

Abstract
Tumor cell vaccines have been successful at inducing immunity in naïve mice, but only in a few reports has vaccination alone induced regression of established tumors and, generally, only when they are very small. Clinically, vaccinations alone may not be able to cause regression of established human cancers, which tend to be weakly immunogenic. We hypothesized that pharmacologic ex vivo amplification of a vaccination-induced immune response with subsequent adoptive immunotherapy (AIT) to tumor-bearing animals would be more effective in treatment of these animals than vaccination alone. The 4T1 and 4T07 mammary carcinomas are derived from the same parental cell line, but 4T1 is much less immunogenic and more aggressive than 4T07. Vaccination with either 4T1, 4T1-IL-2, or 4T07-IL-2 was not effective as treatment for established 4T1 tumors. However, 4T1 or 4T07-IL-2-vaccine-sensitized draining lymph node (DLN) cells, activated ex vivo with bryostatin 1 and ionomycin and expanded in culture, induced complete tumor regressions when adoptively transferred to 4T1 tumor-bearing animals. This was effective against small tumors as well as more advanced tumors, 10 days after tumor cell inoculation. Furthermore, as would be required for this approach to be used clinically, vaccine-DLN cells obtained from mice with established progressive 4T1 tumors (inoculated 10 days before vaccination) also induced regression of 4T1 tumors in an adoptive host. In none of these experiments was exogenous IL-2 required to induce tumor regression. The response to tumor cell vaccine can be amplified by ex vivo pharmacologic activation of sensitized T cells, which can then cure an established, weakly immunogenic and highly aggressive tumor that was resistant to vaccination alone.
Keywords: Bryostatin 1, Ionomycin, Adoptive immunotherapy, Cyclophosphamide, 4T1
Introduction
Recent advances in cellular immunology, molecular biology, and gene therapy have contributed to the development of multiple new vaccine approaches to cancer treatment. These vaccines include genetically modified tumor cells, antigen-loaded dendritic cells, and purified tumor antigens, all designed to amplify the host's initial antitumor response [8, 9, 14, 15, 21, 22, 29, 30, 31, 38, 47]. Although there have been some encouraging results when these vaccines have been used to protect against a tumor challenge or to treat small established tumors, larger tumors have been more resistant to active specific immunotherapy. In some model systems, amplification of the response to vaccines with the addition of systemic cytokines has increased antitumor activity and tumor regressions, but sometimes at the cost of increased adverse effects and toxicity. In clinical trials, ex vivo antitumor activity has been demonstrated after tumor cell vaccination, but vaccines have had only limited success inducing tumor regressions, even with the addition of cytokine infusions or insertion of cytokine genes [14, 16, 18, 34, 38, 46].
Adoptive immunotherapy (AIT) with tumor-sensitized lymph node T cells has long been known to be able to induce regression of small or early tumors in mice [13]. However, this has generally only been shown for immunogenic tumors and usually requires administration of systemic interleukin-2 (IL-2) in vivo. We have previously shown that adoptive transfer of tumor-sensitized lymphocytes, activated with bryostatin 1 and ionomycin (B/I) and expanded in culture with low dose IL-2, induced tumor-specific regression of small, established tumors and 3-day lung metastases [39, 42]. These results were obtained with moderately immunogenic sarcoma, melanoma, and mastocytoma tumor models. Bryostatin 1 activates protein kinase C (PKC) and ionomycin increases intracellular calcium; together these agents mimic critical intracellular signaling events leading to T-cell activation [7, 40]. Recently, we reported that adoptive transfer of pharmacologically activated vaccine-draining lymph node (VDLN) lymphocytes could induce regression of a moderately immunogenic murine mammary carcinoma, 4T07 [11].
The experiments reported here take advantage of the 4T1 and 4T07 tumor cell lines, which were both derived from the same spontaneously arising murine mammary carcinoma, 410.4 [1]. Both 4T1 and 4T07 are highly tumorigenic but, in contrast to 4T07, 4T1 is a poorly immunogenic, highly aggressive, metastasizing cancer. Our aim was to use the 4T1 tumor model to test the hypothesis that adoptive immunotherapy can amplify the antitumor effects of a weakly immunogenic and otherwise ineffective tumor vaccine (4T1 or 4T07-IL-2) in the treatment of this weakly immunogenic cancer (4T1). If so, then adoptive immunotherapy (AIT) with VDLN cells might induce tumor regressions that could not be achieved with vaccination alone. In order to demonstrate the clinical utility of this approach, we also performed AIT with B/I-activated VDLN cells against large established tumors, and with T cells sensitized in a tumor-bearing animal with metastatic disease. In both scenarios, we were able to cure established 4T1 tumors.
Materials and methods
Mice
Virus-free BALB/c mice (Charles River Laboratories, Cambridge, MA) were used, between 8 and 12 weeks of age, caged in groups of six or fewer, and provided with food and water ad libitum. All guidelines of Virginia Commonwealth University, which conform to the American Association for Accreditation of Laboratory Animal Care and the US Department of Agriculture recommendations for the care and humane experimental use of animals, were followed.
Tumor cell lines
For the study, 4T1 and 4T07 mammary tumor cell lines and their IL-2 transduced counterparts (4T1-IL-2, 4T07-IL-2) were kindly provided by Dr Jane Tsai at the Michigan Cancer Foundation, Detroit, Michigan. Cells were maintained in Dulbecco's Modified Essential Medium (DMEM) with 10% heat-inactivated fetal calf serum (FCS) (Hyclone, Logan, UT), 1-mM sodium pyruvate, 100-U/ml penicillin, and 100-μg/ml streptomycin (Sigma, St Louis, MO). G418 (Sigma) 600 μg/ml was added to 4T1-IL-2 and 4T07-IL-2 cultures to maintain selective pressure in favor of the transfected tumor cells. Meth A sarcoma (ATCC, Rockville, MD) and CT26 colon carcinoma (ATCC, Rockville, MD) were maintained in RPMI 1640 medium with 10% heat-inactivated FCS, 1-mM sodium pyruvate, 0.1-mM nonessential amino acids, 2-mM l-glutamine, 100-U/ml penicillin, 100-μg/ml streptomycin, 10-mM Hepes buffer, and 5×10-5 M 2-mercaptoethanol (Sigma) (complete RPMI). All cells were incubated in 250-ml T-flasks (PGC, Gaithersburg, MD) at 37oC in humidified air with 5% CO2. Tumor cells were harvested for inoculation of mice with 0.05% trypsin-EDTA (Fisher, Pittsburgh, PA).
Vaccine Therapy
BALB/c mice were inoculated subcutaneously (s.c.) in the shaven left flank with 5×104 4T1 cells. After 4 days, either 2×106 irradiated (10,000 rads [100 Gy]) 4T1 or 4T1-IL-2 cells, or 1×106 live 4T07-IL-2 cells were injected s.c. into the opposite flank. In some experiments, a single dose of cyclophosphamide (CYP 100 mg/kg) (Mead Johnson, Princeton, NJ) was administered intraperitoneally (i.p.) 24 h prior to vaccine injection, so that appropriate comparison with our AIT protocol would be possible. While 4T1 and 4T1-IL-2 cells for vaccination required radiation to avoid tumor growth at the vaccination site and metastases, 4T07-IL-2, in contrast, regressed spontaneously, due to its greater immunogenicity.
Sensitization of draining lymph nodes
Naïve BALB/c mice were inoculated in one hind footpad with either 2x106 irradiated (100 Gy) 4T1-IL-2 cells or 1×106 viable 4T07-IL-2 cells. Other cells used for LN sensitization included 1×106 viable Meth A or 1×106 viable 4T1 cells. In some experiments, mice were initially inoculated in the flank with 1×104 live 4T1 cells and then were vaccinated in the contralateral footpad 10 days later with 1×106 viable 4T07-IL-2 cells (tumor-bearing donors). Ten days after footpad vaccination, mice were euthanized by CO2 asphyxiation, and popliteal lymph nodes draining the vaccination site were harvested under sterile conditions. Although we have shown that both flank and footpad inoculations sensitized regional DLN (inguinal and popliteal, respectively), we used footpad inoculation and popliteal nodes here for AIT because, in our experience, cell yields are higher [10].
Ex vivo bryostatin 1 and ionomycin activation and expansion
DLN were dispersed into single cell suspensions, washed and resuspended in complete RPMI for activation with bryostatin 1 and ionomycin (B/I). A portion of the lymphocyte suspensions in some experiments was suspended in complete RPMI containing 40 IU/ml of rIL-2 (Chiron, Emeryville, CA) in 250-ml T flasks, without B/I activation, and cultured for 4 days prior to adoptive transfer. DLN cells intended for B/I activation were resuspended at 1x106 cells/ml and incubated for 18 h with 5-nM bryostatin 1 (kindly provided by the National Cancer Institute, Bethesda, MD), 1-μM ionomycin (Calbiochem, San Diego, CA), and 80-IU/ml rIL-2 at 37°C in humidified air with 5% CO2 (B/I-activated). After incubation, cells were washed three times with warm (37°C) complete RPMI and cultured for 7–10 days with 40 IU/ml of rIL-2. Cells were split to 1x106 cells/ml and refed with medium and 40-IU/ml rIL-2 every other day. After 7–10 days of expansion in culture, cells were washed with serum-free medium and the appropriate number of cells in 0.5 ml was injected via the tail vein.
AIT treatment of established tumors
Mice were inoculated s.c. into the shaven left flank with 1×104 or 5×104 4T1 cells in 0.05-ml DMEM. For AIT, the indicated number of lymphoid cells in 0.5 ml of RPMI 1640, was administered i.v. 4 or 10 days after 4T1 flank inoculation. Mice were given a single dose of cyclophosphamide (CYP, 100 mg/kg, i.p.) 24 h prior to AIT (day 3 or 9).
Protection/"memory" experiments
Mice with complete tumor regressions after AIT ("treated") in some experiments were kept for an additional month and then rechallenged s.c. with 5×104 4T1 tumor cells in the opposite flank. Alternatively, naïve mice received 70×106 sensitized, B/I-activated, expanded DLN cells i.v. One month later, these mice and age-matched controls were inoculated s.c. with 4T1 (5×104) tumor cells into the shaven left flank. Tumors were measured serially as described below.
Tumor measurements
In all experiments, tumor growth was monitored with measurements twice a week of perpendicular diameters. When the "tumor area" (product of two diameters) was greater than 120 mm2 or if the mouse appeared ill, the animal was euthanized by CO2 asphyxiation. Complete tumor regression is defined as the absence of a measurable tumor on two consecutive measurements. Generally, when this occurred, tumors never regrew.
Cytokine assays
Tumor-sensitized or non-sensitized DLN cells after B/I-activation and in vitro expansion were cultured in complete RPMI (2×106 cells/ml) either alone or with irradiated (10,000 rads [100 Gy]) 4T1, 4T07, or Meth A cells in 24-well plates. The responder to stimulator ratio was 10:1. In some experiments, T cells were stimulated with splenocytes pulsed with AH1 peptide (SPSYVYHQF, from Sigma Genosys, The Woodlands, TX), 5:1. The spleen of a naïve BALB/c mouse was harvested and prepared into a mononuclear cell suspension by Ficoll-Hypaque gradient centrifugation. After mitomycin C treatment, the splenocytes (1×106/ml) were pulsed with 100-μg AH1 per ml of cells for 4 h at 37°C. Excess peptide was washed away, and the loaded splenocytes were added to T cells in 24-well plates. Unpulsed splenocytes served as a negative control. After 24 h, supernatants were harvested and stored at −20°C until assayed for IFN-γ by ELISA (Endogen, Cambridge, MA). Absorbance was read by an ELISA reader (Molecular Devices) set at 450 and 550 nm, and pg/ml of IFN-γ was calculated by comparison with an IFN-γ standard curve.
Clonogenic Assay
Mice inoculated with 1×104 4T1 cells in the left flank were used to determine the pattern and kinetics of spontaneously arising metastases (regional or distant). Mice with 4T1 flank tumors were euthanized at days 14, 18, and 25. The inguinal lymph nodes, one of the regional lymphatic basins draining the left flank, and the lungs were harvested from five mice at each time point. The lymph nodes were dispersed into a single cell suspension and cultured in serial dilution with DMEM containing 60-μM thioguanine (Sigma, St Louis, MO). Harvested lungs were diced into 1 mm3 pieces and placed in a PBS solution containing 1-mg/ml collagenase IV and 6-U/ml elastase, rotating at medium speed in 4°C for 1 h. The solution and lung pieces were then passed through a 70-μ filter and rinsed with DMEM. The cells were then cultured in serial dilution with DMEM + 60-μM thioguanine. Colony forming units were counted after 10 days of uninterrupted incubation (37°C).
Statistical analysis
Results of tumor measurements are presented as the means and standard errors (SE) of "tumor area" in each treatment group. Differences in tumor growth among different treatment groups were assessed by analysis of variance (ANOVA) and Tukey-Kramer honestly significant difference test (Tukey's HSD) using JMPIN software (SAS Institute, Cary, NC). Each experiment included at least six mice per group and was repeated at least twice. An α<0.05 was used throughout to determine significant differences.
Results
Established 4T1 tumors do not respond to vaccination alone
Use of genetically modified tumor cells has been one of the strategies reported in the literature for improving vaccine efficacy. IL-2 gene transfected 4T1 (4T1-IL-2) and its more immunogenic variant, 4T07-IL-2, were used as vaccines for the treatment of established 4T1 tumors. Cyclophosphamide was administered on the day prior to vaccination, also with the intention of augmenting vaccine efficacy and for appropriate comparison to our AIT protocol [4, 6, 24]. Mice with 4-day 4T1 flank tumors were either untreated (control), treated with CYP alone (on day 3), or with CYP + vaccine (Fig. 1). Vaccines used included 4T07-IL-2, irradiated 4T1-IL-2, and irradiated wild-type (wt) 4T1. Tumor growth curves in all treated groups were similar to controls [F(2,15)=1.20, p=0.33]. Additionally, vaccination without CYP pretreatment as well as multiple weekly vaccinations were ineffective (data not shown).
Fig. 1A–C.

Tumor vaccines are ineffective treatment for established 4T1 tumors. Mice with 4-day 4T1 flank tumors were either untreated (control), were treated with CYP (100 mg/kg, i.p.) alone, or CYP + vaccine. A Vaccinated with 2×106 irradiated 4T1 cells, s.c.; B vaccinated s.c. with 2×106 irradiated 4T1-IL-2 cells; C vaccinated with 1×106 live 4T07-IL-2 cells, s.c. Mean tumor area (± SE) was charted over time for each group. Numbers to the right indicate the number of mice with complete tumor regression per total number of mice in each group. None of the tumor vaccines had any significant effect on 4T1 tumor growth compared with that of control and CYP-alone groups [F(2,15)=1.20, p =0.33]
Regression of established 4T1 tumors is induced by 4T1-sensitized, B/I-activated lymphocytes
Popliteal DLN cells sensitized by 4T1 were treated in vitro with B/I for activation resulting in eight-fold expansion in culture. Adoptive transfer of these lymphocytes into mice bearing 4-day 4T1 flank tumors induced complete tumor regression in all six mice, whereas control mice and mice treated only with CYP exhibited progressive tumor growth (Fig. 2).
Fig. 2.

AIT with 4T1-sensitized, B/I-activated and expanded lymphocytes induced regression of 4-day 4T1 tumors. Mice with 4-day 4T1 flank tumors were either untreated (control), treated with CYP (100 mg/kg, i.p.) alone, or with CYP + AIT (10×106 cells) with 4T1-sensitized, B/I-activated and expanded lymphocytes. Mean tumor area (± SE) was charted over time for each group. Numbers to the right indicate the number of mice with complete tumor regression per total number of mice in each group. All mice treated with CYP + AIT exhibited complete tumor regressions, compared with no tumor regressions seen in control or CYP alone [F(2,16)=125.71, p<0.0001]
CYP + AIT with 4T07-IL-2–sensitized, B/I-activated and expanded lymphocytes induces regression of small and large established 4T1 tumors
Mice with established 4-day 4T1 flank tumors either were not treated (control) or were treated with CYP alone, CYP + AIT with 4T07-IL-2–sensitized, B/I-activated, expanded DLN lymphocytes or AIT alone (Fig. 3). All mice treated with CYP + AIT exhibited complete tumor regression, compared with no tumor regressions seen in control, CYP-alone, or AIT-alone groups [F(3,20)=47.28, p<0.0001]. Figure 4 shows the results of AIT with 4T07-IL-2–sensitized, activated, expanded DLN lymphocytes used to treat larger, 10-day 4T1 flank tumors. All mice receiving CYP + AIT had complete regression of their 10-day flank tumors, which was significantly different from the control and CYP-alone groups [F(2,15)=85.43, p<0.0001]. These results imply that AIT can be an effective treatment for even advanced stage tumors.
Fig. 3.

CYP + AIT with 4T07-IL-2 DLN cells, after B/I-activation and 10-day expansion, induced 4T1 tumor regressions in all mice treated. Mice with established 4-day 4T1 flank tumors either were not treated (control) or were treated with CYP alone, CYP + AIT with 4T07-IL-2–sensitized, activated, expanded DLN lymphocytes (10×106 cells), or AIT alone. Mean tumor area (± SE) was charted over time for each group. Numbers to the right indicate the number of mice with complete tumor regression per total number of mice in each group. All mice treated with CYP + AIT exhibited complete tumor regressions, compared with no tumor regressions seen in control, CYP-alone, or AIT-alone groups [F(3,20)=47.28, p<0.0001]
Fig. 4.

AIT is effective in treatment of larger, 10-day tumors. Mice with established 10-day 4T1 flank tumors either were not treated (control), were treated with CYP alone, or with CYP + AIT (50×106 cells) with 4T07-IL-2–sensitized, activated, expanded DLN lymphocytes. Mean tumor area (± SE) was charted over time for each group. Numbers to the right indicate the number of mice with complete tumor regression per total number of mice in each group. All mice treated with CYP + AIT exhibited complete tumor regressions, compared with no tumor regressions seen in control or CYP-alone groups [F(3,20)=47.28, p<0.0001]
Treatment of mice with 4T1 flank tumors with AIT using DLN cells sensitized in a tumor-bearing animal
Clinically, vaccine-sensitized T cells will not be able to be obtained from "naïve" humans. Rather, this approach will require that patients with established tumors be vaccinated and their T cells then harvested for AIT. To determine whether AIT would be successful in a more clinically relevant animal model, mice bearing 10-day 4T1 flank tumors were vaccinated in the contralateral footpad with 4T07-IL-2 cells. By the end of the 10-day sensitization period, the donor mice had 20-day 4T1 tumors, which measured on average 114.71 mm2 (SE = 7.76 mm2). Their popliteal DLN were harvested and activated per protocol and then used for AIT to treat 4-day 4T1 bearing mice. Figure 5 shows that despite sensitization in mice with advanced and probably metastatic disease (see below), CYP + AIT resulted in complete tumor regression in five of the seven mice treated. Groups treated with CYP + AIT with 4T07-IL-2–sensitized DLN from either naïve mice or tumor-bearing mice fared similarly, and both were significantly better than the control or CYP-alone groups [F(5,33)=19.52, p<0.0001].
Fig. 5.

AIT with 4T07-IL-2–sensitized DLN cells from 4T1 tumor-bearing donors. Mice with established 4-day 4T1 flank tumors either were untreated (control), or were treated with CYP alone, CYP + AIT (25×106 cells) with 4T07-IL-2–sensitized DLN cells from tumor-free donors, or CYP + AIT (25×106 cells) with 4T07-IL-2–sensitized DLN cells from 4T1 tumor-bearing donors. Mean tumor area (± SE) was charted over time for each group. Numbers to the right indicate the number of mice with complete tumor regression per total number of mice in each group. Groups treated with CYP + AIT with 4T07-IL-2–sensitized DLN from either tumor-free mice or tumor-bearing mice were not significantly different. However, these groups did significantly better than the control or CYP-alone groups [F(5,33)=19.52, p<0.0001]
To determine at what point in time 4T1 tumors spontaneously metastasize, mice with 4T1 flank tumors were euthanized at different times after s.c. inoculation, and the presence or absence of metastatic disease in the regional draining lymph nodes and in their lungs was evaluated using clonogenic assays. Of five mice with 14-day 4T1 tumors, two had evidence of metastases in their inguinal LN and three had lung metastases. Overall, by day 14, four of five mice had some form of metastases (regional and/or distant). By day 18 after tumor inoculation, 100% of the animals had evidence of lung metastases.
Adoptive transfer of 4T07-IL-2–sensitized DLN lymphocytes provides long-term immunologic memory and resistance to future 4T1 tumor challenge
To test whether immunologic memory developed in mice cured of 4-day 4T1 tumors by CYP + AIT, successfully treated mice ("prior AIT") and age-matched controls were challenged with 4T1 tumor cells 1 month after the complete regression of the initial tumor (Fig. 6). Mice in which 4T1 tumors had been cured by CYP + AIT were, indeed, completely resistant to growth of 4T1 injected 1 month later. Although the previous rechallenge experiments suggested that AIT results in long-term immunologic memory and resistance to tumor re-growth, it could be argued that immunization of host T cells by exposure to tumor was responsible for this result. To determine whether adoptively transferred B/I-activated lymphocytes are actually responsible for the long-term immunologic memory after AIT, their ability to confer resistance to tumor challenge in otherwise naïve mice was tested. Naïve mice that received adoptive transfer of 4T07-IL-2–sensitized, B/I-activated, expanded lymphocytes ("pre-treated") and age-matched controls were challenged s.c. with 4T1 tumor cells 1 month after adoptive transfer (Fig. 7). Pretreated mice were resistant to 4T1 tumor growth, which was significantly different from tumor growth in the control group [F(1,10)=198.32, p<0.0001].
Fig. 6.

Mice exhibiting complete 4T1 tumor regressions after treatment with B/I-activated 4T07-IL-2–sensitized lymphocytes, were resistant to a second 4T1 tumor challenge; 4T1 tumor-bearing mice successfully treated with CYP + AIT (prior AIT) and age-matched controls were challenged with 4T1 tumor cells 1 month after complete regression of initial tumor. Mean tumor area (± SE) was charted over time for each group. Numbers to the right indicate the number of mice with complete tumor regression per total number of mice in each group. In mice previously cured with AIT, 4T1 tumor growth was significantly different from tumor growth in age-matched controls [F(1,10)=373.36, p<0.0001]
Fig. 7.

Adoptive transfer of 4T07-IL-2–sensitized DLN lymphocytes protects naïve mice from future 4T1 tumor challenges. Naïve mice that had received 70×106 adoptively-transferred 4T07-IL-2–sensitized, B/I-activated, expanded DLN cells (pretreated) and age-matched controls were challenged, s.c., 1 month later with 4T1 tumor cells. Mean tumor area (± SE) was charted over time for each group. Numbers to the right indicate the number of mice with complete tumor regression per total number of mice in each group. Mice pretreated with AIT were resistant to 4T1 tumor growth, compared with growth in the control group [F(1,10)=198.32, p<0.0001]
In vivo antigen-specific T-cell sensitization is necessary for antitumor immunity
In other models, we have shown that antigen-specific in vivo priming of lymphocytes is a critical component of antitumor immunity [40]. To demonstrate this with the 4T1 model, popliteal LNs from naïve mice and LNs from the contralateral limb of sensitized mice (i.e., LN not draining the site of vaccination) were harvested and treated in vitro with B/I. These lymphocytes failed to expand in culture and were insufficient in number for AIT experiments. This was in spite of using four times the number of naïve LNs as 4T07-IL-2–sensitized LNs. These lymphocytes were, however, able to be compared with 4T07-IL-2–sensitized, B/I-activated lymphocytes for their in vitro IFN-γ response to tumor antigen. IFN-γ production was measured by ELISA in response to stimulation with nothing, irradiated 4T1, irradiated 4T07, or irradiated Meth A sarcoma cells for 24 h. Figure 8 shows that only the in vivo tumor-sensitized lymphocytes were able to release IFN-γ in response to antigen stimulation [F(11,23)=133.256, p<0.0001]. We also measured in vitro IFN-γ production by 4T1-sensitized, B/I-activated, expanded lymphocytes, using the same stimulators and conditions. The results were then compared with IFN-γ production by Meth A–sarcoma-sensitized, B/I-activated and expanded lymphocytes. 4T1-sensitized DLN cells produced high levels of IFN-γ in response to 4T1 or 4T07, but not Meth A cells (Fig. 9). Conversely, lymphocytes from Meth A–sensitized DLN activated with B/I, released IFN-γ only when restimulated with irradiated Meth A cells and not with 4T1 or 4T07 cells. This finding also correlates with the antigen specificity requirements seen in vivo, as described next.
Fig. 8.
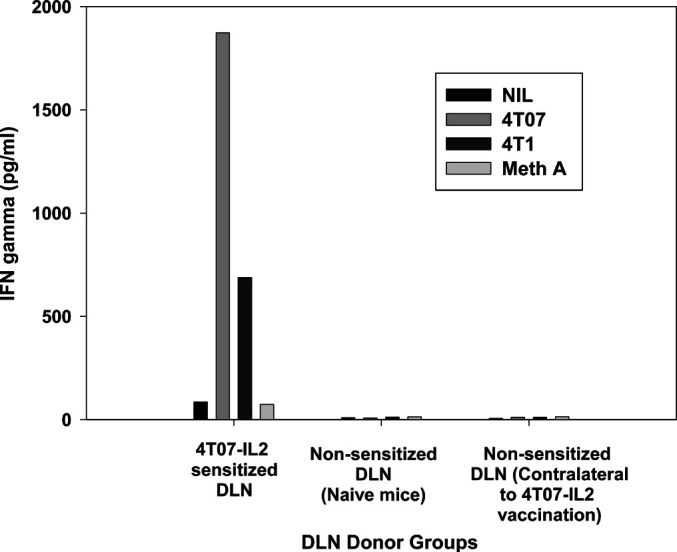
Comparison of in vitro IFN-γ production between sensitized and non-sensitized lymphocytes. Non-sensitized DLN cells from naïve mice and from the contralateral limbs of 4T07-IL-2–sensitized mice were compared with 4T07-IL-2–draining lymph node cells for their IFN-γ production response to 24-h stimulation with either nothing, irradiated 4T1, irradiated 4T07, or irradiated Meth A cells. Values are means of duplicate ELISA wells
Fig. 9.
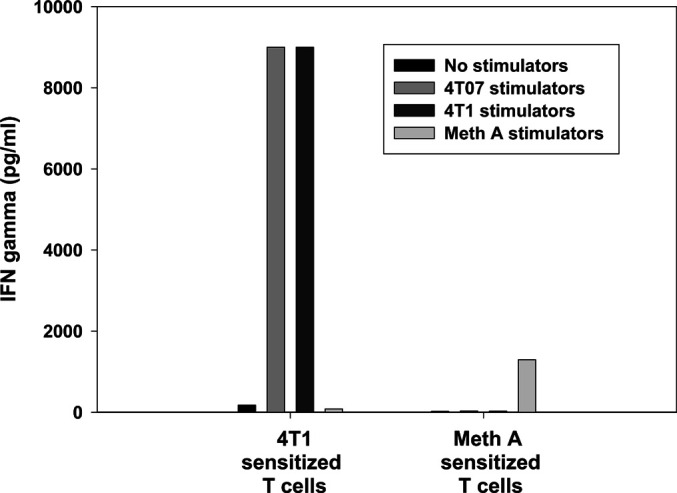
4T1-sensitized lymphocytes exhibit in vitro antigen-specific immunologic activity against 4T07 and 4T1 tumor cells. Lymphocytes that were 4T1-sensitized, B/I-activated, and expanded, released IFN-γ in response to 4T07 or 4T1, but not Meth A tumor cells whereas only Meth A tumor cells stimulated Meth A–sensitized, activated and expanded DLN cells to produce IFN-γ. Values are means of duplicate ELISA wells
It would be expected that vaccination with whole tumor cells would lead to sensitization of a polyclonal T-cell population due to the presence of multiple antigens on the tumor cells' surface. One of the antigens thought to be presented by 4T1 is the 9 amino acid AH1 peptide, which has also been shown to be expressed by CT26 colon carcinoma cells [23, 33]. To delineate further the specificity of 4T07-IL-2–sensitized T cells, we measured the amount of IFN-γ released in response to stimulation with 4T1, CT26, and AH1-pulsed splenocytes from naïve mice. Figure 10 shows that T cells sensitized in vivo with 4T07-IL-2, then activated with B/I and expanded in culture, released IFN-γ in response to 4T1, CT26, and especially to AH1-pulsed splenocytes. In contrast, stimulation with Meth A cells or unpulsed splenocytes did not stimulate IFN-γ release above background levels, once again demonstrating the antigen-specific nature of these T cells.
Fig. 10.

In vitro IFN-γ release in response to AH1-containing antigens. Lymphocytes that were 4T07-IL2–sensitized, B/I-activated, and expanded, released IFN-γ in response to 4T1, CT26, and AH1-pulsed splenocytes. Stimulation with Meth A or unpulsed splenocytes resulted in IFN-γ release that was not higher than background levels. Values are means of duplicate ELISA wells
Successful AIT using vaccine-sensitized, B/I-activated lymphocytes is dependent on antigen-specific sensitization
AIT with T cells primed in vivo with Meth A sarcoma cells did not result in any regressions of 4T1 tumors, despite in vitro activation with B/I and expansion in culture (Fig. 11A). Similarly, adoptive transfer of 4T07-IL-2–sensitized and activated T cells had no effect on established 4-day Meth A sarcomas (Fig. 11B). Conversely, AIT with 4T07-IL-2–sensitized cells completely cured 4T1 tumors (Fig. 11A) and AIT with Meth A–sensitized lymphocytes completely cured Meth A tumors (Fig. 11B; [F(3,22)=30.21, p<0.0001]), confirming the importance of antigen-specific sensitization.
Fig. 11A, B.

Successful AIT is dependent on the specificity of the initial T-cell-sensitizing antigen. A Mice with 4-day 4T1 tumors were either untreated, treated with CYP alone, or with CYP + AIT (15×106 cells) using 4T07-IL-2–sensitized, B/I-activated and expanded lymphocytes, or with Meth A–sensitized, B/I-activated and expanded lymphocytes. All mice treated with CYP + AIT with 4T07-IL-2–sensitized DLN exhibited complete tumor regression, which was significantly different from all other groups [F(3,22)=30.21, p<0.0001]; B mice with 4-day Meth A tumors were either untreated, treated with CYP alone, or with CYP + AIT (15×106 cells) using 4T07-IL-2–sensitized, B/I-activated and expanded lymphocytes or Meth A–sensitized, B/I-activated and expanded lymphocytes. In contrast to control, CYP alone, and CYP + AIT with 4T07-IL-2 DLN groups, all mice treated with CYP + AIT with Meth A–sensitized DLN had complete tumor regression [F(3,21)=9.88, p<0.0004]. Mean tumor area (± SE) was charted over time for each group. Numbers to the right indicate the number of mice with complete tumor regression per total number of mice in each group
Greater antitumor activity is seen with B/I activation of sensitized T cells
Next, to evaluate the importance of B/I activation, we eliminated exposure to B/I and cultured the VDLN cells in IL-2 alone. There was a marked reduction in the ability of these lymphocytes to grow in culture compared with cells activated with B/I (Fig. 12). In fact, these cells could not be maintained in culture beyond 3 to 4 days, after which all the cells were dead. Adoptive transfer of these 4T07-IL-2–sensitized but B/I-untreated lymphocytes to animals bearing 4-day 4T1 flank tumors resulted in temporary tumor regression, but this effect was not durable, resulting in prompt tumor regrowth in four of five mice (Fig. 13). This weak effect is particularly striking when one considers that it took a 21-fold greater number of donor animals to treat the same number of mice when B/I was omitted.
Fig. 12.

Cell expansion profile following ex vivo lymphocyte activation with B/I versus lymphocytes cultured in low-dose IL-2 only. DLN cells sensitized with 4T07-IL-2 were either pulsed with B/I and expanded in low-dose IL-2 (as described in "Material and methods") or cultured with low-dose IL-2 alone. Cells were counted every 2–3 days and the fold increases in cell number were calculated
Fig. 13.

Comparison of AIT with B/I-activated, expanded lymphocytes versus IL-2-only cultured lymphocytes for the treatment of 4-day 4T1 tumors. Mice with 4-day 4T1 flank tumors were either untreated (control), treated with CYP (100 mg/kg, i.p.) alone, or with CYP + AIT (15×106 cells) using 4T07-IL-2–sensitized DLN cells that were either pulsed with B/I and expanded in low-dose IL-2 (as described in "Material and methods") or cultured with low-dose IL-2 alone. Mean tumor area (± SE) was charted over time for each group. Numbers to the right indicate the number of mice with complete tumor regression per total number of mice in each group. All mice treated with CYP + AIT using B/I-activated DLN cells exhibited complete tumor regression, which was significantly different from all other groups [F(3,22)=29.36, p<0.001]. There is a nonsignificant difference between the group treated with CYP + AIT using nonactivated DLN cells (cultured in IL-2 alone) and the control or CYP-alone groups [F(2,16)=3.39, p=0.06]
Discussion
One of the primary goals of cancer immunotherapy is the development of effective vaccines. To reach this goal, a number of different approaches have been proposed, such as the use of genetically modified tumor cells or the use of dendritic cells pulsed with tumor-associated antigens (TAAs). Recent data suggest that both 4T1 and CT26 colon carcinoma cells express a TAA, AH1 peptide, derived from gp70, an endogenous murine leukemia virus envelope glycoprotein [23, 33]. A dendritic cell vaccine pulsed with this immunodominant epitope, AH1, was shown to protect naïve mice against CT26 tumor challenge, but cure of mice with established CT26 tumors has not been reported [33]. We were able to demonstrate antigen-specific IFN-γ release by B/I-activated 4T07-IL-2–sensitized T cells in response to 4T1, CT26, and AH1-pulsed splenocytes, supporting the reports that 4T1 and CT26 share the AH1 antigen. However, our preliminary studies showed that vaccination alone with AH1 peptide-pulsed dendritic cells protected mice against CT26, but was ineffective against tumor challenge with 4T1 (data not shown). This difference in protection may be due to differences in the level of AH1 expression in the two cell lines or perhaps, due to the greater immunogenicity of the CT26 tumor cell. Reduction of CT26 lung metastases with more complex dendritic-cell–based vaccines was also recently reported, but none of the mice were cured [19]. Although these vaccination techniques have demonstrated some success in protecting against a future tumor challenge, they have not been able to treat established murine or human cancers successfully. Other poorly immunogenic murine tumor models have also been relatively resistant to successful treatment with genetically engineered vaccines [8].
The 4T1 mammary carcinoma is a particularly aggressive and weakly immunogenic tumor that spontaneously metastasizes, and therefore, mimics many of the weakly immunogenic or nonimmunogenic human cancers. The poor immunogenicity of 4T1 carcinomas is evidenced by the inability of a triply transfected 4T1 tumor cell vaccine to induce complete tumor regression [30]. Pulaski et al. have shown that 4T1 pulmonary metastases after surgical excision of the primary tumor could be decreased by multiple weekly vaccinations with 4T1 tumor cells transfected with three genes, forcing expression of MHC class II, CD80, as well as a bacterial toxin, Staphylococcal aureus enterotoxin B superantigen, but once again these mice were not cured [30]. Others have studied treatment of 4T1 tumors with allogeneic cellular therapy [25]. Although allo-sensitized DBA/2 spleen cells, mismatched for minor histocompatibility antigens, could eliminate 4T1 cells in the lungs of BALB/c mice, this could only be shown by secondary transfer of lung cells to new hosts, since the treatment induced lethal graft-versus-host disease [26]. Furthermore, syngeneic BALB/c splenocytes from donors similarly immunized three times with irradiated 4T1 cells had no antitumor effect in this model. Not surprisingly, such a weakly immunogenic tumor does not initiate a potent enough immune response to cure an already established tumor. Here we show that even vaccination with IL-2 gene transfected 4T07 or 4T1 cells, with or without pretreatment with cyclophosphamide, cannot induce an effective antitumor immune response. Cyclophosphamide has been shown to augment delayed-type hypersensitivity responses induced by subsequent vaccination [4, 6, 24], but this did not make the tumor vaccines effective for treatment of established 4T1 tumors. 4T1 and 4T07-IL-2 tumor cells can, however, sensitize T cells to become antigen specific effectors; in vitro treatment of these same lymphocytes with B/I can significantly amplify the weak immunologic activity induced by vaccination alone, resulting in clinically evident tumor regression. In the context of AIT, particularly of late tumors, cyclophosphamide may augment the therapeutic effect by reversing the immunosuppressive effect of the tumor, as well as by providing "room" for expansion of the infused lymphocytes [41]. A direct effect of cyclophosphamide on the tumor may also have contributed to the therapeutic efficacy of the regimen used here. Overall, however, the effects of cyclophosphamide alone contributed only a minor antitumor effect, as demonstrated by the absence of a significant difference between the CYP-alone and control groups in all of the experiments. Even the small effect that was seen might be attributable to the immunomodulatory effects of cyclophosphamide, rather than to a direct antitumor effect [4, 6, 45]
Our data show that when used in an AIT protocol, regression of even a tumor as aggressive as 4T1 can be achieved. Not only were all six of six mice cured of their established tumors, but they were also able to reject a second challenge of 4T1, administered 1 month after treatment, suggesting that AIT provides the host with long-term immunologic memory. The induction of memory T cells from adoptively transferred DLN cells is further supported by the ability of AIT with vaccine-sensitized, B/I-activated lymphocytes to protect naïve mice against a 4T1 tumor challenge 1 month later, even though the mice were never exposed to tumor antigen prior to the challenge. When the more immunogenic and genetically modified cell type (4T07-IL-2) was used for vaccination, AIT with B/I-activated and expanded vaccine-draining lymphocytes can induce regression and cure of even large and advanced 4T1 tumors (tumor areas of 9–11 mm2 at the time of treatment). Successful treatment of larger and more advanced tumors is one of the most difficult goals of immune therapy.
To further demonstrate clinical applicability, we used tumor-bearing animals as DLN donors. These mice had 20-day 4T1 tumors in the flank by the time their VDLN were harvested. We have shown that five of five mice bearing 4T1 flank tumors had distant metastatic disease by day 18 after tumor inoculation. Therefore, despite sensitization within a metastatic and possibly immunosuppressive milieu, ex vivo activation with B/I, expansion and subsequent adoptive transfer into syngeneic hosts with 4-day 4T1 tumors resulted in complete tumor regression.
We also provide evidence that pharmacologic activation with bryostatin 1 and ionomycin is a necessary step for the development of lymphocytes with this potent antitumor activity. We have previously shown in the 4T07 tumor model that vaccine-sensitized DLN cells transferred directly, without in vitro activation, had no significant antitumor effect [11]. Pharmacologic treatment with B/I activates intracellular signaling pathways downstream from the T-cell receptor and therefore, mimics the T-cell activation events triggered by specific tumor cells or antigens, or by monoclonal antibodies to the TCR complex (e.g., anti-CD3). Ex vivo B/I activation and expansion in low-dose IL-2 prior to adoptive transfer of 4T07-IL-2–sensitized lymphocytes significantly amplified the potential antitumor activity of these "pre-effector" cells. However, sensitized lymphocytes cultured with IL-2 alone, without B/I exposure, did not expand in vitro, and their in vivo antitumor effects were short lived, resulting in tumor regrowth. The use of bryostatin 1 and ionomycin allows efficient ex vivo activation without the need for autologous tumor cells, monoclonal antibodies, or a known antigen.
Another critical component of a successful AIT protocol is in vivo priming of T cells using an antigen-specific sensitizing agent (i.e., vaccine). Pulsing non-sensitized lymphocytes from either naive mice or mice vaccinated in the contralateral limb did not result in demonstrable antitumor activity [40]. Lack of in vitro IFN-γ production by non-sensitized T cells in response to tumor antigen stimulation also highlights the need for prior antigen-specific in vivo T-cell sensitization. As expected, the antitumor activity of activated and expanded lymphocytes was highly dependent on the specificity of the initial antigen encounter. Thus, 4T07-IL-2–sensitized T cells have no effect on Meth A sarcomas and vice versa. However, T cells sensitized with 4T07-IL-2 do recognize the common shared surface antigens of the related 4T1 tumor cells.
T cells which secrete IFN-γ in response to tumor antigen have been associated with antitumor activity in vivo [3, 5, 32, 36, 48]. Based on our data, comparison of the in vitro and in vivo activities of DLN cells sensitized with either 4T07-IL-2 or 4T1, also suggests a correlation between the release of IFN-γ by B/I-activated and expanded T cells in response to tumor and their ability to mediate tumor regression in vivo. We have shown previously, in a murine sarcoma model, that administration of anti-IFN-γ antibodies abrogated the effect of AIT with B/I-activated T cells [43]. In agreement with that report and the current results, it has recently been reported, using a novel method for separating IFN-γ-producing T cells from nonproducers, that surface-bound IFN-γ+ T cells, but not IFN-γ- cells, were cytolytic and mediated rejection of CT26 colon carcinomas in BALB/c mice [5]. In contrast, Peng et al. have demonstrated that adoptively transferred T cells can induce in vivo regression of sarcoma even if both the adoptively transferred T cells and the adoptive host lack the capacity to produce IFN-γ [27]. Others have also shown a dichotomy between the requirement for IFN-γ release when comparing vaccine-induced immunity and adoptive immunotherapy of a murine melanoma [44]. More recently, it has been reported that the likelihood of successful adoptive immunotherapy of 4T1 mammary carcinoma was not greater using 4T1-sensitized T cells from STAT6-/- mice, even though the cells from these mice exhibited almost 25-fold greater IFN-γ release in response to tumor than similarly sensitized cells from wild-type mice [17]. Thus, despite our results shown here, it may well be that IFN-γ is not the critical mediator of antitumor immunity in the 4T1 model. Nevertheless, it does seem clear that T-cell production of this type 1 cytokine does at least correlate with in vivo antitumor efficacy.
Although this approach does involve ex vivo manipulation of cells, it is certainly no more cumbersome than dendritic cell approaches or the recently proposed use of T cells which have been genetically modified to express a particular TcR [20, 35]. Furthermore, we have shown elsewhere that adoptive transfer of 4T07-sensitized T cells can successfully mediate tumor regression after only 3 days in culture, eliminating the need for long-term culture and expansion of cells [11]. In fact, this approach might be combined with dendritic cell–based vaccines or with other vaccines to maximize antitumor efficacy. Based on these results, a series of clinical trials of adoptive immunotherapy with B/I-activated vaccine-DLN is being planned for patients with high-risk melanoma and breast cancer.
Acknowledgement
This work was supported by R01 CA48075 and T32 CA09573 grants from the NCI, NIH, and DHHS.
References
- 1.Aslakson Cancer Res. 1992;52:1399. [PubMed] [Google Scholar]
- 2.Awwad Immunol. 1988;65:87. [Google Scholar]
- 3.Barth J Exp Med. 1991;173:647. doi: 10.1084/jem.173.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bass Cancer Immunol Immunother. 1998;47:1. doi: 10.1007/s002620050498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Becker Nature Med. 2001;7:1159. doi: 10.1038/nm1001-1159. [DOI] [PubMed] [Google Scholar]
- 6.Berd Cancer Res. 1982;42:4862. [PubMed] [Google Scholar]
- 7.Cantrell Annu Rev Immunol. 1996;14:259. doi: 10.1146/annurev.immunol.14.1.259. [DOI] [PubMed] [Google Scholar]
- 8.Chamberlain Cancer Res. 1996;56:2832. [PMC free article] [PubMed] [Google Scholar]
- 9.Chang Int J Cancer. 2000;86:725. doi: 10.1002/(SICI)1097-0215(20000601)86:5<725::AID-IJC19>3.3.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 10.Chin Ann Surg Oncol. 2002;9:94. doi: 10.1245/aso.2002.9.1.94. [DOI] [PubMed] [Google Scholar]
- 11.Chin J Surg Res. 2001;98:108. doi: 10.1006/jsre.2001.6181. [DOI] [PubMed] [Google Scholar]
- 12.Chin Surg Forum. 2001;52:262. [Google Scholar]
- 13.Chou J Immunol. 1988;140:2453. [PubMed] [Google Scholar]
- 14.Dranoff, J Clin Oncol. 1998;16:2548. doi: 10.1200/JCO.1998.16.7.2548. [DOI] [PubMed] [Google Scholar]
- 15.Egilmez Cancer Res. 2000;60:3832. [PubMed] [Google Scholar]
- 16.Jaffee Ann N Y Acad Sci. 1999;886:67. doi: 10.1111/j.1749-6632.1999.tb09401.x. [DOI] [PubMed] [Google Scholar]
- 17.Jensen SM, Hu H-M, Fox BA. Increased IFN-γ secretion by adoptively transferred T cells does not correlate with enhanced anti-tumor therapy. Proceedings of the AACR. 2001;42:502. [Google Scholar]
- 18.Jiang Cancer Biother Radiopharm. 2000;15:495. doi: 10.1089/cbr.2000.15.495. [DOI] [PubMed] [Google Scholar]
- 19.Kershaw Cancer Res. 2001;61:7920. [PMC free article] [PubMed] [Google Scholar]
- 20.Kessels Nature Immunol. 2001;2:957. doi: 10.1038/ni1001-957. [DOI] [PubMed] [Google Scholar]
- 21.Li J Exp Med. 1996;183:639. doi: 10.1084/jem.183.2.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lohr Mol Ther. 2000;2:195. doi: 10.1006/mthe.2000.0114. [DOI] [PubMed] [Google Scholar]
- 23.Luznik L, Slansky JE, Jalla S, Borrello I, Levitsky HI, Pardoll DM, Fuchs EJ. Successful therapy of metastatic cancer using tumor vaccines in mixed allogeneic bone marrow chimeras. Blood. 2003;101:1645. doi: 10.1182/blood-2002-07-2233. [DOI] [PubMed] [Google Scholar]
- 24.Maguire J Invest Dermatol. 1967;48:39. doi: 10.1038/jid.1967.6. [DOI] [PubMed] [Google Scholar]
- 25.Morecki Cancer Res. 1998;58:3891. [PubMed] [Google Scholar]
- 26.Morecki J Immunother Emphasis Tumor Immunol. 2001;24:114. doi: 10.1097/00002371-200103000-00005. [DOI] [Google Scholar]
- 27.Peng J Immunol. 2000;165:7116. doi: 10.4049/jimmunol.165.12.7116. [DOI] [PubMed] [Google Scholar]
- 28.Proietti J Clin Invest. 1998;101:429. doi: 10.1172/JCI1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pulaski Cancer Res. 1998;58:1486. [PubMed] [Google Scholar]
- 30.Pulaski Cancer Res. 2000;60:2710. [PubMed] [Google Scholar]
- 31.Rodolfo J Immunol. 1996;157:5536. [PubMed] [Google Scholar]
- 32.Shu J Immunol. 1994;152:1277. [PubMed] [Google Scholar]
- 33.Slansky Immunity. 2000;13:529. doi: 10.1016/s1074-7613(00)00052-2. [DOI] [PubMed] [Google Scholar]
- 34.Soiffer Proc Natl Acad Sci USA. 1998;95:13141. [Google Scholar]
- 35.Stanislawski Nature Immunology. 2001;2:962. doi: 10.1038/ni1001-962. [DOI] [PubMed] [Google Scholar]
- 36.Tsung J Immunol. 1997;158:3359. [PubMed] [Google Scholar]
- 37.Tsung J Immunol. 1998;160:1369. [PubMed] [Google Scholar]
- 38.Tuting J Mol Med. 1997;75:478. doi: 10.1007/s001090050133. [DOI] [PubMed] [Google Scholar]
- 39.Tuttle J Surg Res. 1992;52:543. doi: 10.1016/0022-4804(92)90126-k. [DOI] [PubMed] [Google Scholar]
- 40.Tuttle Cancer Res. 1992;52:548. [PubMed] [Google Scholar]
- 41.Tuttle Ann Surg Oncol. 1994;1:53. doi: 10.1007/BF02303541. [DOI] [PubMed] [Google Scholar]
- 42.Tuttle J Immunother Emphasis Tumor Immunol. 1992;12:75. [Google Scholar]
- 43.Tuttle Cancer Res. 1993;53:833. [PubMed] [Google Scholar]
- 44.Winter J Immunol. 2001;166:7370. doi: 10.4049/jimmunol.166.12.7370. [DOI] [PubMed] [Google Scholar]
- 45.Wise Cancer Immunol Immunother. 1988;27:191. doi: 10.1007/BF00205439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wittig Hum Gene Ther. 2001;12:267. doi: 10.1089/10430340150218404. [DOI] [PubMed] [Google Scholar]
- 47.Zitvogel J Immunol. 1995;155:1393. [PubMed] [Google Scholar]
- 48.Zou Int Immunol. 1995;7:1135. doi: 10.1093/intimm/7.7.1135. [DOI] [PubMed] [Google Scholar]