

Abstract
Purpose.
Immunotherapy holds promise as a new strategy for the eradication of residual cells in acute myeloid leukaemia (AML). Leukaemic antigen presenting cells (APCs) combining optimal antigen presentation and tumour antigenicity could be used as potent T cell activators. For clinical purposes it is desirable to culture APCs under serum-free conditions. Therefore, we compared morphological, immunophenotypical and functional outcome of the serum-free culture of AML-APCs to their serum-enriched culture.
Methods.
AML blasts (n=19) were cultured in the presence of either a cytokine mix or calcium ionophore (CI) for 14 and 2 days, respectively, in FCS-containing medium (FCS), StemSpan serum-free medium (SP) and CellGro serum-free medium (CG). After culture relative yields were calculated and immunophenotypic analysis of APC markers was performed. The mixed leukocyte reaction (MLR) was used to determine T cell stimulating capacity.
Results.
Serum-free culture of AML-APCs resulted in comparable morphology, relative yields and immunophenotype to serum-enriched culture. By comparing both serum-free media we observed a trend towards a more mature phenotype of CI-cultured AML-APCs in SP. MLR showed that serum-free cultured cells have equal T cell stimulatory capacity in comparison with serum-enriched culture.
Conclusion.
These data show that the serum-free culture of AML-APCs is feasible and that these APCs are comparable to serum-enriched cultured AML-APCs with regard to morphological, immunophenotypical and functional characteristics. These AML-APCs are suitable for the development of active specific immunisation protocols which meet the criteria for good clinical practise (GCP).
Keywords: Acute myeloid leukaemia, Immunotherapy, Antigen presenting cells, Serum-free culture
Introduction
Despite intensive chemotherapy-based approaches including stem cell transplantation, we are only able to cure at best 40–50% of the patients with acute myeloid leukaemia (AML) [13]. Although complete remission can be achieved in approximately 50–85% of the patients, relapse due to minimal residual disease (MRD) is still a common event [14]. Further intensification of chemotherapy regimes would probably lead to irreversible non-haematological toxicity and subsequently an increase in treatment-related mortality. Therefore, the development of new treatment strategies which control the outgrowth of residual leukaemia cells would be of great value. Recent data indicate that immunotherapy directed at generating autologous anti-leukaemia responses, could contribute to the eradication of MRD cells.
Although considerable progress has been made in identifying relevant tumour antigens, for the majority of human cancers it remains unclear which antigens represent the most important tumour rejection antigens [23]. Therefore, most cancer vaccine approaches are based on using tumour cells themselves as a source of antigen [20]. Dendritic cells (DCs) are believed to be the most potent antigen presenting cells (APCs) [1, 8]. Owing to their capacity to activate naïve T cells, DCs loaded with antigens in the form of peptides or tumour cell lysates [17], DCs fused with whole tumour cells [12] or DCs transducted with RNA encoding tumour antigens [2, 9], have been used for active specific immunisation strategies. Since leukaemic blasts have proven to be able to differentiate into DC-like leukaemic-APCs they provide the unique opportunity to generate antigen presenting cells harbouring the full range of potential, still unidentified tumour antigens [5, 6, 7, 19, 24]. These APCs are potentially suitable for vaccination purposes. AML-APCs can be cultured in the presence of various combinations of cytokines such as GM-CSF, SCF, Flt3-L, IL-3, IL-4 and TNF-α. However, this culture method is time-consuming since it takes approximately 2 weeks for the generation of AML-APCs. Interestingly, it is possible to generate AML-APCs in a much shorter time frame by incubation with calcium ionophores thereby by-passing receptor-mediated signalling [10, 27, 28]. APCs cultured for 2 days with calcium ionophore showed, immunophenotypically and functionally, a more mature stage than those cultured with a combination of cytokines indicating that this culture method could provide an appropriate alternative for the generation of APCs [29]. The medium used to culture AML-APCs has usually included fetal calf serum (FCS), fetal bovine serum (FBS) or autologous human serum. This is undesirable for a number of reasons. Firstly, APCs could take up, process and present soluble irrelevant antigens present in these sera. Indeed, it has been shown that APCs cultured in the presence of FCS were able to induce strong cytotoxic T cell activity against bovine antigens [15, 21]. Secondly, animal-derived culture supplements may constitute a risk of adverse allergic reactions and may give rise to infectious disease if used for clinical vaccination purposes. Finally, autologous sera contain various identified and unidentified growth factors and tumour-derived suppressive factors that affect differentiation and maturation thereby making it impossible to standardise culture conditions. Previous studies documented the possibility of serum-free culture of AML-APCs, however, the influence of the use of either serum-enriched medium or serum-free medium on AML-APC characteristics has not been compared before [6, 7, 19]. Therefore, in the present study, we generated AML-APCs under serum-free conditions and compared these APCs to AML-APCs cultured in medium containing FCS with respect to morphological, immunophenotypical and functional characteristics. We demonstrate that AML-APCs, that can function as potent inducers of T-cell reactivity, can be successfully produced under serum-free conditions and can thus be used for active specific immunisation strategies that comply with good clinical practise (GCP) demands.
Materials and methods
Culture of antigen presenting cells
After informed consent, peripheral blood samples or bone marrow mononuclear cells for culture or cryopreservation were obtained from 19 patients with AML by Ficoll-Hypaque density gradient centrifugation (Amersham Pharmacia Biotech, Uppsala, Sweden). Fresh or frozen-thawed mononuclear cells were cultured at a concentration of 0.5×106 cells/ml in 6-well plates (Corning Costar, Corning, NY) in the following three different culture media:
RPMI-1640 (Gibco-BRL, Life Technologies, Paisley, Scotland), supplemented with 20% fetal calf serum (FCS, Gibco-BRL), 100 U/ml penicillin (Gibco-BRL) and 100 μg/ml streptomycin (Gibco-BRL) (n=17)
StemSpan SFEM serum-free medium (SP) (Stemcell Technologies, Vancouver, Canada) supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin (n=19)
CellGro serum-free medium (CG) (CellGenix, Freiburg, Germany) supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin (n=13).
Differentiation of AML cells to antigen presenting cells was induced by adding a combination of cytokines or calcium ionophore (CI) A23187 (Sigma, St. Louis, MO). The following cytokines were used in the cytokine culture: GM-CSF 250 U/ml (specific activity 1×107 U/mg protein, Pepro Tech, Rocky Hill, NJ), TNF-α 50 U/ml (specific activity 1×108 U/mg protein, Boehringer Mannheim, Germany), IL-3 20 ng/ml (specific activity 1×106 U/mg protein, Pepro Tech), SCF 50 ng/ml (specific activity 1×105 U/mg protein, Pepro Tech), Flt3-L 50 ng/ml (specific activity 2×105 U/mg protein, Pepro Tech) and IL-4 250 U/ml (specific activity 1×108 U/mg protein, Serva, Heidelberg, Germany). Medium exchange with fresh cytokine-supplemented medium was routinely performed in cases of overgrown culture or medium exhaustion determined by evaluation of cell density by microscopy and medium colour, in general every 3–4 days. After 14 days at 37˚C in a 5% CO2 humidified incubator, cells were harvested. Adherent cells were harvested by incubating the wells with 5 mM EDTA (Sigma) in RPMI-1640 for 15 min at 37˚C. In the CI-culture, cells were cultured for 2 days with A23187 375 ng/ml and IL-4 250 U/ml.
Cell counts and viability
Cell numbers and viability were determined by Trypan blue dye exclusion (0.2 g/ml, Merck, Darmstadt, Germany). Viability was further analysed by incubation with syto-16 3 nM (Molecular Probes, Eugene, OR) PSC833 2 μM (kind gift of Novartis, Basel, Switzerland) and 7-amino-actinomycin D 2 μM (7-AAD, ViaProbe, Pharmingen, San Diego, CA) for 45 min at 37°C, as described previously by Schuurhuis et al [25]. Flow cytometric analysis identified viable (syto-16+, 7-AAD−), apoptotic (syto-16dim, 7-AAD−) and dead (syto-16−, 7-AAD+) cells. Relative yield was calculated to determine the number of AML-APCs cultured and was defined as the number of viable cells after culture divided by the number of viable cells before culture, multiplied by the percentage of CD40+ cells after culture [29].
Morphological analysis
APC morphology was characterised by May-Grünwald-Giemsa stained cytospins which were prepared before and after each culture. Characteristic dendritic cell morphology was defined as an eccentric position of the nucleus and large irregular shaped, elongated processes and cytoplasmatic projections.
Immunophenotypic analysis
At the start and end of the culture period, AML blasts and AML-APCs were analysed by performing four-colour flow cytometric analysis on a FACS-Calibur flow cytometer (Becton Dickinson, San Jose, CA). Cells were preincubated with human immunoglobulin 6 mg/ml (CLB, Amsterdam, The Netherlands) for 5 min in order to block aspecific Fc receptor-mediated binding. Cells were analysed for the surface expression of the following markers: FITC-labelled CD86 (Pharmingen), CD1a (CLB), CD54 (DAKO, Glostrup, Denmark), PE-labelled CD40 (Immunotech, Marseille, France), CD80 (Becton Dickinson), CD83 (Immunotech), PerCP-labelled CD45 (Becton Dickinson), anti-HLA-DR (Becton Dickinson), APC-labelled CD34 (Becton Dickinson), CD38 (Becton Dickinson), CD14 (Becton Dickinson). Isotype controls used were FITC-labelled IgG1 (DAKO), IgG2b (CLB), PE-labelled IgG1 (Becton Dickinson), IgG2b (DAKO), PerCP-labeled IgG1 and IgG2a (both Becton Dickinson) and APC-labelled IgG1 and IgG2b (both Becton Dickinson). Results were calculated as the percentage of positive cells compared to the appropriate isotype control. Analysis of the results was done using CellQuest software (Becton Dickinson). Necrotic and dead cells identified by syto-16/7-AAD staining were excluded from the analysis. The mean fluorescence index (MFI) was calculated to determine the intensity of marker expression, defined as the ratio of the mean channel peak fluorescence intensity of the specific antibody and the mean channel peak fluorescence intensity of its isotype control.
Functional analysis
To assess the ability of the cultured AML-APCs to stimulate allogeneic T-cell proliferation, a mixed leukocyte reaction (MLR) was performed in a 96-well round-bottomed plate (Costar). Peripheral blood mononuclear cells isolated from a buffy-coat from one donor were used as responder cells at a fixed concentration of 5×104 per well. The use of one PBMC donor was chosen to enable comparison between the MLR of different APC cultures. Non-cultured AML blasts and cultured AML-APCs were irradiated at 30 Gy. After co-culture of the stimulator cells at different stimulator/responder (S/R) ratios with peripheral blood mononuclear cells for 5 days, each well was pulsed with 0.4 μCi [3H] thymidine (Amersham Pharmacia Biotech, Buckinghamshire, UK) for 5 h. Co-cultures were harvested onto a fibre glass filter mat and analysed for the incorporation of [3H] thymidine in a liquid scintillation counter (Wallac, Turku, Finland).
Statistical analysis
Within one patient sample, we analysed the difference of marker expression and MFI of cultured AML-APCs between the three different culture media. We used the paired sample Student's t-test (two-tailed) to compare the differences between culture media and culture methods. P-values of <0.05 were considered to be significant.
Results
Viability and relative yield of AML-APCs
Blast cells from 19 patients with AML were cultured in serum-enriched medium and 2 different serum-free media in the presence of either a cytokine mix or CI (FCS-cytokine culture n=17, SP-cytokine culture n=19, CG-cytokine culture n=13, FCS-CI-culture n=14, SP-CI-culture n=17, CG-CI-culture n=8). Patient characteristics are described in Table 1. Patient samples were randomly selected for different culture methods based on availability. After 14 days of culture with cytokines, viability of cells in serum-enriched medium and both serum-free media were comparable. However, in the CI-culture, cells cultured for 2 days in CellGro medium were significantly less viable compared to cells cultured in FCS-containing medium (mean %±SEM) (CG: 25.0±6.2 vs. FCS: 52.9±9.8, n=7, P=0.014) and StemSpan medium (CG: 23.1±5.7 vs. SP: 48.9±10.9, n=8, P=0.011).
Table 1.
. Patient characteristics
| UPN1 | Sex (M/F)2 | Age3 | FAB4 | Material5 | Blasts (%)6 | Cytogenetics |
|---|---|---|---|---|---|---|
| 1 | M | 49 | M0 | BM | 85 | 92 XXYY, der (15;15)(q10;q10) |
| 2 | M | 55 | M1 | BM | 86 | del (9) (q11;q22) |
| 3 | M | 76 | M1 | BM | 82 | Normal |
| 4 | M | 72 | M1 | BM | 72 | Normal |
| 5 | F | 61 | M1 | PBMC | 83 | del (11) (q13;q25) |
| 6 | M | 69 | M2 | BM | 45 | Normal |
| 7 | M | 24 | M3 | BM | 86 | t(15;17) |
| 8 | F | 46 | M4eo | BM | 68 | inv (16) |
| 9 | F | 35 | M4eo | PBMC | 51 | inv (16) del (7) (q22) |
| 10 | F | 33 | M4eo | PBMC | 34 | Monosomy (7) |
| 11 | F | 22 | M4eo | BM | 70 | inv (16) |
| 12 | F | 34 | M4eo | BM | 31 | inv (16) |
| 13 | F | 69 | M5a | BM | 84 | no metaphases |
| 14 | M | 37 | M5a | BM | 95 | Normal |
| 15 | F | 46 | M5a | BM | 97 | t(9;11) (p22;q23) |
| 16 | M | 38 | M5a | BM | 84 | Normal |
| 17 | F | 54 | M5a | BM | 97 | Normal |
| 18 | F | 63 | RAEB-t | BM | 23 | Normal |
| 19 | M | 77 | Unclassified | BM | 95 | Not tested |
1Unique patient number.
2 M male, F female.
3Age at diagnosis (years).
4French-American-British classification.
5 BM bone marrow, PBMC peripheral blood mononuclear cells.
6Percentages of blasts were scored in May-Grünwald-Giemsa stained bone marrow smears or PBMC before gradient cell separation.
The relative yields of AML-APCs cultured in the three different media and supplemented with either the cytokine mix or calcium ionophore are shown in Fig. 1. No significant differences in relative yields were found between serum-enriched medium and serum-free media in the cytokine cultures. Furthermore, no significant differences were found between the two cultures with serum-free media. However, in accordance with the viability results, in the CI-culture a significant difference was found between medium containing FCS and CellGro serum-free medium in favour of FCS-containing medium (CG: 16.7±5.5 vs. FCS: 29.5±4.4, P=0.015). Moreover, StemSpan serum-free medium showed a significantly better relative yield compared to CellGro serum-free medium (CG: 15.1±5.1 vs. SP: 35.5±11.6, P=0.049). No significant differences between StemSpan serum-free medium and medium containing FCS were found.
Fig. 1.
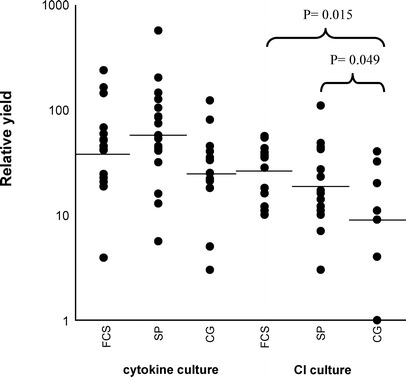
. Relative AML-APC yield: the number of viable cells after culture divided by the number of viable cells before culture, multiplied by the percentage of CD40+ cells after culture. AML blasts were cultured in the presence of cytokines or calcium ionophore (CI) in FCS-containing medium (FCS, FCS-cytokine culture n=17, FCS-CI-culture n=14), StemSpan serum-free medium (SP, SP-cytokine culture n=19, SP-CI-culture n=17), and CellGro serum-free medium (CG, CG-cytokine culture n=13, CG-CI-culture n=8). Horizontal lines represent the mean relative yield. The yield of AML-APCs cultured with CI in CellGro medium was significantly less as compared to AML-APCs cultured in FCS-containing medium and StemSpan medium (P=0.015, P=0.049, respectively).
Morphology
Cultured AML-APCs displayed pronounced morphological changes compared to uncultured blasts. Cells showed a variable increase in size with an eccentric position of the nucleus, cytoplasmatic protrusion and elongated processes. With respect to the comparison of serum-enriched cultured AML-APCs with serum-free cultured AML-APCs we did not observe any major differences in the position of the nucleus or the dendritic veils. As we observed previously, cells cultured in the presence of CI showed a higher cytoplasm/nucleus ratio compared to cytokine-cultured AML-APCs [29].
Immunophenotyping
Before culture AML blasts showed minimal or no expression of CD40, CD80, CD83, and CD86. Uncultured blast cells did show expression of CD54 (mean %±SEM) (21.6±5.8) and HLA-DR (52.9±7.1). At the end of culture, cells showed a significant increase in the percentages of CD40 and CD80 for both culture methods with or without FCS (Fig. 2). In the CI-culture, CD54, CD83 and CD86 were also significantly up-regulated. Despite up-regulation of these surface molecules in the cytokine culture with FCS-containing medium and StemSpan medium, this did not reach significance. In the cytokine culture with CellGro medium no up-regulation could be shown for CD54 and CD83. In accordance with previous observations a more mature phenotype is observed after culture with CI, as measured by a significant increase in maturation markers CD40, CD54 and CD83 and an increased activation state as measured by an increased expression of CD80, CD86 and HLA-DR [29]. Overall, the immunophenotyping results with serum-free media were similar to the results generated in serum-enriched conditions. By comparing the differences between the media within one patient sample, the only statistically significant difference between serum-enriched and serum-free media, in favour of FCS as compared to CG was the increase in the percentage of CD40 in the CI-culture (11.7±4.8, P=0.05). Comparison of both serum-free media showed significantly more up-regulation of CD80 and CD83 in StemSpan medium compared to CellGro medium, in the CI-cultures and cytokine cultures, respectively (12.4±4.9, P=0.04; 2.1±0.9, P=0.03, respectively).
Fig. 2A.
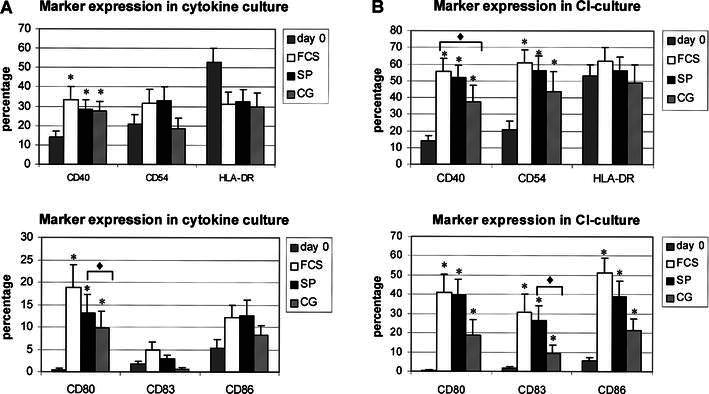
. Immunophenotype of AML cells before and after culture with cytokines in FCS-containing medium (FCS, n=17), StemSpan serum-free medium (SP, n=19) and CellGro serum-free medium (CG, n=13). Mean percentage and the standard error of the mean are depicted. Cultured AML-APCs showed a significant upregulation of CD40 and CD80 in serum-enriched and serum-free media (asterisk). AML-APCs cultured in StemSpan medium showed a significantly higher upregulation of CD83 in the cytokine culture (P=0.03) as compared to cells cultured in CellGro medium (diamond). B Immunophenotype of AML cells before and after culture with calcium ionophore (CI) in FCS-containing medium (FCS, n=14), StemSpan serum-free medium (SP, n=17) and CellGro serum-free medium (CG, n=8). Mean percentage and the standard error of the mean are depicted. Cultured AML-APCs showed a significant upregulation of CD40, CD54, CD80, CD86 and CD83 in serum-enriched and serum-free media(asterisk). AML-APCs cultured in the presence of CI in FCS-containing medium showed significantly higher expression of CD40 as compared to cells cultured in CellGro medium (P=0.05) (diamond). AML-APCs cultured in StemSpan medium showed a significantly higher upregulation of CD80 as compared to CellGro medium in the CI-culture (P=0.04) (diamond).
The mean fluorescence index (MFI) was calculated to determine the intensity of marker expression on the surface of the AML-APCs. Table 2 shows that the MFI of AML-APCs cultured in serum-free media did not differ from the MFI of AML-APCs cultured in serum-enriched medium. The intensity of the expression of CD83 in the cytokine culture was significantly higher in StemSpan medium compared to CellGro (0.2±0.07, P=0.01). Although not reaching significance, a trend could be observed in the CI-culture towards a lower MFI if cells were cultured in CellGro medium as compared to cells cultured in FCS-containing medium and StemSpan serum-free medium.
Table 2.
Mean fluorescence index of DC markers after immunophenotypic analysis of uncultured AML blasts and cultured AML-APCs in three different media
| MFI cytokine culture | ||||||||||||
| CD40 | CD54 | CD80 | ||||||||||
| Day 0 | FCS | SP | CG | Day 0 | FCS | SP | CG | Day 0 | FCS | SP | CG | |
| Mean | 4.5 | 15.7 | 13.1 | 11.7 | 3.1 | 7.7 | 7.7 | 5.2 | 0.8 | 5.2 | 4.4 | 3.7 |
| SEM | 1.1 | 2.8 | 1.9 | 2.8 | 0.5 | 1.8 | 1.6 | 0.8 | 0.1 | 1.1 | 0.8 | 1.4 |
| Median | 3.2 | 10.8 | 11.7 | 9.1 | 2.1 | 5.7 | 6.1 | 3.8 | 0.8 | 3.2 | 2.8 | 1.6 |
| CD83 | CD86 | HLA-DR | ||||||||||
| Day 0 | FCS | SP | CG | Day 0 | FCS | SP | CG | Day 0 | FCS | SP | CG | |
| Mean | 0.9 | 1.1 | 1.7a | 1.0 | 2.7 | 3.6 | 3.4 | 3.6 | 15.8 | 9.0 | 14.3 | 15.9 |
| SEM | 0.1 | 0.1 | 0.5 | 0.1 | 0.6 | 0.6 | 0.6 | 0.9 | 4.5 | 2.1 | 2.9 | 6.5 |
| Median | 0.9 | 0.9 | 1.2 | 0.9 | 1.4 | 2.8 | 2.6 | 2.0 | 12.8 | 5.6 | 11.3 | 9.6 |
| MFI CI-culture | ||||||||||||
| CD40 | CD54 | CD80 | ||||||||||
| Day 0 | FCS | SP | CG | Day 0 | FCS | SP | CG | Day 0 | FCS | SP | CG | |
| Mean | 4.5 | 47.2 | 47.3 | 24.2 | 3.1 | 37.3 | 36.7 | 13.0 | 0.8 | 23.8 | 25.2 | 7.9 |
| SEM | 1.1 | 12.5 | 11.0 | 6.4 | 0.5 | 11.1 | 10.4 | 4.0 | 0.1 | 8.5 | 8.1 | 2.8 |
| Median | 3.2 | 25.1 | 28.5 | 16.0 | 2.1 | 17.7 | 17.9 | 8.6 | 0.8 | 8.3 | 8.4 | 4.1 |
| CD83 | CD86 | HLA-DR | ||||||||||
| Day 0 | FCS | SP | CG | Day 0 | FCS | SP | CG | Day 0 | FCS | SP | CG | |
| Mean | 0.9 | 7.8 | 8.4 | 2.6 | 2.7 | 27.4 | 28.9 | 6.8 | 15.8 | 40.9 | 38.4 | 29.0 |
| SEM | 0.1 | 3.2 | 2.8 | 0.8 | 0.6 | 8.1 | 11.1 | 1.9 | 4.5 | 13.3 | 11.6 | 15.7 |
| Median | 0.9 | 3.3 | 3.0 | 1.9 | 1.4 | 16.8 | 10.9 | 6.5 | 12.8 | 24.3 | 19.3 | 13.4 |
a P <0.05 as compared to CG
CI calcium ionophore.
FCS FCS-containing medium (cytokine culture n=17, CI culture n=14).
SP StemSpan serum-free medium (cytokine culture n=19, CI culture n=17).
CG CellGro serum-free medium (cytokine culture n=13, CI culture n=8).
T-cell stimulatory capacity
The ability of cultured cells to present antigens to naïve T cells was determined in the allogeneic mixed leukocyte reaction. We calculated the ratio between the [3H] thymidine incorporation by the AML-APCs and the uncultured blasts. Cultured AML-APCs proved to have an increased capacity to stimulate T-cell proliferation compared to uncultured blasts (mean fold increase ± SEM) (FCS-cytokine culture: 3.4±1.4, n=14; SP-cytokine culture: 2.4±1.0, n=18; CG-cytokine culture: 3.2±1.2, n=10; FCS-CI culture: 2.3±0.5, n=13; SP-CI culture: 2.7±0.5, n=16; CG-CI culture: 2.1±0.7, n=8) (Fig. 3A). Although the results varied greatly between patients no significant differences were detected between the serum-enriched medium and serum-free media as outlined in Fig. 3B. In the CI-culture, APCs cultured in StemSpan medium were significantly better stimulators of T-cell proliferation compared to AML-APCs cultured in CellGro medium (CG: 2.0±0.8, SP: 3.5±0.8, P=0.002) (Fig. 3A,B).
Fig. 3A.
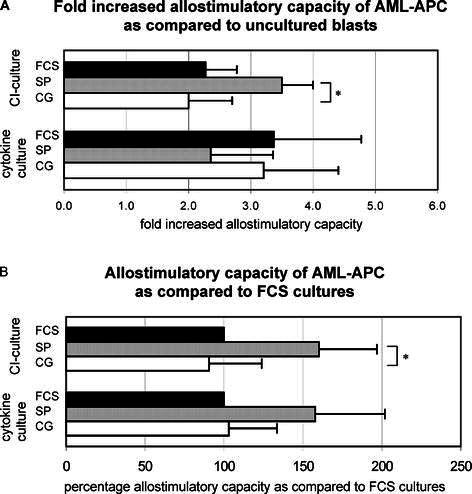
Fold increase in allostimulatory capacity of AML-APCs as compared to AML blasts. Non-cultured AML blasts and cultured AML-APCs were irradiated and co-cultured with peripheral blood mononuclear cells for 5 days at 1:5 stimulator/responder (S/R) ratio. For each patient, the ratio between the [3H] thymidine incorporation by the AML-APCs and the uncultured blasts was calculated. Mean fold increased T cell stimulatory capacity and SEM are depicted. No significant differences between serum-free cultured and serum-enriched cultured AML-APCs were found. CI-cultured AML-APCs cultured in SP were significantly better stimulators of T cell proliferation as compared to AML-APCs cultured in CG medium (P=0.002) (asterisk). B Allostimulatory capacity of serum-free cultured AML-APCs as compared to FCS cultures. Non-cultured AML blasts and cultured AML-APCs were irradiated and co-cultured with peripheral blood mononuclear cells for 5 days at 1:5 S/R ratio. For each patient, the difference between serum-free cultures and standard FCS cultures, set at 100%, was calculated. Mean differences (%) and SEM are depicted. CI-cultured AML-APCs cultured in SP were significantly better stimulators of T-cell proliferation as compared to AML-APCs cultured in CG medium (P=0.049) (asterisk). Considering the total group of patients, negative controls did not show [3H] thymidine incorporation [counts per minute (cpm), median, range] (responder cells: 483, 68–1088; stimulator cells 132, 53–290) whereas significantly higher responses were observed in the co-cultures (uncultured blasts: 2,723, 312–18,720, P=0.004; FCS-cytokine culture: 5,934, 1,583–21,000, P=0.001; SP-cytokine culture: 4,003, 576–24,000, P=0.002; CG-cytokine culture: 4,235, 1,290–9,299, P=0.001; FCS-CI culture: 10,098, 699–38,184, P=0.005; SP-CI culture: 13,761, 2,168–44,565, P=0.002; CG-CI culture: 3,230, 621–15,018, P=0.03). FCS FCS-containing medium (cytokine culture n=14, CI culture n=13), SP StemSpan serum-free medium (cytokine culture n=18, CI culture n=16), CG CellGro serum-free medium (cytokine culture n=10, CI culture n=8). Note In Fig. 3A, UPN 15 is left out from the analysis of the cytokine culture in SP medium since no control blast culture could be performed.
Discussion
In this report we present data on the generation of AML-APCs under serum-free conditions for use in clinical vaccination protocols which meet GCP criteria. Serum-free media have been used for the generation of antigen presenting cells from apheresis cells [26], haematopoietic progenitors [22], monocytes [11] but also from leukaemic blasts [3, 4, 6, 7]. However, the studies done by Cignetti et al. [7] and Choudhury et al. [6], were not designed to compare serum-free culture methods to serum-enriched culture methods. Furthermore, the studies performed by Bruserud et al. focussed on the investigation of AML blast functions such as proliferative responses and cytokine secretion after culture in serum-free medium with IL-3 and GM-CSF or IL-3, SCF, GM-CSF and G-CSF, rather than dendritic cell characteristics [3, 4]. We provide evidence that culture of AML-APCs in serum-free medium is feasible and, more importantly, that these APCs are comparable to AML-APCs cultured in serum-enriched medium with regard to morphological, immunophenotypical and functional characteristics.
In a recently conducted phase-I pilot study on CML-APC vaccination in advanced stage disease, we were able to detect strong DTH responses representing autologous CML-specific T-cell responses [18]. In this study CML-APCs were cultured in FCS-containing medium. It would be desirable to culture leukaemia-derived APCs in serum-free medium since fetal calf serum contains xenoantigens and carries the risk of introducing infectious agents.
For AML, functional APCs were obtained using various combinations of cytokines such as GM-CSF, Flt-3L, IL-3, IL-4, TNF-α and CD40L [5, 6, 7, 19, 24]. An interesting finding is that AML blasts are able to respond to calcium ionophores thereby by-passing receptor mediated signalling, providing a time and cost-effective approach for the generation of AML-APCs [10]. Recently, we showed that AML-APCs cultured with the calcium ionophore A23187 resulted in a significantly more mature phenotype and were significantly more potent stimulators of T cell proliferation compared to cytokine cultured AML-APCs [29]. Therefore, in the present study we focused on the comparison of serum-free and serum-enriched cultures of AML-APCs in the presence of either a cytokine mix or a calcium ionophore.
Our results demonstrate that it is possible to generate comparable numbers of AML-APCs with similar morphology under serum-free conditions compared to serum-enriched conditions. With regard to immunophenotypical characteristics, we showed that AML-APCs generated in serum-free media are comparable to AML-APCs cultured in serum-enriched media. Despite the heterogeneous nature of AML the majority of the AML cells were able to differentiate into AML-APCs as evidenced by the up-regulation of adhesion, activation and co-stimulatory molecules. Considering the differences in percentages of up-regulation of these markers, no major differences were found between AML-APCs cultured in serum-enriched medium and those cultured in serum-free medium. In the CI-culture, the increase in the percentage of CD40 was significantly less in AML-APCs cultured in CellGro medium in comparison with AML-APCs cultured in FCS-containing medium. This led to a significantly lower relative yield and less stimulatory capacity to induce T-cell proliferation. For AML-APCs cultured in StemSpan serum-free medium, these differences could not be detected.
The intensity of marker expression on the cell surface was determined by calculating the mean fluorescence index. Except for the significantly higher MFI of CD83 of cytokine-cultured cells with StemSpan medium in comparison with CellGro medium, no significant differences were found between the MFI of AML-APCs cultured in the three different media. Our preference for the use of StemSpan medium over CellGro medium in the CI-culture is based on the clear trend we observed towards a more mature phenotype of AML-APCs cultured in the presence of CI in StemSpan medium as compared to CellGro medium.
With their high expression levels of co-stimulatory and adhesion molecules and their ability to produce cytokines such as IL-12, dendritic cells are potent inducers of T cell activation [1, 16]. The results of the MLR show that cells cultured in serum-free medium have an equal capacity to induce T cell proliferation compared to cells cultured in serum-enriched medium.
The possibility to culture functionally mature AML-APCs under serum-free conditions has potential therapeutic implications. Since leukaemic antigen presenting cells have shown to be able to generate autologous anti-leukaemia responses, these cells can be used for the development of active specific immunisation protocols which meet the criteria for GCP, thereby providing an effective approach to eliminate minimal residual disease in AML.
References
- 1.Banchereau Nature. 1998;392:245. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 2.Boczkowski Cancer Res. 2000;60:1028. [PubMed] [Google Scholar]
- 3.Bruserud J Hematother. 1999;8:63. doi: 10.1089/106161299320587. [DOI] [PubMed] [Google Scholar]
- 4.Bruserud J Hematother Stem Cell Res. 2000;9:923. doi: 10.1089/152581600750062372. [DOI] [PubMed] [Google Scholar]
- 5.Charbonnier Eur J Immunol. 1999;29:2567. doi: 10.1002/(SICI)1521-4141(199908)29:08<2567::AID-IMMU2567>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 6.Choudhury Blood. 1997;89:1133. [PubMed] [Google Scholar]
- 7.Cignetti Blood. 1999;94:2048. [PubMed] [Google Scholar]
- 8.Hart Blood. 1997;90:3245. [PubMed] [Google Scholar]
- 9.Heiser J Clin Invest. 2002;109:409. doi: 10.1172/JCI200214364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koski Blood. 1999;94:1359. [PubMed] [Google Scholar]
- 11.Koski Eur J Immunol. 2001;31:3773. doi: 10.1002/1521-4141(200112)31:12<3773::AID-IMMU3773>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 12.Kugler Nat Med. 2000;6:332. doi: 10.1038/73193. [DOI] [PubMed] [Google Scholar]
- 13.Löwenberg J Clin Oncol. 1997;15:3496. [Google Scholar]
- 14.Löwenberg N Engl J Med. 1999;341:1051. [Google Scholar]
- 15.Mackensen Cancer Immunol Immunother. 2000;49:152. doi: 10.1007/s002620050614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mellman Cell. 2001;106:255. doi: 10.1016/s0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]
- 17.Nestle Nat Med. 1998;4:328. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 18.Ossenkoppele Leukemia. 2003;98:in press. [Google Scholar]
- 19.Panoskaltsis Leuk Res. 2002;26:191. doi: 10.1016/S0145-2126(01)00104-7. [DOI] [PubMed] [Google Scholar]
- 20.Pardoll Nat Med. 1998;4:525. doi: 10.1038/nm0598supp-525. [DOI] [PubMed] [Google Scholar]
- 21.Porgador J Exp Med. 1995;182:255. doi: 10.1084/jem.182.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramadan Clin Exp Immunol. 2001;125:237. doi: 10.1046/j.1365-2249.2001.01605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Renkvist Cancer Immunol Immunother. 2001;50:3. doi: 10.1007/s002620000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson Br J Haematol. 1998;103:763. [PubMed] [Google Scholar]
- 25.Schuurhuis Bone Marrow Transplant. 2001;27:487. doi: 10.1038/sj.bmt.1702809. [DOI] [PubMed] [Google Scholar]
- 26.Tarte Leukemia. 2000;14:2182. doi: 10.1038/sj.leu.2401925. [DOI] [PubMed] [Google Scholar]
- 27.Waclavicek Br J Haematol. 2001;114:466. doi: 10.1046/j.1365-2141.2001.02970.x. [DOI] [PubMed] [Google Scholar]
- 28.Westers Blood. 2001;98:121a. [Google Scholar]
- 29.Westers TM, Stam AGM, Scheper RJ, Regelink JC, Nieuwint AWM, Schuurhuis GJ, Loosdrecht AA van de, Ossenkoppele GJ (2002) Rapid generation of antigen presenting cells from leukaemic blasts in acute myeloid leukaemia. Cancer Immunol Immunother 52:17 [DOI] [PMC free article] [PubMed]