

Abstract
Vaccination with hybrids comprising fused dendritic cells (DCs) and tumor cells is a novel cancer immunotherapy approach designed to combine tumor antigenicity with the antigen-presenting and immune-stimulatory capacities of DCs. For clinical purposes, we have incorporated a large-scale process for the generation of clinical-grade DCs together with novel electrofusion technology. The electrofusion system provides for ease and standardization of method, efficient DC–tumor cell hybrid formation, and large-quantity production of hybrids in a high-volume (6-ml) electrofusion chamber. In addition, we have evaluated DC electrofusion with a variety of allogeneic human tumor cell lines with the rationale that these tumor cell partners would prove a ready, suitable source for the generation of DC–tumor cell hybrid vaccines. The DC production process can generate 6×108 to 2×109 DCs from a single leukapheresis product (~180 ml). As determined by FACS analysis, electrofusion of 6×107 total cells (1:1 ratio of DC and tumor cells) resulted in a consistent average of 8–10% DC–tumor cell hybrids, irrespective of the tumor type used. Hybrids were retained in the population for 48 h postfusion and following freezing and thawing. Upon pre-irradiation of the tumor cell partner for vaccine purposes, the overall fusion efficiency was not altered at doses up to 200 Gy. Evaluation of DC–tumor cell hybrid populations for their ability to stimulate T-cell responses demonstrated that electrofused populations are superior to mixed populations of DCs and tumor cells in generating a primary T-cell response, as indicated by IFN-γ release. Moreover, hybrids comprising HLA-A*0201 DCs and allogeneic melanoma tumor cells (Colo 829 cell line) stimulated IFN-γ secretion by antigen-specific CD8+ T cells, which are restricted for recognition of a melanoma gp100 peptide antigen (gp100209–217) within the context of the DC HLA haplotype. Maturation of the DC-Colo 829 cell hybrid population served to further improve this T-cell gp100-specific response. Overall, our results are promising for the large-scale generation of electrofused hybrids comprising DCs and allogeneic tumor cells, that may prove useful in human vaccine trials.
Keywords: Cancer, Dendritic cells, Electrofusion, Hybrids, Immunotherapy
Introduction
Dendritic cells (DCs) are highly potent antigen-presenting cells that are gaining status as a preferred adjuvant for cancer vaccine immunotherapy (for reviews, see [2, 27, 32]). DCs play a central role in generation of immune effector cells, including naïve T cells, CD4+ T-helper cells, CD8+ cytotoxic T lymphocytes (CTLs), NK and NKT cells [2, 27]. Tumor vaccine strategies have included loading DCs with defined tumor-associated antigen (TAA) peptides, whole protein, or nucleic acid (RNA or DNA) (for review, see [36]). Other approaches have focused on inducing immunity against both known and unidentified TAAs. These include pulsing DCs with whole tumor cell products, such as RNA, necrotic cell lysates, apoptotic bodies, and peptides eluted from major histocompatibility complexes (MHCs) [36].
Vaccines comprising fused hybrids of DCs and tumor cells provide a further DC-based strategy for immunizing against whole cell TAAs, both known and unknown. DC–tumor cell hybrids are considered to have the distinct advantage of providing for efficient TAA delivery within the context of optimal DC antigen processing, presentation, and immune stimulation. In animal models, immunization with DC–tumor cell hybrids can effectively provide antitumor protection or eradicate established disease [1, 6, 9, 11, 21, 22, 25, 29, 34]. Hybrids comprising human DCs and tumor cell lines or primary human tumor cells have been shown to induce in vitro CTL responses against autologous tumor cell types [3, 4, 5, 7, 8, 12, 30]. In all of these studies, fused DC-tumor hybrids induce greater antitumor CTL responses than mixed cultures of DCs and tumor cells. Moreover, DC-tumor hybrids appear more effective than DC-pulsing with TAAs tumor apoptotic bodies [4] or necrotic cell lysates [4, 5].
To date, relatively few clinical trials have been performed employing DC–tumor cell hybrids. Although these studies have not demonstrated overt clinical efficacy, the hybrid cell vaccines appear safe and well-tolerated and capable of inducing T-cell immunity with at least some clinical responses [17, 18, 23]. In light of these encouraging findings, further investigations of this therapeutic strategy are warranted.
In the majority of in vitro and in vivo studies, polyethylene glycol (PEG) has been used to induce DC–tumor cell fusion. However, PEG fusion is hampered by toxicity, a lack of reproducibility, and difficulties in standardization that make this method problematic for translation to the clinical setting. Electrofusion has been used as an alternative for producing DC–tumor cell hybrids in preclinical studies [5, 11, 16, 22, 25, 26, 28, 29] and in a clinical trial of renal cell cancer [23]. For clinical application, electrofusion has several advantages over PEG fusion: (1) fewer cell manipulations, (2) the ability to standardize the method using controlled electrical parameters, (3) less reliance on individual technical skills, and (4) the potential for developing an enclosed system avoiding chance contamination.
The process of electrofusion involves multiple steps [25, 37]. The first is the close alignment of cells under an alternating current electrical field (dielectrophoresis). The second step is delivery of direct current electrical fields that destabilize cell membranes and begin the process of fusion of adjacent cell membranes. After the fusion pulse, a postfusion alignment using alternating current can be incorporated as a means to help maintain the relative proximal position of the cells [37]. The final phase of the electrofusion process is maturation of the fusion complex with mixing of cytoplasmic contents resulting in hybrid cells [24, 37]. Until recently, DC–tumor cell electrofusion has been performed using improvised electroporation cuvettes or other types of relatively small chambers [5, 16, 22, 25, 28].
We have applied a novel, concentric (coaxial) electrofusion chamber of large volume (6 ml), specifically designed for clinical development. Recently, a similar type of electrofusion apparatus has been described by one other group [11, 26]. With clinical translation as our primary consideration, we have used a clinically compatible process for the production of large numbers of DCs [10, 31] and tested DC electrofusion with a variety of allogeneic human tumor cell lines. Our rationale for using tumor cells derived from cell lines is based on several problems encountered in generating autologous tumor cells. These include tumor availability, generation of adequate numbers of tumor cells, and the logistics of certifying each patient vaccine lot. Allogeneic tumor cell lines that can be readily cultured in the laboratory may well prove suitable for DC-tumor hybrid vaccines as such cell lines are widely known to express TAAs common to primary tumors [14]. In addition, allogeneic tumor cells have been successfully validated in animal vaccine models and have shown promise in clinical trials [13, 14, 15, 35]. The present studies demonstrate the reproducible generation of heterologous hybrids of DCs and several different tumor cell lines derived from a variety of tumor types. Overall, our methodologies incorporate several features important for the clinical application of DC–tumor cell vaccines.
Materials and methods
Dendritic cells and cell culture
DCs were generated from leukapheresis product obtained as peripheral blood mononuclear cell (PBMC) separations from healthy donors after written informed consent, and study approval by the University of Arizona Institutional Review Board. Large-scale production of DCs was performed as previously described [31]. Briefly, PBMCs were cultured in gas-permeable hydrophobic bags (Baxter, Glendale, CA, USA) at 5×106 cells/ml in 200-ml AIM-V medium (Gibco, Paisley, Scotland) supplemented with 1% l-glutamine (Gibco), 500 IU/ml GM-CSF (Immunex, Seattle, WA, USA) and 50 ng/ml IL-13 (Sanofi-Synthelabo, Paris, France) for 7 days at 37°C in 5% CO2. Fresh cytokines were added on day 4 of culture. On day 7, DCs were purified from the cell suspension according to size using an elutriator system (Beckman Coulter, Fullerton, CA, USA). DCs were frozen in liquid nitrogen at 2.5×107 cells/ml in 0.9% NaCl (Baxter) containing 10% DMSO (Sigma, St Louis, MO, USA), 4% human serum albumin (Baxter). Cells thawed at 4–8 weeks postfreezing were >90% viable.
The A549 human lung cancer, U251 human glioma, MCF-7 human breast cancer, and Colo 829 human melanoma cell lines were obtained from the American Type Culture Collection (Rockville, MD, USA). The A549, Colo 829, and MCF-7 cell lines were maintained in RPMI 1640 medium (Gibco, Grand Island, NY, USA) containing 10% heat-inactivated fetal bovine serum (FBS; Omega Scientific, Tarzana, CA, USA). The U251 cell line was maintained in Dulbecco’s modified essential medium (DMEM; Fisher, Tustin, CA, USA) containing 10% heat-inactivated fetal bovine serum, 1% glutamine, 1% penicillin-streptomycin, 1% sodium pyruvate, and 1% MEM nonessential amino acids (Gibco).
For clonogenic assays of irradiated Colo 829 cells, control cells and populations receiving 25, 50, and 200 Gy of irradiation were plated in duplicate at 2×106 cells in 75-cm2 tissue culture flasks. Cultures were maintained for 2 weeks and fed every 3 days with RPMI 1640 containing 10% FBS. Colonies were stained using Diff-Quik (VWR, West Chester, PA, USA) and counted.
Electrofusion and culture of DC–tumor cell hybrids
DCs were thawed and washed twice in AIM-V medium. Tumor cells (~70% confluent) were detached using a 0.02% EDTA solution (Sigma, St Louis, MO, USA), washed in phosphate-buffered saline (PBS) and irradiated (50 Gy). In some cases, cells were pre-stained with individual membrane dyes for detection of fusion hybrids by FACS analysis. DCs were pre-stained for 30 min with 2.25 μM of the “red” fluorescent CMFTR CellTracker dye (Molecular Probes, Eugene, OR, USA), and tumor cells were pre-stained for 30 min with 1.25 μM of the “green” fluorescent CMFDA CellTracker dye (Molecular Probes, Eugene, OR, USA). The DCs and tumor cells were then mixed at a 1:1 ratio, pelleted, and washed three times in Cytofusion Medium C, an isotonic sorbitol solution containing magnesium and calcium (Cyto Pulse Sciences, Columbia, MD, USA). The cells were resuspended in 3 ml of Cytofusion Medium C (2×107 cells/ml, 6 x107 cells total) and placed in a 6-ml coaxial electrofusion chamber (4-mm gap distance) (Cyto Pulse Sciences, Columbia, MD, USA). Electrofusion was performed using a PA-101 generator (Cyto Pulse Sciences). Cells were fused by first aligning using an alternating current at 100 V/cm (30 s) and then 187.5 V/cm (10 s) followed by four direct current pulses at 2,000 V/cm (40 μs each, 0.125-s pulse intervals, 0.8-MHz). After the fusion pulses, an alternating current at 112.5 V/cm (55 s) was applied to help maintain cell contact [37]. Following fusion, the cells were incubated in the chamber for 30 min at room temperature. An equal volume of RPMI 1640 medium was then added, and the cells were incubated for a further 15 min. Mixed populations were treated identically but without experiencing electrofusion. Cell populations were then either immediately analyzed for hybrid formation or placed in culture for later analysis. Populations were cultured at 1×106 cells/ml in RPMI 1640 containing 10% FBS, GM-CSF (500 IU/ml), and IL-13 (50 ng/ml). Mature DC–hybrid cultures were generated by a further 24-h culture in a cocktail mixture containing GM-CSF (500 IU/ml), IL-13 (50 ng/ml), 10 ng/ml TNF-α (R&D Systems, Minneapolis, MN, USA), 10 ng/ml IL-1β (R&D Systems), 15 ng/ml IL-6 (R&D Systems), 1 μg/ml PGE2 (Sigma), and 25 μg/ml poly I:C (Sigma) [20, 31]. The percentage of multinucleated cells was determined by cytocentrifugation followed by dye staining with Diff-Quik (VWR, West Chester, PA, USA) and counting of multinucleated cells. The percentages of heterologous hybrids were determined by FACS analysis. Cell viabilities were assessed by dye exclusion staining using trypan blue (Sigma).
Monoclonal antibodies and FACS analysis
The DC phenotype was assessed by FACS analysis using fluorescent-conjugated mouse monoclonal antibodies recognizing HLA-DR, CD40, CD14, CD3, CD80, CD86, CD83, and CD25 (BD Pharmingen, San Diego, CA, USA). Heterologous DC–tumor cell hybrids were detected by FACS analysis with the DC parent immune stained using a phycoerythrin (PE)-conjugated anti-HLA-DR antibody or pre-stained with the CMFTR CellTracker dye (see above). The tumor cell parent was detected by intracellular staining using a fluorescein isothiocyanate (FITC)-conjugated anticytokeratin antibody (for detection of the epithelial-derived tumor cells) or by FACS detection of cells pre-stained with the CMFDA CellTracker dye (see above). Intracellular anticytokeratin antibody staining was performed by first treating the cells with CytoPerm/Cytofix buffer (BD BioSciences, San Jose, CA, USA), followed by incubation with a FITC-conjugated anticytokeratin antibody (Sigma) and washing in the presence of Perm/Wash buffer per manufacturer’s procedures (BD BioSciences). Appropriate isotype antibodies were used as controls for all immune staining procedures. FACS analysis was performed on a FACScan (Becton Dickinson, San Jose, CA, USA) using Cell Quest software (Becton Dickinson) or WinMDI software (J. Trotter, Scripps Institute, La Jolla, CA, USA).
Immune stimulatory function of DC–Colo 829 cell hybrid populations
DC–tumor cell hybrids were evaluated for stimulation of autologous PBMCs. PBMCs were isolated from whole blood of autologous DC donors by centrifugation through Ficoll-Hypaque (Pharmacia, Peapeck, NJ, USA), as previously described [33]. PBMCs (1×105 cells) were stimulated weekly for 3 weeks with 1×104 cells of a DC–Colo 829 electrofused population, DCs plus Colo 829 mixed population, or Colo 829 cells only. Cells were cultured in a 96-well U-bottom plate (VWR, South Plainfield, NJ, USA) with X-Vivo 15 medium (BioWhittaker, Walkersville, MD, USA) supplemented with 5% autologous serum. IL-7 (10 IU/ml; R&D Systems, Minneapolis, MN, USA) and IL-2 (50 IU/ml; Chiron, Emeryville, CA, USA) were added on the 1st day of each weekly stimulation. On days 3 and 5 of culture, IL-2 was added when one half of the media was replaced with fresh media. The stimulated PBMC cultures were subsequently tested against Colo 829 tumor cells or DCs for activation-induced IFN-γ release. PBMCs were plated in triplicate with target cells in 96-well plates (1×105 PBMCs and 1×104 target cells per well). After incubating for 48 h, supernatants were removed and stored at –20°C. Samples were thawed and analyzed for human IFN-γ by ELISA using a human IFN-γ antibody set, as described by the manufacturer (BD Pharmingen, San Diego, CA, USA). Mean values (pg/ml) were calculated and standard deviations determined.
Antigen-specific stimulation of T cells by DC–Colo 829 cell hybrid populations was evaluated using the CD8+ CTL line H3.1, which is specific for the melanoma antigen gp100 and recognizes the antigenic gp100209–217 peptide (ITDQVPFSV) when displayed in the context of the HLA-A*0201 MHC complex [19]. H3.1 CTLs (1×105 cells) were cocultured in triplicate with 1×104 electrofused or mixed DC–Colo 829 populations, DCs only, or Colo 829 cells only. After 48 h, supernatants were removed and tested for IFN-γ release by ELISA (see above).
Results
Generation of DC–tumor cell hybrids
DCs were generated from healthy donor leukapheresis product after 7 days of culture with GM-CSF and IL-13 in hydrophobic bags followed by elutriation [10, 31]. This technology is a single-use disposable system that provides built-in controls for large-scale production of DCs. As determined by FACS forward and side scatter, the initial day 0 leukapheresis sample contains mainly smaller cells typical of the high numbers of lymphocytes present in a leukapheresis sample, while the day 7 elutriated product comprises larger cells representative of DCs (Fig. 1A). Immune phenotyping by FACS analysis demonstrated that the day 7 elutriated DCs are characteristic of immature DCs with low-level detection of the monocyte marker CD14, high expression of HLA-DR (MHC II), CD40, and the costimulatory molecule CD80 and absence of the maturation marker CD83 (Fig. 1B). Only low levels of CD3+ T cells are present (14%), indicative of the purity of the DC population. This phenotype is representative of 15 separate DC isolations. When initiating cultures from approximately 180 ml of leukapheresis product, relatively large numbers of DCs are obtained (6×108 to 2×109 DCs) with yields ranging between 9–14% of the leukapheresis PBMC population. For DC-tumor electrofusion studies, DCs derived from five different leukapheresis donors were used.
Fig. 1.

A Forward and side scatter FACS analysis of day 0 leukapheresis sample and day 7 elutriated DCs. B Marker phenotype analysis of day 7 elutriated DCs. Cells were immune stained either with fluorescent-conjugated monoclonal antibodies directed against the indicated markers (dark line) or fluorescent-conjugated isotype control antibody (gray line). The percentage positivity for each marker is indicated
Figure 2A shows multinucleated cells generated by electrofusion of DCs with A549 human lung cancer cells or MCF-7 human breast cancer cells. At 45 min postfusion, large multinucleated cells with up to six nuclei were observed. Upon visual counting, the percentage of hybrid cells with >1 nucleus ranged between 25–35% of the population. Dual immunofluorescent antibody staining using anti-HLA-DR antibody for the detection of the DC parent and anticytokeratin antibody for the detection of the epithelial tumor cell parent clearly confirmed the presence of heterologous fusion hybrids comprising both DCs and tumor cells, in this case A549 cells (Fig. 2B).
Fig. 2A, B.

Photomicrographs of mixed and electrofused populations. A Diff-Quik dye staining of mixed and fused DC-tumor cell populations. Mixed and electrofused populations were cytocentrifuged and dye stained. B Dual immunofluorescent staining of DC-A549 hybrid cell product. Electrofused cells were cytocentrifuged and immune stained using an anti-HLA-DR-PE monoclonal antibody (upper panel) and an anticytokeratin-FITC monoclonal antibody (lower panel). Arrow indicates a large, fused DC-tumor hybrid cell positive for both HLA-DR and epithelial keratin filaments
FACS analysis was subsequently used to determine total percentage of DC-tumor hybrids at 45 min postfusion. Dot-plot FACS analyses are shown; the upper right quadrant represents dual-positive cells (Fig. 3). Background positive cells were observed in the mixed population, which most likely arise due to cell-cell clumping. Total percentage hybrid formation was calculated by subtracting the percentage of dual-positive cells present in the upper right quadrant of the mixed population from that observed in the electrofused population. For the DC-A549 electrofusion, 10% hybrid formation was observed upon dual immunofluorescent staining with anti-HLA-DR antibody and anticytokeratin antibody. Samples comprising electrofused DCs and MCF-7 cells contained 13% DC–MCF-7 hybrid cells at 45 min postfusion. Electrofusion of DCs and Colo 829 melanoma cells or U251 glioma cell partners resulted in 9% and 10% DC–tumor cell hybrids, respectively. For these populations, DCs were pre-stained with the CMFTR dye while the tumor cells were pre-stained with the CMFDA dye. As summarized in Table 1, the DC-tumor hybrid efficiency at 45 min postfusion averaged between 8% and 10% for each tumor type tested.
Fig. 3.

FACS analyses of electrofused DC–tumor cell populations at 45 min postfusion. DC–A549 cell populations were immune stained with the indicated antibodies postfusion. For DC–MCF-7 cell, DC–Colo 829 cell, and DC–U251 cell electrofusions, DCs were pre-stained with CMFTR dye, and tumor cells were pre-stained with CMFDA dye. Percentages of dual-positive cells present in the upper right quadrant are indicated. For calculating the percentage of DC-tumor hybrids, the percentage of dual-positive cells present in the upper right quadrant of the mixed population is substracted from that observed in the eletrofused population
Table 1.
Fusion efficiency of DC–tumor cell hybrids. Percentages were determined by FACS analysis at 45 min postfusion
| DC–tumor cell fusion | Average % hybrid | Range (%) |
|---|---|---|
| DC-A549 (n=10) | 9 | 8–11 |
| DC–Colo 829 (n=10) | 8 | 6–15 |
| DC–MCF-7 (n=3) | 10 | 9–13 |
| DC-U251 (n=2) | 9.5 | 9–10 |
In our studies, tumor cells were routinely irradiated at 50 Gy prior to DC electrofusion so as to inhibit tumor cell proliferation. Growth-inhibited tumor cells would be required for clinical application of the fusion vaccine product. Using a 2-week clonogenic assay, 50 Gy was a reasonable dose when testing Colo 829 cells for residual growth following irradiation. While cells receiving no irradiation grew to a high density over a 2-week period postirradiation, a relatively low number of residual proliferating cells were observed in Colo 829 populations receiving 25 Gy (average of 920 colonies / 106 irradiated cells plated) with no detectable colonies in cultures receiving 50 and 200 Gy. The effects of irradiation on electrofusion were examined in DC–tumor cell populations. Comparable hybrid efficiencies were observed for nonirradiated Colo 829 and populations receiving doses of 50 or 200 Gy prior to electrofusion with DCs (Fig. 4). Similar results were observed for DC–A549 cell electrofusions (data not shown).
Fig. 4.

DC electrofusion of nonirradiated and irradiated Colo 829 cells. DCs were pre-stained with the CMFTR dye, and Colo 829 cells were pre-stained with the CMFDA dye prior to irradiation and electrofusion
Viability and maintenance of hybrids at 48 h postfusion
Important for the clinical application of DC-tumor hybrids is the fate of hybrids at later times postfusion, both with respect to the retention of hybrids and viability. When examined at 48 h postfusion, DC–Colo 829 populations continued to show a similar percentage of hybrids as seen at the earlier 45-min time point (Fig. 5A). As determined by trypan blue dye exclusion, cell viabilities at both time points were similar for the mixed and electrofused cultures. At 45 min postfusion, cell viability for each population was in the range of 90–95%, and at 48 h the range was 85–90%. These results indicate that the electrofusion process itself did not selectively cause cell death.
Fig. 5.

A FACS analysis of DC–Colo 829 populations at 45 min and 48 h postfusion. Colo 829 cells were pre-stained with CMFDA dye prior to electrofusion. The DC marker, HLA-DR, was detected by immune staining postfusion. Mixed and electrofused populations were analyzed at 45 min postfusion and after 48 h of culture. Cell populations were cultured in RPMI 1640 containing 10% FCS. B Analysis of DC-tumor hybrids after freeze/thaw. FACS analysis of mixed and electrofused populations was performed at 45 min postfusion and after freezing for 1 week and thawing. Colo 829 cells were pre-stained with CMFDA prior to fusion. The DC marker, HLA-DR, was detected by immune staining postfusion
A further consideration for clinical trial development is whether hybrid populations can be frozen and stored to provide multiple, sequential doses of the hybrid cell product. At 45 min postfusion, both electrofused and mixed populations of DC–Colo 829 cells were frozen in liquid nitrogen. One week after freezing, cells were thawed and evaluated by FACS analysis. Trypan blue dye staining indicated that the mixed and electrofused populations were ~90% viable at 45 min postfusion and remained at this value after freezing and thawing. As determined by FACS analysis, similar numbers of hybrids (8–9%) were observed in electrofusion products before and after freezing (Fig. 5B). Comparable results have been obtained for DC–MCF-7 and DC-A549 hybrid populations (data not shown).
T-cell immune responses to DC–Colo 829 cell hybrid populations
To determine whether DC-tumor hybrids are capable of enhancing primary T-cell responses, autologous PBMCs were stimulated weekly for 3 weeks with electrofused DC–Colo 829 populations, mixed populations, or Colo 829 cells only. The stimulated PBMC populations were then tested for their ability to respond to tumor cell target as indicated by secretion of IFN-γ. The experimental design is that of a mixed lymphocyte reaction (MLR) in that the tumor cell line used for this analysis is allogeneic to the normal donor from whom the DCs and PBMCs were derived. When tested against Colo 829 target cells, the greatest amount of IFN-γ secretion was observed for those PBMCs previously stimulated with the DC–Colo 829 electrofused population (Fig. 6A). Mixtures of DCs and Colo 829 cells, as well as Colo 829 cells only, were significantly less capable of stimulating PBMCs that recognized the Colo 829 target cells. When the PBMC populations were tested against DC targets, the detected levels of IFN-γ were less than 100 pg/ml, indicating only a limited response to autologous DCs. The results suggest that electrofused hybrids are capable of generating an enhanced allogeneic response.
Fig. 6A–C.
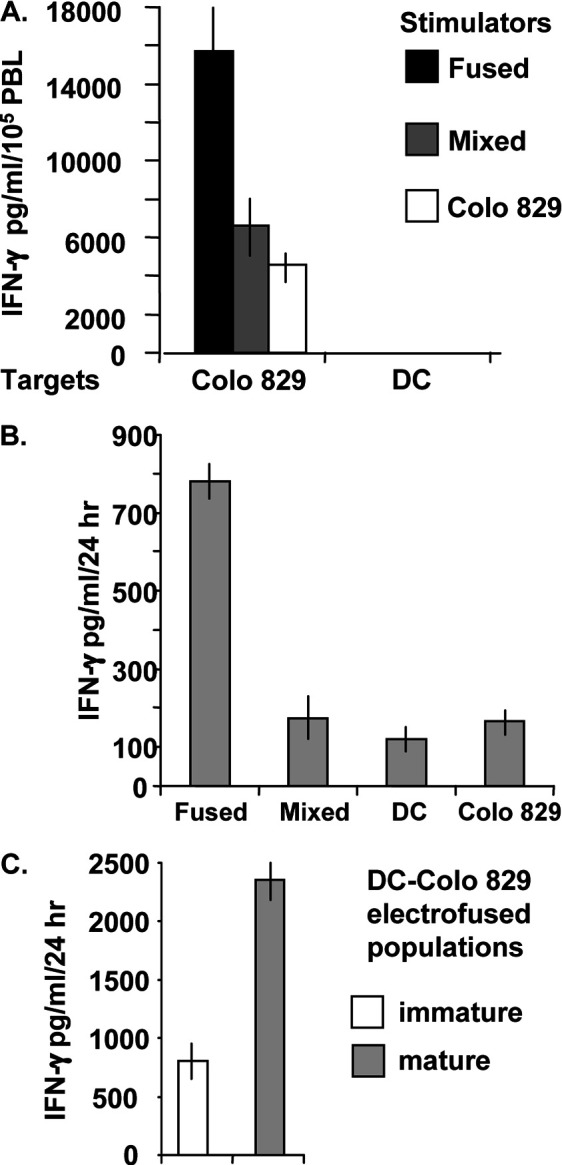
Induction of T-cell responses against DC–Colo 829 hybrid populations. A Autologous PBMC responses. Autologous PBMCs (105 cells) were stimulated weekly for 3 weeks in the presence of DC–Colo 829 electrofused cells, mixed DC–Colo 829 cells, or Colo 829 cells only (104 cells). The stimulated PBMC cultures were tested against Colo 829 tumor cells or DCs for activation-induced IFN-γ release (ratio 105 PBMCs to 104 target cells). Supernatants were analyzed by IFN-γ ELISA at 24 h postincubation. B Induction of gp100 antigen-specific H3.1 CTL response by electrofused DC–Colo 829 hybrid populations. H3.1 CTLs (1×105 cells) were cocultured with the indicated targets (104 cells). The electrofused population indicated 8% DC–Colo829 fusion hybrids as determined by FACS analysis (data not shown). After 24 h, supernatants were removed and tested for IFN-γ production by ELISA. C Comparison of gp100 antigen-specific H3.1 CTL responses induced by immature or mature DC–Colo829 hybrid populations. H3.1 CTLs (1×105 cells) were cultured with 1×104 cells from either an immature hybrid DC–Colo 829 population or the same population exposed to maturation agents for 24 h prior to coculture with H3.1 CTLs. Supernatants were removed after 24 h and tested for IFN-γ production by ELISA. For all samples, mean values and standard deviations of triplicate samples are shown
To examine whether DC-tumor hybrids are capable of augmenting specific antitumor antigen T-cell responses, stimulation of the gp100-specific CTL line (H3.1) by DC–Colo 829 cell hybrids was evaluated. H3.1 CTLs specifically recognize antigenic gp100 peptide (gp100209–217) when displayed in the context of the HLA-A*0201 major histocompatibility complex; both IFN-γ production and cytotoxicity against melanoma cells are observed upon gp100 peptide stimulation of this cell line [19]. DCs were derived from an HLA-A*0201(+) donor and fused with Colo 829 melanoma cells, which were determined to be gp100(+) (data not shown). In addition, Colo 829 cells are HLA-A*0201(-) (data not shown) and would not be expected to stimulate H3.1 CTLs. Antigen-specific stimulation would only occur via the processing of the gp100 expressed by the DC–Colo 829 hybrid cells and subsequent display of antigenic gp100 peptide in the context of the DC-derived HLA-A*0201 complex. At 45 min postfusion, the CTLs were added to the electrofused population, mixed cells, DCs, or Colo 829 tumor cells. After 24 h, supernatants were removed and tested for the presence of IFN-γ by ELISA. The highest level of IFN-γ secretion was observed for CTLs cultured in the presence of hybrid cells and was approximately fourfold to fivefold higher than the levels expressed by CTLs cocultured with mixed cells, DCs only, or Colo 829 cells only (Fig. 6B).
Thus far, our studies have involved the use of immature DCs. Because mature DCs are more potent inducers of effector T cells [2, 27, 32], an electrofused DC–Colo 829 population that was matured for 24 h in the presence of a maturation cocktail was compared with immature fused cells for induction of H3.1 CTL responses. A significant increase in IFN-γ secretion was induced by the mature hybrid population relative to the immature hybrid population (Fig. 6C). As determined by FACS analysis, 7% of the mature hybrids expressed the DC maturation marker CD83, indicative that DC–Colo 829 fused cells are competent for maturation (Fig. 7A). As determined by CD83 staining, residual immature DCs (CD83−) were seen in both mixed and electrofused populations exposed to maturation cocktail (lower left quadrant). The overall fusion efficiency was 12%, as determined by our more standard method of FACS analysis using anti-HLA-DR antibody for the detection of the DC parent (data not shown). Assessment of other markers of maturation demonstrated increased expression of CD25 and CD86, along with CD83 (Fig 7B), further indicating that electrofusion itself did not alter the overall maturation capacity of DCs within the population. In addition, mature electrofused populations are competent for expression of IL-12p70 and display increased levels of HLA-DR (data not shown).
Fig. 7.

A FACS detection of mature DC–Colo 829 hybrid cells expressing the maturation marker CD83. Colo 829 cells were pre-stained with the CMFDA membrane dye. Mixed and electrofused populations were cultured for 24 h with or without maturation cocktail and immune stained with an anti-CD83 monoclonal antibody followed by FACS analysis. B Phenotype of immature and mature electrofused DC–Colo 829 populations. Following electrofusion, cell populations were cultured for 24 h in the presence or absence of maturation cocktail. The cells were subsequently immune stained with the indicated fluorescent-conjugated monoclonal antibodies. Gray line isotype control; dark line antibody
Discussion
Theoretically, the use of DC-tumor fusion hybrids for cancer vaccine therapy is highly appealing. Unlike other types of DC-based vaccines, the hybrids have the capacity for sustained expression of the entire tumor antigen repertoire within the context of the immune stimulatory capacity of the DCs. However, a major issue for the application of DC–tumor cell hybrid vaccines is the development of compatible and reproducible procedures that can be readily translated to the clinical setting. Of equal concern is the availability of sufficient tumor material for the preparation of tumor cells. In this study we have integrated several features important for the clinical development of DC–tumor cell hybrid vaccines: (1) a large-scale process for the generation of DCs, (2) an electrofusion system that permits fusion of large numbers of DCs and tumor cells, and (3) the application of tumor cells derived from allogeneic cells lines.
Our method for generating large quantities of DCs from leukapheresis product is suitable for clinical applications [10, 31], and the development of cancer vaccine trials applying this DC cellular product is in progress. In our study, the percentage of observed heterologous DC–tumor cell hybrids generated by electrofusion ranged between 8% and 10% irrespective of the tumor type used. These results demonstrate that the electrofusion process, as delivered by this system, is consistent and can be standardized for clinical purposes. Our findings of similar electrofusion efficiencies between DCs and cells derived from different tumor cell types is possibly related to the similar physical sizes of the tumor cells used in this study (~15 μm in diameter). This parameter is one factor known to influence electrical field–induced cell fusion [24, 37].
The observed efficiency of electrofusion is within the range of that reported by others when using smaller types of fusion chambers [16, 23, 25, 28, 29]. The electrofusion system described here is superior in providing large-scale, controlled production of hybrids. Recently, one other group has reported success in applying a similar large-scale, concentric fusion chamber for the generation of DC-tumor hybrids [11, 26]. Based on previous clinical trials applying DC–tumor cell hybrid vaccines [17, 18, 23], our system would be appropriate for the generation of at least one batch of vaccine product.
Several enrichment techniques have been used to further increase the percentage of hybrids, including FACS cell sorting [12] and plastic adherence for the selection of DC-tumor hybrids [11, 26]. We have attempted the adherence procedure with the notion that this method could be readily adapted for clinical studies, but there was no clear indication of DC–tumor cell enrichment (data not shown). This reported method does not appear to be easily standardized and is highly variable [26].
In our study, we evaluated several parameters for the clinical application of DC–tumor cell hybrids. Following freezing and thawing of electrofused populations, hybrids are retained with high overall viability, indicating that batches of electrofused cells can be frozen for future vaccine use. In addition, we demonstrate that tumor cell irradiation does not affect overall DC–tumor cell hybrid generation when delivering doses up to 200 Gy. Our preliminary titering of radiation dose indicated that >25 Gy would be required to inhibit Colo 829 cell growth, and a dose of 50 Gy was selected for our electrofusion studies. Early after electrofusion (45 min), the cell viabilities of both mixed and electrofused fused populations are relatively high (90–95%). After 48 h in culture, DC–tumor cell hybrids are retained with only a slight decrease in viability (cell viabilities at 85–90%). These findings portend well for the in vivo longevity of a DC–tumor cell hybrid vaccine produced in our described system.
Previous studies have applied preirradiated tumor cells or irradiated DC–tumor cell hybrids to inhibit residual tumor cell growth in fusion vaccine preparations; radiation doses have varied between 30 Gy and 200 Gy [11, 17, 18, 25, 29]. However, the effect of the radiation dose has not been directly addressed. Increased dosages of irradiation would induce higher levels of apoptosis, and high-level cell death has been observed in populations of Colo 829 cells over time after exposure to 200 Gy (data not shown). Because DCs are capable of processing apoptotic tumor cells and presenting tumor-associated antigens [36], the degree of tumor cell apoptosis in a DC–tumor cell population could well mask the true antitumor vaccine efficacy of the hybrid. Determination of an appropriate radiation dose would be necessary, especially as tumor cells are well known to display variability in their radiosensitivity.
Evaluations of T-cell responses in vitro suggest that electrofused hybrid populations are better than mixed cultures in eliciting antitumor cell effects. As determined by IFN-γ release, DC–Colo 829 melanoma cell hybrids display an increased capacity for stimulating primary, autologous T cells that recognize the parental Colo 829 cells relative to stimulating with a mixed population of DCs and Colo 829 tumor cells, or Colo 829 tumor cells alone. The observed T-cell response is presumably an allogeneic response against the Colo 829 cells. We were unable to demonstrate whether this response included the generation of primary TAA-specific T cells (data not shown). Only sparse numbers of antigen-specific T cells would be generated in the primary PBMC populations, and the allogeneic response would be expected to dominate. Not withstanding, this finding demonstrates the functional ability of DC-tumor hybrids to induce primary T-cell immune responses.
With respect to antigen-specific T-cell responses, DC–Colo 829 melanoma cell hybrids stimulate IFN-γ release by CD8+ T cells (H3.1 CTLs) that are specific for an immunodominant gp100 peptide epitope displayed in the context of the DC-derived MHC I HLA-A*0201 molecule. This result suggests that melanoma TAAs are processed and displayed by the DC–Colo 829 cell hybrid. Moreover, this finding emphasizes the utility of allogeneic tumor cell lines for the generation of antitumor immunity by a DC–allogeneic tumor cell vaccine. Beyond the benefit of cell lines serving as a source of tumor cells that express a broad repertoire of known TAAs, an additional advantage is that the processing and display of allogeneic molecules themselves would provide additional immune stimulatory signals, enhancing antigen-specific immune responses [14, 35].
As would be expected, maturation of the DC–Colo 829 hybrid population induces improved anti-gp100 T-cell responses as compared to an immature hybrid population. The cell hybrids appeared to mature based on expression of the DC CD83 marker. Moreover, the hybrid population displayed other markers of DC maturation (CD25 and CD86), indicative that the electrofusion process did not alter the maturation capacity of the DCs. In a recent report, hybrids comprising melanoma cells and DCs matured prior to fusion were shown to stimulate IFN-γ production by both CD8+ and CD4+ gp100-specific T cells, suggesting that processed gp100 antigenic epitopes are displayed in the context of both MHC I and MHC II molecules derived from the DC hybrid parent [26]. However, in this previous study, mature and immature hybrid populations were not directly compared. Furthermore, the DC–tumor cell hybrids, as well as residual DCs in the population, appeared to lose CD83 expression [26]. Because immature DCs are more effective in the processing of antigen while mature DCs are characterized by improved antigen display and effector T-cell costimulation [2, 27, 32], maturation of the DC-tumor hybrids postfusion (as described in our study) is a more reasonable approach than using DCs that are matured prior to tumor cell fusion. Additional investigation is required to determine whether DC maturation is more beneficial when applied prefusion or postfusion. Beyond questions of the maturation status of the DC–tumor cell hybrid vaccine, further aspects for improved clinical efficacy include those under consideration for DC-based vaccine trials in general (appropriate dosage, schedule of administration, and route of inoculation) [2, 27, 32].
Finally, although DC–tumor cell hybrids are assumed to have an enhanced ability to generate antitumor T-cell responses, other types of DC-based vaccines have shown efficacy in animal models and humans. Yet, reports have indicated that DC–tumor cell hybrids are more efficient at stimulating CTL activity than are DCs pulsed with apoptotic or necrotic (lysate) tumor cells [4, 5]. Taken together, our findings are encouraging for the large-scale production of DC-tumor hybrid vaccines and their application in the treatment of cancer.
Acknowledgements
The authors thank Drs E. Wang and M. Panelli (Immunogenetics Laboratory, Department of Transfusion Medicine, National Institutes of Health, Bethesda, MD) for the generous gift of the H3.1 CTL cell line. This work was supported in part by the Arizona Disease Control Research Commission (Contract #30005), the Department of Defense (Grant Number DAMD17–01–1-0473), The Brain Tumor Society, and the Arizona SPORE in GI Cancer (NIH/NCI CA95060).
References
- 1.Akasaki J Immunother. 2001;24:106. [PubMed] [Google Scholar]
- 2.Brossart Exp Hem. 2001;29:1247. doi: 10.1016/S0301-472X(01)00730-5. [DOI] [PubMed] [Google Scholar]
- 3.Chan Immunol Lett. 2002;3:101. doi: 10.1016/S0165-2478(02)00078-0. [DOI] [PubMed] [Google Scholar]
- 4.Galea-Lauri Cancer Immunol Immunother. 2002;51:299. doi: 10.1007/s00262-002-0284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goddard Clin Exp Immnol. 2002;131:82. doi: 10.1046/j.1365-2249.2003.02047.x. [DOI] [Google Scholar]
- 6.Gong Nat Med. 1997;3:558. doi: 10.1038/nm0597-558. [DOI] [PubMed] [Google Scholar]
- 7.Gong Proc Natl Acad Sci U S A. 2000;97:2715. doi: 10.1073/pnas.050587197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gong J Immunol. 2000;165:1705. doi: 10.4049/jimmunol.165.3.1705. [DOI] [PubMed] [Google Scholar]
- 9.Gong Blood. 2002;99:2512. doi: 10.1182/blood.V99.7.2512. [DOI] [PubMed] [Google Scholar]
- 10.Goxe Immunol Invest. 2000;29:319. doi: 10.3109/08820130009060870. [DOI] [PubMed] [Google Scholar]
- 11.Hayashi Clin Immunol. 2002;104:14. doi: 10.1006/clim.2002.5224. [DOI] [PubMed] [Google Scholar]
- 12.Holmes J Immunother. 2001;24:122. doi: 10.1097/00002371-200103000-00006. [DOI] [Google Scholar]
- 13.Hsueh J Clin Oncol. 2002;20:4549. doi: 10.1200/JCO.2002.01.151. [DOI] [PubMed] [Google Scholar]
- 14.Jaffee Ann N Y Acad Sci. 1999;886:67. doi: 10.1111/j.1749-6632.1999.tb09401.x. [DOI] [PubMed] [Google Scholar]
- 15.Jaffee J Clin Oncol. 2001;19:145. doi: 10.1200/JCO.2001.19.1.145. [DOI] [PubMed] [Google Scholar]
- 16.Jantscheff Cancer Immunol Immunother. 2002;51:367. doi: 10.1007/s00262-002-0295-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kikuchi Cancer Immunol Immunother. 2001;50:337. doi: 10.1007/s002620100205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krause J Immunother. 2002;25:421. doi: 10.1097/00002371-200209000-00006. [DOI] [PubMed] [Google Scholar]
- 19.Lee J Immunol. 1998;161:4183. [PubMed] [Google Scholar]
- 20.Lee AW, Truong T, Bickham K, Fonteneau JF, Larsson M, Da Silva I, Somersan S, Thomas EK, Bhardwaj N (2002) A clinical grade cocktail of cytokines and PGE(2) results in uniform maturation of human monocyte-derived dendritic cells: implications for immunotherapy. Vaccine 20[Suppl 4]:A8 [DOI] [PubMed]
- 21.Li Cancer Immunol Immunother. 2001;50:456. doi: 10.1007/s002620100218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linder Euro J Clin Invest. 2002;32:207. doi: 10.1046/j.1365-2362.2002.00968.x. [DOI] [Google Scholar]
- 23.Marten Hum Gene Ther. 2003;14:483. doi: 10.1089/104303403321467243. [DOI] [PubMed] [Google Scholar]
- 24.Neil Methods Enzymol. 1993;220:174. doi: 10.1016/0076-6879(93)20082-e. [DOI] [PubMed] [Google Scholar]
- 25.Orentas Cell Immunol. 2001;213:4. doi: 10.1006/cimm.2001.1864. [DOI] [PubMed] [Google Scholar]
- 26.Parkhurst J Immunol. 2003;170:5317. [Google Scholar]
- 27.Schuler Curr Opin Immunol. 2003;15:138. doi: 10.1016/S0952-7915(03)00015-3. [DOI] [PubMed] [Google Scholar]
- 28.Scott-Taylor Biochim Biophys Acta. 2000;1500:265. doi: 10.1016/S0925-4439(99)00108-8. [DOI] [PubMed] [Google Scholar]
- 29.Siders Mol Ther. 2003;7:498. doi: 10.1016/S1525-0016(03)00044-3. [DOI] [PubMed] [Google Scholar]
- 30.Soruri Cancer Immunol Immunother. 2001;50:307. doi: 10.1007/s002620100198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spisek Cancer Immunol Immunother. 2001;50:417. doi: 10.1007/s002620100215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steinman J Clin Invest. 2002;109:1519. doi: 10.1172/JCI200215962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trevor Cancer Immunol Immunother. 2001;50:397. doi: 10.1007/s002620100214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J Immunol. 1998;161:5516. [PubMed] [Google Scholar]
- 35.Ward Cancer Immunol Immunother. 2002;51:351. doi: 10.1007/s00262-002-0286-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou J Immunother. 2002;25:289. doi: 10.1097/00002371-200207000-00001. [DOI] [PubMed] [Google Scholar]
- 37.Zimmermann Biochim Biophys Acta. 1982;694:227. doi: 10.1016/0304-4157(82)90007-7. [DOI] [PubMed] [Google Scholar]