

Abstract
B-lymphoma cells express a highly tumor-specific antigen, monoclonal Ig, which is a promising target for immunotherapy. Previous work has demonstrated that B-lymphoma cells spontaneously process their endogenous monoclonal Ig and present variable (V) region peptides (Id-peptides) on their MHC class II molecules to CD4+ T cells. Id-specific CD4+ T cells protect mice against B-lymphoma cells in the absence of anti-idiotypic antibodies. The molecular mechanism by which Id-specific CD4+ T cells kill B-lymphoma cells is hitherto unknown. We here demonstrate in an Id-specific T-cell receptor (TCR)–transgenic mouse model that Id-specific CD4+ T cells induce apoptosis of Fas+ B-lymphoma cells in vitro by FasLigand (FasL)–Fas interaction. Moreover, the rare B lymphomas that had escaped rejection in TCR-transgenic mice had down-regulated their sensitivity to Fas-mediated apoptosis. Although these results suggest that FasL-Fas interaction is important, Id-specific CD4+ T cells could eliminate Id+ B-lymphoma cells in vivo by other mechanisms, since three independent ways of blocking FasL-Fas–mediated killing failed to abrogate tumor protection in TCR-transgenic mice. These results suggest that there are several redundant pathways by which Id-specific CD4+ T cells eliminate Id+ B-lymphoma cells in vivo, of which FasL-Fas interaction is only one.
Keywords: Apoptosis, B lymphoma, CD4+ T cells, Idiotype, Immunotherapy, TCR-transgenic mouse model
Introduction
Traditionally, cytotoxic CD8+ cells have been thought to be the most important T cells in immunotherapy of cancer. However, CD4+ T cells are also effective against tumors [1–3] and are receiving increasing attention [4]. Apparently, both Th1 and Th2 cells are effective [5–8]. CD4+ T cells have been reported to kill tumor cells by a variety of mechanisms like induction of cytotoxic eosinophils and macrophages [5], through an eotaxin and STAT6-dependent mechanism [7], perforin [8], and through IFN-γ–mediated [9, 10] or IL-4–mediated [11] destruction of tumor vasculature.
Fas (CD95) is one of several members of the TNF receptor family and is expressed on the cell surface of a number of cell types including B-lymphoma cells. When Fas on a target cell is ligated by FasLigand (FasL), caspases are activated, and the target cell undergoes apoptosis [12]. Activated T cells, including cytotoxic Th1 cells, transiently express FasL on their cell membrane and kill Fas+ targets [13]. Circumstantial evidence suggests that FasL-Fas interaction is of importance in immunosurveillance of tumors. Thus, several reports have described mutations in the CD95 gene in different human lymphomas and leukemias, suggesting that tumors escape elimination by modulation of Fas expression or function [14–20]. Moreover, tumor cells that express FLIP, an inhibitor of caspase-8 involved in Fas-mediated apoptosis, have increased tumorigenicity [21, 22]. Finally, patients with autoimmune proliferative syndrome, caused in 80% of cases by a deficiency in the Fas gene, have an increased incidence of lymphomas. However, in these cases [14, 15, 17–19, 21, 22], the effector cells involved in generation of Fas− tumor variants have not been defined. We have here explored the role of FasL-Fas in CD4+ T-cell–mediated killing of B-lymphoma cells in an established tumor model.
B-lymphoma cells produce a highly tumor-specific antigen, monoclonal Ig, that expresses unique antigenic determinants called idiotopes (Id) in its variable (V) regions [23]. Immunization with monoclonal Ig can induce Id-specific antibodies [24] and Id-specific T cells [25], and early animal experiments suggested that both these arms of adaptive immunity could have an anti-B-lymphoma effect [26, 27]. Id vaccination has moved into clinical trials of follicular B-lymphoma patients [28–31], but there is a need to better define the mechanism of action in order to improve its efficacy. Recent experiments in mouse models indicate that Id-specific antibodies alone [32] and Id-specific CD4+ T cells alone [33] can confer tumor protection, suggesting that in many cases of B lymphomas, humoral and cellular Id-specific immunity probably work in synergy to afford protection.
Id-specific CD4+ T cells could cure mice of B lymphomas even if they were transferred as late as 10–17 days after tumor cell injection [33]. These experiments were performed in a T cell receptor (TCR)–transgenic model in which MHC class II–restricted Id-specific CD4+ T cells recognize a unique CDR3 Id-peptide of a particular λ2315 L chain expressed in transfected B-lymphoma cells. The B-lymphoma cells express the λ2315 L chain in their B-cell receptor (BCR) and at the same time, they constitutively present the Id-peptide on their class II molecules to cloned Id-specific CD4+ cells which become activated [34, 35]. Although Id-specific Th1 and Th2 cells kill B-lymphoma cells in 51Cr-release assays and growth inhibition assays [2, 6], and in apoptosis assays [33], the molecular mechanism of cytotoxicity is unknown. It is clear, however, that naïve Id-specific CD4+ T cells are unable to kill B-lymphoma cells while previously stimulated effector memory CD4+ T cells (Th1>Th2) are effective [33]. We here demonstrate that Id-specific Th1 cells induce apoptosis in Fas+ Id+ B-lymphoma cells by FasL-Fas interaction in vitro but that this is only one of several mechanisms of tumor rejection in vivo.
Materials and methods
Mice
Mice transgenic for an Id-specific αβ TCR (Tg46) on a BALB/c background have been described [36], as have Id-specific TCR-transgenic mice homozygous for the scid mutation on a C.B-17 background (IgH-congenic with BALB/c). The Id-specific TCR recognizes residues 91–101 of the λ2315 Ig L chain, presented on the MHC class II I-Ed molecule [37, 38]. Both lines of TCR-transgenic mice are hemizygous for the transgenes and were bred by (BALB/c × TCR-transgenic) and (SCID × TCR-transgenic SCID) crosses, respectively. Mice hemizygous for TCR transgenes and homozygous for the gld mutation, a single mutation in the FasL gene destroying its function [39, 40], were made by first producing F2 mice starting from a (TCR-transgenic × C3H gld) cross. F2 mice homozygous for H-2d and gld were selected and typed for TCR-transgenic status. The line was maintained by crossing TCR-transgenic with nontransgenic mice, yielding TCR-transgenic gld and gld littermates. TCR-transgenic status and MHC haplotypes were determined by staining of blood lymphocytes using the clonotype-specific mAb GB113 [41] and MHC allele-specific mAbs from Pharmingen; gld status was determined by a ligase assay kindly performed by Dr Z. Dembic, Oslo [42]. Mice were of SPF standard. Experiments were performed according to Norwegian governmental and institutional guidelines.
Cell lines
The following cell lines have been described: the mouse T-lymphoma cell line L5178Y and the FasL-transfected derivative called BALB FasL/L5178Y [43]; the Fas-negative L1210 mouse leukemic cell and its Fas-transfected derivative called L1210Fas [44]; the F9A.15.3.19 (F9) cell line which represents A20 BALB/c (H-2d ) B-lymphoma cells transfected with the λ2315 gene [34, 35]; the F9R.3.1 (F9R) cell line which represents F9 insensitive to ligation by FasL—this cell line was obtained by limiting dilution of F9 in the presence of soluble FasL/CD8 (B. Bogen, unpublished data); the F55B17Ny (F55) cell line which represents A20 cells transfected with the vector alone (pSV2neo)—F55B17Ny is a subline of F55B17 [35], selected for high Fas expression. F9 was further retrovirally transduced with the green fluorescent protein (GFP)–expressing bicistronic MSCV retroviral vector into which the long form of human cellular FLIP had been cloned [45]. GFP-positive clones were obtained by limiting dilution (F9MFL7.2, F9MFL10.7). F9 transduced with empty MSCV vector were similarly obtained (F9M7.1, F9M8.18).
Soluble FasL/CD8
The BALB/c FasL-hCD8α/PSG5 vector was constructed by transferring a cDNA insert encoding the BALB/c FasL-hCD8α fusion protein [43] from the pMKITneo vector into the EcoRI-XhoI sites of pSG5 vector (Stratagene, La Jolla, CA, USA). pSG5 was transiently transfected into COS-7 cells using Lipofectamine (Invitrogen, San Diego, CA, USA). Supernatant (SN) obtained 24 h later contained mouse FasL—human CD8 fusion protein. Control mock SN was from COS cells exposed to the transfection procedure in the absence of vector.
[3H]TdR-release assay
This assay was essentially performed as described previously [46]. Briefly, target cells were cultured at 2.5×105/ml in complete tissue culture medium overnight with the addition of 2.5–5 μCi [3H]TdR/ml (5.0 Ci/mmol; Amersham, UK). Labeled cells were washed and placed in microtiter wells (1×104/w) that already contained agonistic Jo2 anti-Fas mAbs or soluble FasL/CD8 (flat-bottomed microtiterplates were used), or FasL/L5178Y transfectant or T cells (round-bottom wells were used, contents were centrifuged at 400 g for 2 min prior to incubation). MFL1 and UC8 mAbs were added to effector cells 10 min prior to adding target cells. Assays were usually run in pentduplicates. Duration of assays is indicated in “Results.” Cells were harvested using a Micromate 196 (Packard, Canberra, Australia). Percentage of thymidine release was calculated as [(count control target cells) − (count apoptotic target cells)]/[count control target cells]×100. Mean ± SEM are given.
Id-specific CD4+ T cells
Naïve Id-specific CD4+ T cells were isolated from lymph node (LN) and spleen of TCR-transgenic or TCR-transgenic SCID mice by anti-CD4-coated magnetic beads and detachment. To obtain Th1 or Th2 cells, LN cells from TCR-transgenic or TCR-transgenic SCID mice (5×105/ml) were cultured with BALB/c spleen cells (2.5×106/ml), synthetic 91–107 Id (λ2315) peptide (1 μg/ml). For Th1 polarization, recombinant mouse IL-12 (2 ng/ml; R&D Systems, Minneapolis, MN, USA) and anti-IL-4 mAb (11B11, 1 μg/ml) were added to the cultures. For Th2 polarization, IL-4 (20 U/ml) was added. T cells were expanded after several days and used after one or two 10-day cycles of stimulation. The Id (λ2315)–specific 7A10B2 Th1 clone has been described previously [38]. Complete tissue culture medium was RPMI 1640 with 10% FCS (Gibco), 1% NEAA (×100), 0.5 mM monothioglycerol, 0.5 ml gentamicin sulfate, and defined supplements.
T-cell proliferation assay
Assays were essentially performed as described previously [36]. A20-derived transfectants were mitomycin C treated [38] and titrated (5×102−5×105). Responder Id-specific T cells were used at 2×104/well (for clones and T-cell lines) or 105/well (for naïve T cells). Cultures were pulsed with [3H]TdR after 48 h (some assays with T-cell clones or T-cell lines) or 72 h and harvested 12 h later. As a positive control, a maximal stimulatory concentration of synthetic 91–107(λ2315) peptide 4 μg/ml was added to a parallel set of cultures. Mean ± SEM are given.
Antibodies and conjugation
The B-cell hybridoma UC8-1B9 producing hamster IgG anti-TNP and the B-cell hybridoma MFL1 producing hamster IgG antimouse FasL have been described previously [43]. Antibodies were affinity purified from SN on protein G-Sepharose, and biotinylated by standard procedures. Purified hamster antimouse Fas mAb Jo2 was from PharMingen (San Diego, CA, USA).
Staining and flow cytometry
The following mAbs were used for staining: biotinylated anti-Cλ2/3 (2B6 [47]), biotinylated hamster anti-TNP (UC8), biotinylated anti-I-Ek (14–4-4S; ATCC, Manassas, VA, USA), and biotinylated anti-H2 Kk (11–4.1; ATCC) were all produced in our laboratory. Biotinylated anti-Fas (Jo2), PE-labeled anti-CD80, PE-labeled anti-CD86, and PE or biotin-labeled isotype-matched control mAbs were from PharMingen. Biotinylated antibodies were detected with streptavidin-Cy-Chrome (PharMingen). Annexin V/propidium iodide staining was performed according to the supplier of the kit (Boehringer Manheim). Stained cells were analyzed on a FACSCalibur (Becton Dickinson, Mountain View, CA, USA).
Tumor challenges, survival curves, statistics
Mice were injected s.c. interscapularly with the indicated number of tumor cells as described previously [2], and monitored weekly for tumor development. Tumor sizes (diameters) were estimated by palpation and by the use of a caliper. Once established, tumors always progressed. A tumor of 3 mm was scored as a tumor take. Percentage of tumor avoidance (survival curves) and statistical comparison between survival curves were calculated by use of Graph Pad Prism 3.0 software (San Diego, CA, USA). In some experiments, 100 μg of the mAbs MFL1 or UC8 in PBS was injected i.p. every 2–3 days for 6 weeks, starting 1 day prior to tumor challenge.
Results
Id+ A20 B-lymphoma cells (F9) express Fas and are sensitive to induction of apoptosis by Fas ligation
In the λ2315 model, Id-specific CD4+ T cells recognize amino acids (aa) 91–101 of the λ2315 L chain derived from the BALB/c MOPC315 myeloma; the Id-peptide is presented on the I-Ed MHC class II molecule [37, 38]. The particular Id-peptide is very rare as its expression depends on mutations in codons 94, 95, and 96 in the BALB/c Vλ2 gene segment [38]. A20 BALB/c B-lymphoma cells transfected with the λ2315 gene spontaneously process the L chain and present the Id-peptide to CD4+ T cells that in turn kill the lymphoma cells [34, 35].
The λ2315 (Id)–transfected A20 (F9) cells express Fas (Fig. 1a). Moreover, F9 cells underwent apoptosis upon ligation of Fas by agonistic hamster antimouse Fas Jo2 mAb (Fig. 1b), mouse FasL-human CD8 fusion protein (Fig. 1c, d), and mouse FasL-transfected L5178Y cells (Fig. 1e, f). Induction of apoptosis was detected in several different apoptosis assays, including release of nucleosomal DNA from [3H]TdR-labeled lymphoma cells ([3H]TdR-release assay) (Fig. 1b, d, e, f) and exteriorization of phosphatidyl serine detected by staining with annexin V (Fig. 1c; and data not shown). Killing could be inhibited by hamster antimouse FasL mAb MFL1 (Fig. 1f). Apoptosis of F9 exposed to the various Fas ligands could easily be detected after 3 h and increased with time (Fig. 1c, d, e; and data not shown). The experiments described above included Fas-transfected L1210 and wild-type L1210 cells as satisfactory positive and negative controls (data not shown). These results demonstrate that λ2315–transfected F9 cells and vector alone (pSVneo)-transfected F55 cells (data not shown) express Fas and quickly undergo apoptosis when exposed to a variety of Fas ligands in several different assays.
Fig. 1a–f.
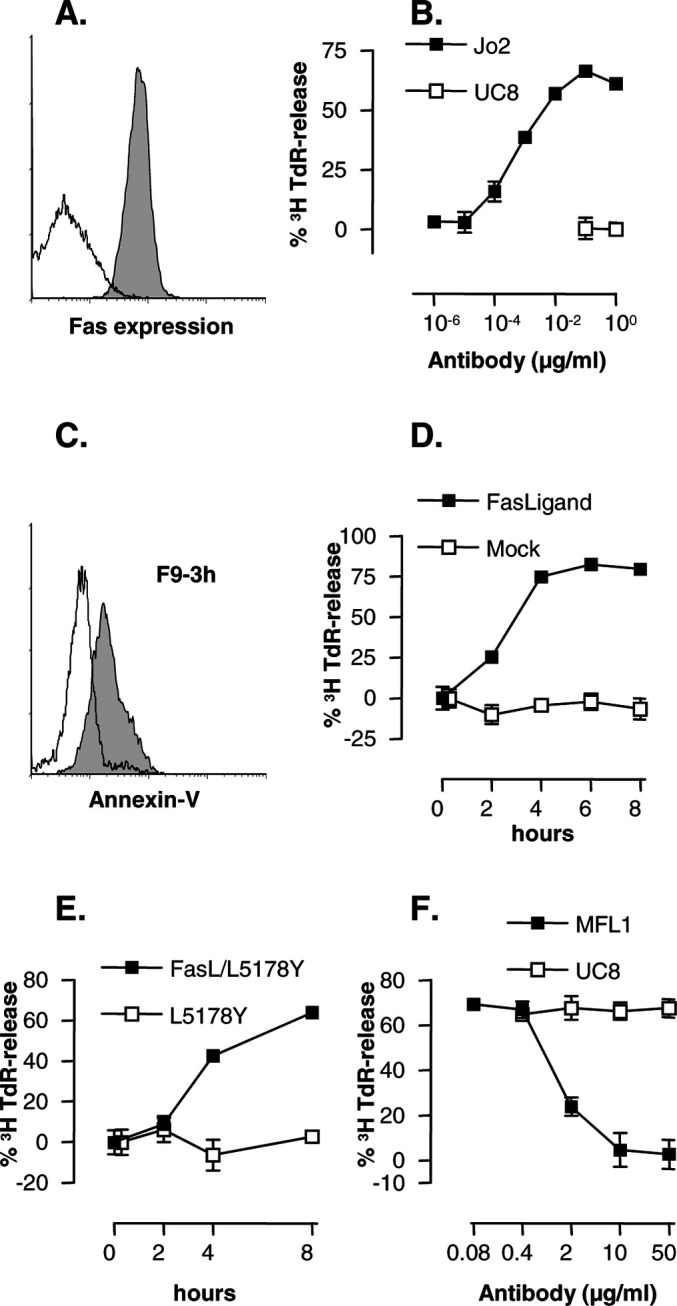
Id+ F9 cells express Fas and rapidly undergo apoptosis when exposed to a variety of ligands that bind Fas. a Staining of F9 cells with anti-Fas mAb Jo2 (shaded histogram) and control mAb UC8 (open histogram). b Anti-Fas mAbs induce apoptosis of [3H]TdR-labeled F9 cells and release of labeled nucleosomal fragments of DNA in a 6-h assay. c Annexin V staining of F9 cells exposed to FasL-CD8 fusion protein (shaded) or SN from mock-transfected COS cells in a 3-h assay (open). d Kinetics of apoptosis of F9 cells induced by FasL-CD8 fusion protein in a [3H]TdR-release assay. e Kinetics of apoptosis of F9 induced by FasL-transfected L5178Y (E/T=10:1) in a [3H]TdR-release assay. f Anti-FasL mAb MFL1 inhibits killing of [3H]TdR-labeled F9 target cells exposed to mouse FasL-transfected L5178Y (E/T=10:1) in a 6-h assay
Id-specific Th1 cells kill Id+ F9 cells in a FasL-dependent manner
We have previously shown that memory, but not naïve, Id-specific T cells kill Id+ F9 cells. Moreover, Th1-polarized cells became increasingly cytotoxic with repeated stimulation, while the opposite was the case for polarized Th2 cells. Killing was Id-specific because T cells did not kill F55 transfected with empty pSV2neo vector alone [33]. Given that F9 was so sensitive to apoptosis induced by Fas-ligation (Fig. 1), we tested if Th1 cells killed F9 cells by a FasL-Fas–dependent mechanism. Indeed, apoptosis induced by the Th1 clone 7A10B2 or by Th1-polarized cells from TCR-transgenic mice was almost completely inhibited by an anti-FasL mAb but not by a control hamster mAb (Fig. 2a, b). Killing effected by Th2-polarized cells was less pronounced and less efficiently inhibited by anti-FasL mAb (Fig. 2c). This suggests that when only T cells and B-lymphoma cells are present in vitro, killing through FasL (T cell)–Fas (lymphoma cell) interaction is of overriding importance for Th1 cells but less so for Th2 cells.
Fig. 2a–c.

Induction of apoptosis of F9 cells by Id-specific Th1 cells is inhibited by anti-FasL mAb. [3H]TdR-labeled F9 target cells were incubated with cloned Id-specific Th1 7A10B2 cells (a), or day 10 Th1-polarized (b), or Th2 polarized (c) cells from Id-specific TCR-transgenic mice in the presence of anti-FasL mAb MFL1 (solid squares) or control hamster mAb UC8 (open squares) or medium (gray column). In c, only the highest mAb concentration (50 μg/ml) was tested. E/T ratio was 10:1 in all experiments
F9 tumors that have escaped rejection in TCR-transgenic SCID mice have reduced sensitivity to FasL
Previous results have shown that TCR-transgenic SCID mice injected with low numbers of F9 cells are completely protected against tumor development [33]. However, when the tumor cell inoculum was increased to 1.25×106 cells, delayed tumors arose in about 30% of mice [33]. Three such F9 tumors arising in TCR-transgenic SCID mice after high-dose challenge were excised, grown as single cell suspensions, and analyzed within 1–2 weeks. Parental F9 served as a positive control while a FasL-resistant variant, F9R (B. Bogen, unpublished data), served as a negative control. One of the ex vivo tumors (7373), and 3/3 clones derived from it by limiting dilution, had completely lost sensitivity to agonistic anti-Fas mAb (data not shown). The two other ex vivo tumors (7372 and 7375) had reduced sensitivity to anti-Fas mAb (data not shown). When 7375 was cloned, one out of two clones was fully sensitive to Fas ligation while the other had a strongly reduced sensitivity. This indicates that reduced but not abolished sensitivity to FasLigation of these two ex vivo tumors was due to clonal heterogeneity. The results were significantly different for nine F9 tumors that arose in SCID mice: ex vivo tumor cell lines were all (9/9) fully sensitive to anti-Fas mAb (data not shown) (p=0.0045). Because the effect of Jo2 anti-Fas mAb depends on FcγRIIb expression on A20 cells, these results could be explained by loss of this receptor in tumor escapees in TCR-transgenic SCID mice. This possibility is ruled out since the 7373 variant was insensitive to FasL/L5178Y effector cells (data not shown). Since Fas-expression in the 7373 escape was not significantly reduced compared to the original F9 (data not shown), the insensitivity to Fas ligation is not due to failure of Fas-expression but rather some signaling defect.
Anti-FasL mAb does not abrogate protection of TCR-transgenic SCID mice against F9
Because anti-FasL MFL1 mAb completely inhibited Th1 killing of F9 in vitro (Fig. 2), we next proceeded to test if injection of 100 μg MFL1 mAb every 2–3 days for 6 weeks, starting the day before F9 injection, could abrogate the protection against an F9 challenge in TCR-transgenic SCID mice (Fig. 3). In agreement with previous results [33], about two thirds of SCID mice injected with 1.25×105 F9 cells developed tumors, while TCR-transgenic SCID mice were 100% protected, either in the presence or absence of the control UC8 anti-TNP hamster IgG mAb (Fig. 3; p=0.0016). Anti-FasL mAb MFL1 did not abrogate tumor protection against F9, because TCR-transgenic mice were still 100% protected (Fig. 3). This result suggested that Id-specific CD4+ T cells could use other mechanisms than FasL-Fas interaction to eliminate B-lymphoma cells in vivo.
Fig. 3.

Anti-FasL mAb does not inhibit tumor protection mediated by Id-specific CD4+ T cells in vivo. Id-specific TCR-transgenic SCID mice were injected with purified anti-FasL mAb MFL1 (open squares) or control mAb UC8 (solid squares) 100 μg i.p. every 2–3 days for 6 weeks. Mice received 1.25×105 F9 cells 1 day after first injection of mAb and were followed for tumor development. As a control for tumorigenicity, the same number of F9 cells were also injected in SCID mice (open circles)
TCR-transgenic mice homozygous for the gld mutation are resistant against F9 but not against F55
To design an experiment where tumor protection could be tested in the complete absence of FasL in vivo, we bred Id-specific TCR-transgenic mice homozygous for the gld mutation. The gld mutation is a single nucleotide substitution that causes a T to C missense point mutation, changing Phe273 to Leu. This results in a normally expressed, but nonfunctional FasL molecule [39, 40]. The TCR-transgenic gld mice, as well as gld littermates, were established from F2 mice originating from a (TCR-transgenic × C3H gld) cross (see “Materials and methods”). These mice have considerable amounts of background genes from C3H even though they were selected to be H-2d homozygous. It was therefore important to test if F9 was tumorigenic in gld littermates, as tumor cells might be rejected due to responses against BALB/c minor histocompatibiliy antigens not expressed by the challenged mice. As can be seen from Fig. 4a, tumor take in nontransgenic littermate gld mice was 50%, which is less than that obtained in normal BALB/c mice (80%) [2]. This result suggests that BALB/c minor antigens on tumor cells could cause some tumor resistance, nevertheless, tumor take was considerable. By contrast to gld mice, TCR-transgenic gld mice were 100% protected against F9 (Fig. 4a). This level of protection was significant and was Id-specific because gld and TCR-transgenic gld littermates were equally susceptible to tumor induction by the Id-negative F55 cells (Fig. 4b). These results conclusively demonstrate that FasL is not required for Id-specific protection against F9 in vivo.
Fig. 4a–c.

FasL-deficient Id-specific TCR-transgenic mice are protected against Id+ F9, but not against Id− F55 B-lymphoma cells. FasL-deficient TCR-transgenic gld and nontransgenic gld littermates were H-2d but had a mixture of BALB/c and C3H background genes. Mice were challenged with 1.25×105 F9 (a) or F55 (b) cells and monitored for tumor development. c Polarized Id-specific Th1 cells from FasLigand deficient mice (Th1gld, solid squares) are less cytotoxic against Id+ F9 cells than are Id-specific Th1 cells from FasL sufficient mice (Th1, open squares)
This somewhat surprising result prompted us to test whether polarized Th1 cells generated from TCR-transgenic gld mice were cytotoxic against F9 in vitro. A number of experiments with polarized T-cell lines demonstrated that FasL-deficient Th1 cells were cytotoxic but much less so than Th1 cells from FasL-expressing TCR-transgenic SCID mice (Fig. 4c). We conclude that TCR-transgenic gld Th1 cells kill F9 in a FasL-independent manner. It may be that in the absence of FasL, other yet-undefined killing mechanisms become more pronounced.
B-lymphoma cells transduced with human FLIPL are resistant to induction of apoptosis by several different Fas ligands and are resistant to killing by an Id-specific Th1 clone in vitro
The experiments of Figs. 3 and 4 exclusively focused on FasL-Fas interaction as a mechanism for killing of B-lymphoma cells. However, because several different TNF–TNF receptor family members like FasL-Fas, TNF-TNF-receptor, and TRAIL-DR5 converge on signaling through caspase-8 [48], we sought to more broadly inhibit apoptosis effected through the caspase-8 checkpoint. To this end, we investigated if the long isoform of human cellular FLIP (cFLIPL , FLICE [FADD (Fas-associated death domain)–like IL-1β converting enzyme]-inhibitory protein), shown to inhibit FasL-Fas induced apoptosis in mice [49], could inhibit killing by Id-specific Th1 cells when expressed in F9 cells. FLIPL contains two DEDs and a caspase-like domain with significant homology to caspase-8/caspase-10. FLIP prevents association of caspase-8 with the adaptor molecule FADD through DED-DED interactions and thus inhibits apoptosis. FLIPL is the most potent form of FLIP, probably because of the additional ability to heterodimerize to the protease domain of caspase-8 and to preclude activation of caspase-8. In addition, other binding partners have been reported, such as TRAF-1 and TRAF-2, which are components of the TNF-R1 and TNF-R2 signaling complexes (reviewed in [50]). The TRAIL-R signaling pathway could be even more effectively blocked by FLIP than the Fas signaling pathway [51]. F9 cells were transduced with either the retroviral MSCV vector alone (clones F9M7.1 and F9M8.18) or MSCV with the long version of human FLIP (clones F9MFL10.7 and F9MFL7.2). F9 cells transduced with the vector alone were readily killed by FasL-transfected L5178Y cells in a 6-h [3H]TdR-release assay, whereas FLIPL-transduced F9 cells were resistant (Fig. 5a; and data not shown). Similar results were obtained with other Fas ligands, like agonistic anti-Fas mAb and FasL-CD8 fusion protein (data not shown). Next, we tested the killing potential of 7A10B2, an Id-specific Th1 clone which efficiently kills the Id+ λ2315–expressing F9 but not the Id− F55 control transfectant [35]. As expected, 7A10B2 killed F9 cells transduced with MSCV vector alone (Fig. 5b). However, it did not kill F9 cells transduced with FLIPL (Fig. 5b). We next tested if killing of vector-only–transduced F9 by 7A10B2 could be inhibited by an inhibitory anti-FasL mAb MFL1. As expected from Fig. 2, MFL1 completely inhibited killing of vector-only–transduced F9 cells, while an isotype-matched control mAb UC8 did not (data not shown). In conclusion, 7A10B2 Id-specific Th1 cells kill F9 cells in vitro through a FasL-Fas interaction that is completely inhibited by high expression of FLIP.
Fig. 5a–c.

F9 cells transduced with the long form of human FLIP are resistant to apoptosis induced by ligation of Fas. a F9 cells transduced with FLIPL (F9MFL10.7) or MSCV vector alone (F9 M8.18) were [3H]TdR-labeled and used as targets for L5178Y and FasL-transfected L5178Y effector cells in a 6-h [3H]TdR-release assay for detection of apoptosis. b Killing of F9 B-lymphoma cells by Id-specific Th1 cells is inhibited by expression of FLIP. F9 transduced with MSCV vector alone (F9M7.1, F9M8.18) or FLIPL (F9MFL7.2, F9MFL10.7) were [3H]TdR-labeled and used as targets for cloned Id-specific Th1 cells (7A10B2) in a 6-h assay. c Proliferation of TCR-transgenic LN (left) and polarized Th2 cells (right) cultured with FLIPL transfectants (F9MFL10.7, F9MFL7.2) or control-vector-alone transfectants (F9M7.1, F9M8.18) as APC
B-lymphoma cells transduced with FLIPL are potent presenters of Id
The results described above could be explained by FLIPL-transduced F9 cells not being able to stimulate Id-specific T cells. We did a number of experiments to rule out this possibility. Parental F9 and retrovirally transduced F9 with or without FLIP expression displayed approximately the same amount of CD80, CD86, BCR with λ2315 L chains, Fas (CD95), MHC class II I-Ed, and secreted roughly the same amount of λ2315 Ig (data not shown). Thus, they were expected to be equally good stimulators of Id-specific CD4+ T cells. Indeed, retrovirally transduced F9 cells with or without expression of FLIPL were on average equally potent at inducing proliferation of LN cells or Th2 cells from TCR-transgenic mice (Fig. 5c). Thus, expression of FLIP did not influence the ability of F9 B-lymphoma cells to stimulate Id-specific CD4+ T cells.
TCR-transgenic SCID mice are protected against challenges with FLIPL-expressing B-lymphoma cells
We have recently described how Id-specific CD4+ T cells protect TCR-transgenic SCID mice from tumor development after challenge with Id+ F9 cells but not Id− F55 cells [33]. Because Id-specific T cells did not kill FLIP-transduced F9 cells in vitro, we expected that TCR-transgenic mice would not be protected against FLIPL-transduced F9 cells. To our surprise, TCR-transgenic SCID mice proved to be protected against FLIPL-transduced F9 cells to the same extent as F9 transduced with vector alone (Fig. 6a). Moreover, the level of protection closely resembled that previously observed for ordinary F9 cells [33]. Thus, expression of FLIPL did not confer a survival advantage on the lymphoma cells in vivo in Id-specific TCR-transgenic mice. A trivial explanation for this finding could be that FLIPL was lost upon tumor cell growth in vivo. However, when a 1.4-cm FLIPL-transduced F9 tumor was excised from a SCID mouse and cultured for 10 days as a single cell suspension, B-lymphoma cells were still completely resistant to agonistic Jo2 anti-Fas mAb in vitro, demonstrating that FLIPL expression is stable during tumor development in vivo (Fig. 6b). It is, however, difficult to completely rule out the possibility that apoptosis resistance of FLIPL transfectants occasionally could be abrogated in vivo. The results can hardly be explained by T-cell responses against GFP or other antigens since TCR-transgenic SCID mice are recombination deficient and scarcely express endogenous TCR or BCR.
Fig. 6a,b.

TCR-transgenic SCID mice reject F9 cells transduced to express FLIP. a Id-specific TCR-transgenic SCID (lines) or SCID littermates (dots) were injected with the indicated numbers of tumor cells s.c. between the shoulder blades. F9MFL10.7.1 cells express FLIP; F9M8.18 cells have been transfected with vector alone. b A single tumor cell suspension obtained from a SCID mouse with a 1.4-cm s.c. FLIPL F9 tumor (F9MFL10.7.1) was grown in vitro for 10 days and tested for sensitivity to titrated amounts of agonistic anti-Fas mAb Jo2 and isotype-matched control UC8
Discussion
Id-specific Th1 cells killed Fas+ B-lymphoma cells in vitro by a mechanism that could be almost totally attributed to FasL-Fas interaction. This mechanism was apparently also important in vivo because F9 tumors that occasionally grew out in challenged TCR-transgenic mice, or in SCID mice first injected with lymphoma cells and then treated with Id-specific CD4+ T cells, had either totally or partially lost sensitivity to Fas ligation. Selection of low sensitivity variants depended on presence of Id-specific CD4+ T cells because F9 tumors in nontransgenic SCID mice always retained full sensitivity to Fas ligation. Taking these results into account, it was surprising to observe that Id-specific TCR-transgenic mice were protected against Id+ B lymphoma even when the FasL-Fas pathway was blocked. Thus, anti-FasL mAb failed to abrogate protection in Id-specific TCR-transgenic SCID mice as did induced expression of FLIP in Id+ B lymphoma. Moreover, Id-specific TCR-transgenic gld mice that lack expression of functional FasL, nevertheless rejected Id+ lymphoma cells. Collectively, these results suggest that FasL-Fas interaction plays an important role in the elimination of Id+ tumor cells by Id-specific CD4+ T cells. However, if FasL-Fas interaction is blocked, killing may proceed by other pathways.
By what other mechanisms could Id-specific CD4+ T cells eliminate Id+ B-lymphoma cells in vivo? It should be stressed that in vitro, only Id-specific CD4+ T cells and B-lymphoma cells were present, thus limiting any cytotoxic activity of T cells to that exerted directly on B-lymphoma cells, like FasL-Fas interaction. By contrast, in vivo, a multitude of different cell types could be present in the tumor in addition to activated Id-specific T cells, including macrophages, dendritic cells, and neutrophils [33, 52, 53], all of which could contribute to the elimination of lymphoma cells. Indeed, in a murine model using GM-CSF transduced B16 tumor cells, the antitumor effect of CD4+ T cells depended on macrophages and eosinophils in the tumor [5]. Tumor macrophages could, for example, be activated by Th1-produced IFN-γ to increase their production of cytotoxic effector molecules like NO2 [54, 55] as well as proinflammatory cytokines. We recently found that CD4+ T-cell responses to Id+ B lymphoma were accompanied by a massive influx of neutrophils into the tumor; such an inflammatory reaction could contribute to tumor cell eradication [33]. Another possibility is that Id-specific CD4+ T cells inhibit tumor angiogenesis and thus the nutrition of the tumor. Supporting this possibility, it has been shown that IFN-γ produced by activated tumor-specific Th1 cells destroys host-derived endothelial cells, and hence the blood supply of the tumor [9, 10]. Finally, a number of cytotoxic molecules produced by activated Id-specific Th1 cells, like (non-FasL) members of the TNF family and perforin, could be of more importance in vivo than in vitro.
The discussion in the preceding paragraph focuses on Th1 cells in vivo. However, in the Id-specific TCR-transgenic model used herein, an Id+ myeloma induces a mixed Th1/Th2 lymphokine secretion pattern [2]. Moreover, Id-specific Th2 cells killed Id+ lymphoma in a Winn-type assay in vivo [6] as well as in apoptosis assays in vitro [33]. Id-specific Th2 cells could kill Id+ B lymphoma by several mechanisms independent of FasL, some of which might depend on eotaxin and STAT6 [7], and IL-5 and eosinophils [5]. In addition, activated Th2 cells produce IL-4 that could inhibit tumor angiogenesis [11, 56].
Althouth a multitude of mechanisms could operate in CD4+ T-cell–mediated elimination of B-cell lymphoma in vivo, it should be emphasized that all of these potential mechanisms crucially depend on initial activation of Id-specific CD4+ T cells. This is so because only Id-specific TCR-transgenic SCID mice, and not SCID mice, are protected against Id+ B lymphoma [33]. Even more impressive, we have found that all B-lymphoma tumors that escape rejection in TCR-transgenic SCID mice (protection is not 100%) have lost expression of the λ2315 L chain and thus become Id− and unable to stimulate Id-specific T cells (K. Lundin and B. Bogen, manuscript in preparation). Collectively, these results suggest that unknown mechanisms for cytotoxicity against B lymphoma in this model must emanate from—and thus be downstream of—activation of Id-specific CD4+ T cells.
The Id-specific TCR-transgenic model has also been used to study CD4+ T-cell–mediated rejection of Id+ myeloma cells, which, in contrast to B-lymphoma cells, lack MHC class II molecules (reviewed in [57]). Nevertheless, it seems that the initial response of the immune system against these two types of malignancies is similar—i.e., uptake of secreted antigen by APCs, presentation of antigen to CD4+ T cells, and activation of Id-specific T cells (K. Lundin and B. Bogen, manuscript in preparation). Whether FasL-Fas interaction plays any role in killing of MOPC315 multiple myeloma cells remains to be seen.
It has previously been described that A20 B-lymphoma cells transduced to express viral FLIP had increased tumorigenicity in BALB/c mice compared to the parental tumor cells. By contrast, SCID mice were equally susceptible to both [21]. Because SCID mice lack B and T cells, and assuming that T cells are most important in tumor protection, the results suggested that T cells of unknown specificity killed B-lymphoma cells by a caspase-8-dependent mechanism that could be inhibited by FLIP expression. In the present experiments, the effect of FLIP appeared to be split in that FLIP-transduced Id+ A20 (F9) lymphoma cells were resistant against killing by Id-specific CD4+ T cells in vitro, but not in vivo. How could these seemingly conflicting in vivo results of the previous [21] and present experiments be explained? One possibility is that in the previous experiments, CD8+ T cells of some unknown anti-tumor specificity could be important. Such putative anti-tumor CD8+ T cells could to a larger extent kill B-lymphoma cells by a caspase-8-dependent mechanism than do Id-specific CD4+ T cells. Consistent with this idea, our results [6] and also those of others [5, 7, 9, 10] indicate that CD4+ cells are not only directly cytotoxic but indirectly kill tumor cells by recruitment of cytotoxic macrophages and eosinophils, and secretion of IFNγ that destroys tumor vessels.
Deficiency of FasL-Fas interaction has previously been implicated in development of B lymphomas. Thus, lymphomas and leukemias have been described to have mutations in the Fas gene [14–20] suggesting that tumor cells escape immunosurveillance by modulating their sensitivity to FasL. Complementary evidence stems from the observation that patients with Fas deficiency have an increased frequency of lymphomas [58]. Finally, B lymphoma [21] and T lymphoma [22] with experimentally induced expression of FLIP have increased tumorigenicity in mice. However, none of these studies defined FasL+ tumoricidal effector cells, nor were the specificities of putative cytotoxic effector cells investigated. This is not a trivial issue because FasL can be expressed by a number of cell types including T cells and NK cells (reviewed in [59]). The present results show that Id-specific Th1 cells exert a major part of their tumoricidal effect on Id+ B-lymphoma cells through FasL-Fas interaction. Whether Th1 cells are also important in previous studies [14–22, 58] that have incriminated FasL-Fas interaction in tumor protection remains to be seen.
Naïve Id-specific CD4+ T cells do not kill Id+ lymphoma cells, while memory cells do [33]. Thus, T cells have to be stimulated by Id-peptide on MHC class II molecules prior to becoming cytotoxic. Whether the B-lymphoma cells themselves are the primary presenters of Id-peptide [34, 35], or professional APC like dendritic cells that have endocytosed and processed Id+ Ig secreted by lymphoma cells [53], remains to be established. Likewise, whether Id priming of T cells occurs in draining LN or in the tumor, is not known. Anyway, once memory Th1 and Th2 cells are present in the tumor, they appear to kill Id+ B-lymphoma cells by several mechanisms out of which FasL-Fas interaction is a major one exerted by Th1 cells.
Footnotes
Supported by grants from the Norwegian Cancer Society, the Research Council of Norway, and the Multiple Myeloma Research Foundation.
References
- 1.Kahn J Immunol. 1991;146:3235. [PubMed] [Google Scholar]
- 2.Lauritzsen Proc Natl Acad Sci U S A. 1994;91:5700. doi: 10.1073/pnas.91.12.5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagarkatti J Immunol. 1990;144:4898. [PubMed] [Google Scholar]
- 4.Pardoll Curr Opin Immunol. 1998;10:588. doi: 10.1016/S0952-7915(98)80228-8. [DOI] [PubMed] [Google Scholar]
- 5.Hung J Exp Med. 1998;188:2357. doi: 10.1084/jem.188.12.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lauritzsen Scand J Immunol. 1993;37:77. doi: 10.1111/j.1365-3083.1993.tb01668.x. [DOI] [PubMed] [Google Scholar]
- 7.Mattes J Exp Med. 2003;197:387. doi: 10.1084/jem.20021683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishimura J Exp Med. 1999;190:617. doi: 10.1084/jem.190.5.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mumberg Proc Natl Acad Sci U S A. 1999;96:8633. doi: 10.1073/pnas.96.15.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qin Immunity. 2000;12:677. doi: 10.1016/S1074-7613(00)80218-6. [DOI] [PubMed] [Google Scholar]
- 11.Volpert J Exp Med. 1998;188:1039. doi: 10.1084/jem.188.6.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagata Science. 1995;267:1449. [Google Scholar]
- 13.Suda J Immunol. 1995;154:3806. [PubMed] [Google Scholar]
- 14.Gronbaek Blood. 1998;92:3018. [PubMed] [Google Scholar]
- 15.Landowski Leuk Lymphoma. 2001;42:835. doi: 10.3109/10428190109097702. [DOI] [PubMed] [Google Scholar]
- 16.Maeda J Exp Med. 1999;189:1063. doi: 10.1084/jem.189.7.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muschen Cancer Res. 2000;60:5640. [PubMed] [Google Scholar]
- 18.Plumas Blood. 1998;91:2875. [PubMed] [Google Scholar]
- 19.Seeberger Lab Invest. 2001;81:977. doi: 10.1038/labinvest.3780310. [DOI] [PubMed] [Google Scholar]
- 20.Tamiya Blood. 1998;91:3935. [PubMed] [Google Scholar]
- 21.Djerbi J Exp Med. 1999;190:1025. doi: 10.1084/jem.190.7.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Medema J Exp Med. 1999;190:1033. doi: 10.1084/jem.190.7.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lynch Proc Natl Acad Sci U S A. 1972;69:1540. [Google Scholar]
- 24.Sirisinha Proc Natl Acad Sci U S A. 1971;68:3130. doi: 10.1073/pnas.68.12.3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janeway Proc Natl Acad Sci U S A. 1975;72:2357. [Google Scholar]
- 26.Campbell J Immunol. 1990;145:1029. [PubMed] [Google Scholar]
- 27.George J Immunol. 1987;138:628. [PubMed] [Google Scholar]
- 28.Bendandi Nat Med. 1999;5:1171. doi: 10.1038/13928. [DOI] [PubMed] [Google Scholar]
- 29.Hawkins Hum Gene Ther. 1997;8:1287. doi: 10.1089/hum.1997.8.10-1287. [DOI] [PubMed] [Google Scholar]
- 30.Hsu Nat Med. 1996;2:52. doi: 10.1038/nm0196-52. [DOI] [PubMed] [Google Scholar]
- 31.Timmerman Blood. 2002;99:1517. doi: 10.1182/blood.V99.5.1517. [DOI] [PubMed] [Google Scholar]
- 32.Syrengelas J Immunol. 1999;162:4790. [PubMed] [Google Scholar]
- 33.Lundin Blood. 2003;102:605. doi: 10.1182/blood-2002-11-3381. [DOI] [PubMed] [Google Scholar]
- 34.Weiss Proc Natl Acad Sci U S A. 1989;86:282. [Google Scholar]
- 35.Weiss Cell. 1991;64:767. [Google Scholar]
- 36.Bogen Eur J Immunol. 1992;22:703. doi: 10.1002/eji.1830220313. [DOI] [PubMed] [Google Scholar]
- 37.Bogen EMBO J. 1989;8:1947. doi: 10.1002/j.1460-2075.1989.tb03599.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bogen Eur J Immunol. 1986;16:1373. doi: 10.1002/eji.1830161110. [DOI] [PubMed] [Google Scholar]
- 39.Lynch Immunity. 1994;1:131. doi: 10.1016/1074-7613(94)90106-6. [DOI] [PubMed] [Google Scholar]
- 40.Takahashi Cell. 1994;76:969. doi: 10.1016/0092-8674(94)90375-1. [DOI] [PubMed] [Google Scholar]
- 41.Bogen Eur J Immunol. 1990;20:2359. doi: 10.1002/eji.1830201030. [DOI] [PubMed] [Google Scholar]
- 42.Abravaya Nucleic Acids Res. 1995;23:675. doi: 10.1093/nar/23.4.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kayagaki Proc Natl Acad Sci U S A. 1997;94:3914. doi: 10.1073/pnas.94.8.3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rouvier J Exp Med. 1993;177:195. doi: 10.1084/jem.177.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Djerbi Scand J Immunol. 2001;54:180. doi: 10.1046/j.1365-3083.2001.00941.x. [DOI] [PubMed] [Google Scholar]
- 46.Matzinger J Immunol Methods. 1991;145:185. doi: 10.1016/0022-1759(91)90325-A. [DOI] [PubMed] [Google Scholar]
- 47.Bogen Scand J Immunol. 1989;29:273. doi: 10.1111/j.1365-3083.1989.tb01125.x. [DOI] [PubMed] [Google Scholar]
- 48.Aggarwal Nat Rev Immunol. 2003;3:745. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 49.Screpanti J Immunol. 2001;167:2068. doi: 10.4049/jimmunol.167.4.2068. [DOI] [PubMed] [Google Scholar]
- 50.Tschopp Curr Opin Immunol. 1998;10:552. doi: 10.1016/S0952-7915(98)80223-9. [DOI] [PubMed] [Google Scholar]
- 51.Irmler Nature. 1997;388:190. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 52.Dembic Blood. 2001;97:2808. doi: 10.1182/blood.V97.9.2808. [DOI] [PubMed] [Google Scholar]
- 53.Dembic Proc Natl Acad Sci U S A. 2000;97:2697. doi: 10.1073/pnas.050579897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bingle J Pathol. 2002;196:254. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- 55.Stuehr J Exp Med. 1989;169:1543. doi: 10.1084/jem.169.5.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tepper Science. 1992;257:548. doi: 10.1126/science.1636093. [DOI] [PubMed] [Google Scholar]
- 57.Corthay Cancer Immunol Immunother. 2004;53:759. doi: 10.1007/s00262-004-0504-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Straus Blood. 2001;98:194. doi: 10.1182/blood.V98.1.194. [DOI] [PubMed] [Google Scholar]
- 59.Green Proc Natl Acad Sci U S A. 1997;94:5986. doi: 10.1073/pnas.94.12.5986. [DOI] [PMC free article] [PubMed] [Google Scholar]