Immature DC from melanoma patients A, B, C, D and E were cultured with or without irradiated autologous tumour cells, matured with autologous MCM, and used at a range of doses as allostimulators of mononuclear cells from healthy donors a, b and c. Thus, a ratio of 3 (±1) DC:1,000 mononuclear cells was needed for DC from patient A to stimulate a half-maximal response from donor b, irrespective of whether they were co-cultured with autologous irradiated tumour cells.
bMinimal allostimulation was seen with the combination C×c (k>50)
The minimum yield of melanoma cells needed to make the priming course of vaccines was 1.5×106 and 7.5×106 for the low- and high-dose regimens, respectively. This was achieved in all patients except numbers 7 and 12. Excepting these two patients, the range of tumour cell yield was 5×106–4×108. Autologous cell lines were eventually needed in the maintenance phase as the source of tumour cells for patients 4, 11 and 14. An irradiated mixture of these was used as an allogeneic source of tumour antigens in the maintenance phase for patient D19.
Patients
Of the 19 patients enrolled in the trial, 12 were able to complete the six priming vaccinations. As noted above, of the remaining seven, vaccine could not be prepared for two (patients 7 and 12) because the biopsy did not yield sufficient viable tumour cells for vaccine production. Rapid clinical deterioration after a single vaccination (patient 9), development of multiple cerebral metastases requiring dexamethasone and radiotherapy which contravened continued immunotherapy (patients 3, 10, 16), and personal reasons (patient 8), resulted in the withdrawal of the other five patients prior to completion of the priming phase.
Clinical results
Three patients (one high dose, two low dose) had CR of all clinically and radiologically apparent disease (Figs. 1, 2, 3 and 4), durable to date in each case. Patient 5 had para-renal, mediastinal, soft tissue, adrenal and pleuro-pericardial disease, which resolved completely by 19 months after the first vaccination, i.e., during the maintenance phase. He remains disease free and in full-time employment 36+ months after CR. Patient 14 had mesenteric lymph node involvement at the time of initial screening, and a metastasis in the diploic space of the calvarium was disclosed 5 weeks after the first vaccination. Both sites fully resolved 4.5 months after commencement of treatment (duration 39+ months). The third patient (19) with a CR had three lung metastases and a pericardial nodule resolve by the completion of the six priming vaccinations (duration 30+ months).
Fig. 1a, b.

Male, 39 years (patient 5). a CT of the thorax with i.v. contrast. Nodal metastasis 4.8 cm in diameter in the aorto-pulmonary window. b Minor residual soft tissue lesion consistent with fibrosis, <1 cm, 19 months after commencement. CR durable at 36+ months following regression
Fig. 2a–d.

Female, 24 years (patient 14). a 99mTc-hydroxydiphosphonate isotope bone scan. A focal area of increased tracer uptake is present in the right frontal region. b No evidence of the lesion on bone scan 4.5 months following commencement of treatment. c MRI: T1-weighted coronal image of the skull after i.v. gadolinium demonstrates an enhancing bony metastasis in the diploic space of the right frontal bone. d No evidence of the lesion on MRI 4.5 months following commencement of treatment. CR durable at 39+ months
Fig. 3a, b.

Female, 24 years (patient 14). a CT abdomen with i.v. contrast. A 4.2-cm metastasis with heterogeneous enhancement is present in the mesentery to the left of the aorta. b Minor residual fibrosis <1 cm in diameter. CR durable at 39+ months
Fig. 4a, b.

Female, 34 years (patient 19). a CT of the left lung base 1 1/2 months after commencement of treatment. A 1.4-cm lung metastasis not present on baseline CT is demonstrated. b CR with no measurable lesion 1 1/2 months later. CR durable at 30+ months
All patients with CR had low-volume visceral disease at the time of the first vaccination. The largest lesion to completely resolve in this group was a mediastinal lymph node measuring 4×4 cm. In contrast, patients with PD all had visceral masses (adrenal, lung, bone, bowel, liver) of >3 cm, with the exception of patient 4, who had a small residual pancreatic mass remaining following biopsy for vaccine production. All patients with CR had their tumour biopsies for vaccine production taken from deep tissue (Table 1). Importantly, all patients with objective clinical responses achieved that response using uncultured, autologous tumour cells as the source of antigen.
Of patients with a PR, two had resolution of subcutaneous nodules (patient 1: 3/3 nodules) or skin metastases (patient 2: 2/5), and the third (patient 11) had resolution of two enlarged neck lymph nodes (>6 cm). All of these patients later had progression of visceral disease leading to death. Their survival times from the commencement of vaccination were 19.5, 8, and 21 months, respectively.
No significant difference in objective response was seen between the two dosage levels: one CR and one PR were observed in seven patients assigned to high dose, versus two CR and two PR in ten assigned to low dose. There was no correlation between the CD83 status of the vaccine and clinical outcome or patient survival.
Adjuvant therapy was required in six patients who continued treatment following priming. One patient (1) had a single cerebral metastasis which was surgically removed before recommencing the vaccine treatment. Four patients with PD (2, 11, 15 and 17) required regional radiotherapy to local lesions. One patient (4) with a pancreatic metastasis developed biliary obstruction, requiring an internal bypass. Another (patient 5) had a bleeding jejunal metastasis resected while continuing therapy, 5 months post vaccine 1. This patient ultimately had a CR, 14 months later.
Adverse effects
No significant systemic toxic effects occurred, apart from mild flu-like symptoms which were experienced within two days following vaccination, and resolved within 48 h. No evidence of vaccine-related autoimmune disease was seen.
DTH response to irradiated autologous tumour
Sufficient tumour was available to perform DTH tests in 10 of 12 patients who completed the priming schedule, and were therefore considered evaluable for immune responses (Table 1). No reaction was observable in four of these (patients 4, 6, 17, 18: PD, 1: PR), while three (patients 13: PD, 4: PR, 5: CR) had a faint response on one occasion. Three patients (15: PD, 11: PR, 14: CR) had consistently positive reactions, the largest by far being observed in patient 14. Therefore, DTH was not consistently correlated with patients' clinical responses. Only a weak correlation between DTH reaction and survival was seen (Pearson product moment correlation coefficient =0.5, P=0.1).
Haematology and biochemistry
The haematological abnormalities included anaemia in six patients, usually attributable to bleeding lesions. All of these patients underwent leucocyte filtered red cell transfusion. There were no biochemical abnormalities of significance, other than a rising C-reactive protein accompanying progressive disease.
Histopathology
Histological examination of regression in skin nodules showed a peripheral lymphoid response with tumour penetration by activated lymphocytes and macrophages, with occasional tumour necrosis. CD3+, CD43+ and CD45 RO+ cells were plentiful, but B cells (CD20+) were absent. CD68+ macrophages were present or plentiful. These responses were less or absent if the patient deteriorated. Some patients showed plasmacytic peripheral and perivascular responses as well as the T-cell response. Interestingly in one patient (5) who underwent complete regression the initial biopsy used for vaccine production already showed a strong T-cell lymphoid infiltrate; however, another patient (6) with a similar infiltrate of her initial biopsy failed to respond to the vaccine.
S-100B analysis
Plasma for S-100B analysis was available from 14 patients, of whom six responded partially or completely to treatment (Fig. 5). The ROC analysis resulted in an optimal threshold for S-100B of less than or equal to 0.36 μg/ml plasma for predicting an objective response (sensitivity =83.3%, specificity =75%). A test on the significance of S-100B being better than random in predicting response (i.e., on the area under the ROC curve being greater than 0.5) resulted in a P=0.05. A Kaplan-Meier survival analysis comparing patients categorised into two groups according to this threshold value (0.36 μg/ml S-100B) showed a very significant difference in survival (log rank P<0.001), based on all patients assessed (Fig. 6). While no relationship was seen between S-100B levels and the specific sites of disease, as noted above, patients with PD generally had larger visceral tumour masses and higher blood S-100B levels (Fig. 5) than those with CR.
Fig. 5.

Pre-treatment plasma S-100B (μg/ml) for all patients assessed. The threshold for optimum sensitivity/specificity determined by ROC analysis (0.36 μg/ml) is marked (___). Patients 8 and 10 did not complete the initial course of six injections
Fig. 6.

Kaplan-Meier survival analysis comparing patients categorised according to the plasma S-100B threshold which best predicted objective response (0.36 μg/ml), determined by ROC analysis
Discussion
This report documents a phase I/II trial of a DC-based vaccine with a high objective clinical response rate and minimal discomfort for the patient. It is fully autologous, inexpensive to manufacture and administer (compared with most gene therapies and systemic high-dose cytokines), and can be produced within 2 weeks. Importantly, all CRs include deep or visceral disease, and are durable to date, at an average of >2 1/2 years. In previous studies of DC-based therapies, only isolated cases of complete regression of visceral melanoma have been reported [2, 27]. The ability to overcome visceral disease is critical to the successful therapy of advanced disease since the liver, bone and intestine account for over one-quarter of the initial sites of stage IV melanoma metastasis. The three patients with CR had a low volume of disease, and their tumour for vaccine production was harvested from deep metastases. In the three patients with PR, the chief sites of regression were limited to subcutaneous nodules and/or regional lymph nodes, and tumour had been harvested from subcutaneous nodules in two cases (patients 1, 2).
Although the incidence of spontaneous regression in metastatic melanoma is unknown, Bodurtha [7] collected the cases published prior to 1979. His estimate, 1/400, included partial regressions. It is very unlikely that the regressions in the present trial were spontaneous.
An important part of the protocol in a therapy that takes some time to take effect is to perform the normal, ethical treatment of symptomatic lesions. One of the patients (patient 5) with a complete response had resection of a bleeding jejunal mass. It should be noted that numerous other masses in this patient were not removed, and that these slowly (but completely) responded under immunotherapy alone, 14 months subsequently. We therefore consider it most unlikely that the resection was responsible for the subsequent CR.
A feature distinguishing this study from previous reports [2, 24, 27, 33] is that the protocol included continuation of treatment beyond the priming phase. Most significantly, regression of residual metastases finally occurred during that maintenance period in two of three patients who had a CR. The need to periodically restimulate the immune system against cancer has been recognised [13], and another group has reported a CR in a patient receiving post-trial treatment with a DC-based therapy [33]. Since residual micrometastases might persist even after a CR, it is likely that extended treatment will be required to achieve durable remission. However, even when treatment can continue over an extended period, it is not always sufficient to cause an objective tumour response. This is exemplified by patient 15, who continued treatment for 3 1/2 years with slowly progressive disease, and no clinical regression of disease.
Most defined cancer epitopes are from melanoma antigens, and are restricted through a small subset of major histocompatibility types. Recent studies employing synthetic peptides are therefore restricted to a minority of cancer patients. Our report demonstrates the feasibility of using irradiated, autologous tumour cells as the antigen source for the multiple treatments required to achieve complete regression in patients with advanced melanoma. Even in the case of limiting amounts of original biopsy material, cell lines were generated in 74% of patients, and as a result all but one long-term surviving patient has been able to continue treatment, following their clinical response, with autologous tumour cells as the source of antigen. The high proportion of durable CR in our patients is unique for a fully autologous immunotherapy. Murine models have strongly implicated CD4+ T lymphocytes as important effectors of anti-tumour immunity [20]. Using tumour antigens as the source of help for the generation of cell-mediated immunity (rather than foreign proteins, such as keyhole limpet haemocyanin) allows for this potential activity of CD4+ effectors at the tumour site. Importantly, the methods used in our trial could be extended to studies with other malignancies.
We found only a weak correlation between patients' survival and their DTH reaction to the intradermal challenge with irradiated, autologous tumour cells. Most previous immunotherapy trials have used antigenically modified or supplemented tumour cell preparations for DTH testing, which invalidates their use as repeated measures of anti-tumour immunity [6]. Nonetheless, trials in which DTH testing has been performed with unmodified preparations of autologous tumour cells (similar to those used in our study) have shown weak to significant correlations with either regression or survival [5]. The extent to which the immune responses of patients in those trials may have been modified by their intermittent treatment with the cytoablative agent cyclophosphamide is unclear.
The importance of DC maturity in the outcome of immunotherapy or in vitro stimulation of T cells has been highlighted by recent reports [21]. CD83 is a marker of DC maturity; however, no correlation between clinical response and CD83 expression on vaccine cells was seen. Complete clinical responses in patients with advanced melanoma, treated with DC that have not been purposely exposed to maturation factors, have been reported previously [27]. Further, primate studies suggest that alterations in the maturation state of monocyte-derived DC occur spontaneously during migration to the draining lymph nodes, and that the state of maturation prior to injection has no influence on the proportion of DC reaching the nodes [3].
Melanoma cells can secrete immunosuppressive cytokines known to affect DC function [10, 32]. The effect of uncultured, irradiated tumour cells on their autologous dendritic cells was examined for five patients with metastatic melanoma, to determine whether this was a major source of variability in the efficacy of vaccines made by this method. Both enhancement and suppression of allostimulatory responses were observed. These effects were small, and further study is needed to examine whether they are of clinical significance, and to correlate them with the release of particular cytokines by the tumour cells.
The lack of dose response observed in our trial needs to be examined further, because of the small number of patients enrolled. However, coupled with the apparent equal effectiveness of the low and high doses, it appears that small numbers of intradermally injected DC are capable of initiating clinically effective immune responses. Primate studies have suggested that ~1% of these may migrate to the draining lymph nodes [3], their presumed site of activity. If these figures are translated to the doses of DC delivered to patients 5 and 19, as few as 1,000 immigrant tumour antigen-loaded DC may be sufficient to drive the complete eradication of disease.
While numerous potential mechanisms of escape by tumours from immune control have been described [11], it is clear from this study that in many cases the appropriate choice of antigen source and presenting cell can overcome these. However, the finding that only patients with low bulk disease had complete regressions suggests that tumour extent remains a major obstacle to active immune therapies. The serum marker S-100B has been extensively studied, and shown to be of prognostic significance in stage IV melanoma [15], as well as valuable in predicting progression/non-progression of disease following chemotherapy or immunotherapy [14]. Our study shows that pre-treatment levels of S-100B may predict both objective clinical response and survival following DC-based therapy. Therefore, some feature(s) of patients with higher levels of S-100B (e.g., tumour bulk, extent or site of disease) prevent the vaccine from eliciting an effective anti-tumour response. Surgical debulking might improve survival following immunotherapy in this group. Further, other features of the patients' immune system, tumour or vaccine preparation may have prevented CR in half of the objective clinical responses. Analysis of these possibilities should give insights leading to improvements in this group.
The definition of in vitro markers correlating with clinical responses in immunotherapy trials is critical in providing a theoretical underpinning for understanding the mechanism of the treatment. Our data linking pre-treatment plasma S-100B with clinical response are consistent with the hypothesis that tumour bulk and site (visceral involvement) are prime determinants of the immunotherapeutic outcome in patients with advanced metastatic melanoma.
Acknowledgements
This work was supported by grants from the Cooperative Research Centre for Vaccine Technology, the Queensland Cancer Fund, the National Health and Medical Research Council of Australia, Paul and Judy Allen, the Variety Club, the Giraffes, and the QIMR Trust.
We wish to thank Dr. J. Faoagali (Royal Brisbane Hospital) for quality control assistance; Prof. I. Frazer, (Lions Laboratory, Princess Alexandra Hospital, Brisbane) for tissue typing; Dr. K. Taylor (Mater Hospital) for aphereses; Dr. D. Thomas (Mater Hospital branch of the Queensland Radiation Institute); and Mr. D. Nicholas, Immunodiagnostics, Australia, for performing S-100B assays.
References
- 1.Banchereau Annu Rev Immunol. 2000;18:767. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 2.Banchereau Cancer Res. 2001;61:6451. [PubMed] [Google Scholar]
- 3.Barratt-Boyes J Immunol. 2000;164:2487. doi: 10.4049/jimmunol.164.5.2487. [DOI] [PubMed] [Google Scholar]
- 4.Bender J Immunol Methods. 1996;196:121. doi: 10.1016/0022-1759(96)00079-8. [DOI] [PubMed] [Google Scholar]
- 5.Berd J Clin Oncol. 1997;15:2359. doi: 10.1200/JCO.1997.15.6.2359. [DOI] [PubMed] [Google Scholar]
- 6.Berd Cancer Invest. 1988;6:337. doi: 10.3109/07357908809080657. [DOI] [PubMed] [Google Scholar]
- 7.Bodurtha AJ (1979) Spontaneous regression of malignant melanoma. In: Clarke WH, Goldman M, Mastrangelo JM (eds) Human malignant melanoma. Grune & Stratton, New York, p 227
- 8.Boland Proc Natl Acad Sci U S A. 1999;96:14675. doi: 10.1073/pnas.96.26.14675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonfrer Br J Cancer. 1998;77:2210. doi: 10.1038/bjc.1998.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Int J Cancer. 1994;56:755. doi: 10.1002/ijc.2910560524. [DOI] [PubMed] [Google Scholar]
- 11.Ellem Adv Cancer Res. 1998;75:203. doi: 10.1016/s0065-230x(08)60743-5. [DOI] [PubMed] [Google Scholar]
- 12.Fidler IJ (1992) The biology of melanoma metastasis. In: Balch CM, Houghton AN, Milton GM, Sober AJ, Soong SJ. Cutaneous melanoma. Lippincott, Philadelphia, p 112
- 13.Fuchs Semin Immunol. 1996;8:271. doi: 10.1006/smim.1996.0035. [DOI] [PubMed] [Google Scholar]
- 14.Hauschild Br J Dermatol. 1999;140:1065. doi: 10.1046/j.1365-2133.1999.02905.x. [DOI] [PubMed] [Google Scholar]
- 15.Hauschild Oncology. 1999;56:338. doi: 10.1159/000011989. [DOI] [PubMed] [Google Scholar]
- 16.Hauschild Melanoma Res. 1999;9:155. doi: 10.1097/00008390-199904000-00008. [DOI] [PubMed] [Google Scholar]
- 17.Heath Annu Rev Immunol. 2001;19:47. doi: 10.1146/annurev.immunol.19.1.47. [DOI] [PubMed] [Google Scholar]
- 18.Hollinshead Cancer. 1982;49:1387. doi: 10.1002/1097-0142(19820401)49:7<1387::aid-cncr2820490715>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 19.Houghton AN, Herlyn M, Ferrone S (1992) Melanoma antigens. In: Balch CM, Houghton AN, Milton GM, Sober AJ, Soong SJ. Cutaneous melanoma. Lippincott, Philadelphia, p 130
- 20.Hung J Exp Med. 1998;188:2357. doi: 10.1084/jem.188.12.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jonuleit Arch Dermatol Res. 2000;292:325. doi: 10.1007/s004030000144. [DOI] [PubMed] [Google Scholar]
- 22.Lopez Curr Opin Mol Ther. 2002;4:54. [PubMed] [Google Scholar]
- 23.Lusted Science. 1971;171:1217. doi: 10.1126/science.171.3977.1217. [DOI] [PubMed] [Google Scholar]
- 24.Mackensen Int J Cancer. 2000;86:385. doi: 10.1002/(sici)1097-0215(20000501)86:3<385::aid-ijc13>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 25.Miller Cancer. 1981;47:1109. doi: 10.1002/1097-0142(19810301)47:5+<1109::aid-cncr2820471308>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 26.Mukherjee Cancer Gene Ther. 2001;8:580. doi: 10.1038/sj.cgt.7700347. [DOI] [PubMed] [Google Scholar]
- 27.Nestle Nat Med. 1998;4:328. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 28.Peters Adv Exp Med Biol. 1993;329:275. doi: 10.1007/978-1-4615-2930-9_46. [DOI] [PubMed] [Google Scholar]
- 29.Romani J Immunol Methods. 1996;196:137. doi: 10.1016/0022-1759(96)00078-6. [DOI] [PubMed] [Google Scholar]
- 30.Rosenberg Nat Med. 1998;4:321. doi: 10.1038/nm0398-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sallusto J Exp Med. 1994;179:1109. [Google Scholar]
- 32.Steinbrink Blood. 1999;93:1634. [PubMed] [Google Scholar]
- 33.Thurner J Exp Med. 1999;190:1669. doi: 10.1084/jem.190.11.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Eur J Radiol. 1998;27:88. [Google Scholar]
- 35.Velders Crit Rev Immunol. 1998;18:7. doi: 10.1615/critrevimmunol.v18.i1-2.30. [DOI] [PubMed] [Google Scholar]